X-Ray micro-tomography of freeze dried nickel alginate beads and transformation into NiO nanopowders
Zihua
Wang
a,
Girish M.
Kale
*a,
Qingchun
Yuan
b and
Mojtaba
Ghadiri
b
aInstitute for Materials Research, School of Process, Environmental and Materials Engineering, University of Leeds, Leeds, LS2 9JT, UK. E-mail: g.m.kale@leeds.ac.uk; Tel: 0044-113-3432805
bInstitute of Particle Science and Engineering, School of Process, Environmental and Materials Engineering, University of Leeds, Leeds, LS2 9JT, UK.
First published on 16th August 2012
Abstract
A generic ion-exchange mediated sol–gel method has been developed for the production of high purity nickel and other metal oxide nanopowders using sodium alginate precursor. X-Ray micro-tomography (XMT) shows that the nickel ions have been uniformly cross-linked in the alginate structure and remained stable after freeze drying as evidenced by the bright green color of the freeze dried beads. The freeze dried nickel alginate beads exhibit a porous foam-like structure composing of a thin nickel alginate film and a dominant fraction of voids. The porous nickel alginate beads when calcined at 700 °C for 1 h in ambient air yielded spherical nickel oxide nanoparticles of ∼50 nm in size. Powder X-ray diffraction (XRD) confirms that the sample is single phase cubic NiO powders with no trace of impurity. The mean crystallite sizes calculated from XRD analysis using the Rietveld refinement method agree with the morphological features observed by transmission electron microscopy (TEM).
Introduction
Nickel oxide (NiO) is an antiferro-magnetic material with outstanding catalytic and magnetic properties. Due to the chemical and physical properties, NiO nanoparticles have a wide range of applications, such as nickel zinc ferrite (NixZn1−xFe2O4) for magnetic materials,1 nickel sulfamate for electroplating solutions, nickel molybdate for hydro-desulfurization catalysts,2 gold-doped nickel oxide film for transparent electrodes in optoelectronic devices, in cermet anodes for SOFC,3 solar thermal absorber,4 catalyst for O2 evolution,5 gas sensors,6,7 and in lithium-ion batteries.8 Different methods have been reported for the synthesis of NiO nanoparticles, such as solvothermal9 and sol–gel using acetic9 or citric acid.10 The interest in nanoparticles of NiO is mainly due to their high physico-chemical activity in various applications listed above as a result of high surface area and also because of ease with which it can be sintered at low temperatures to high density.11 Nanoparticulate oxide compacts usually have the ability to sinter to high density at much lower temperatures as a result of high surface area and exhibit high fracture toughness.11,12 However, from a commercial perspective, it is important to synthesize nanopowders of NiO in large quantities rapidly, in an environmentally friendly and cost effective manner.Sodium alginate (Na-ALG) is a water soluble linear chain polysaccharide extracted from the cell walls of brown algae. It contains varying amounts of 1,4′-linked β-D-mannuronic acid (M) and α-L-guluronic acid (G) residues covalently linked together in sequence as –GG– or –MM– structures or as –GM– block copolymers. Divalent cations such as Ni2+ can be coordinated into the alginate macromolecules through interaction with the carboxylate groups.13 Since the immobilized nickel cations are widely spaced in the alginate structure they are expected to have limited opportunity to become large crystals during the calcination stage for the formation of metal oxide nanoparticles. Hence it is possible to produce metal oxide nanoparticles through a gelation and calcination process.
In this study, a generic method developed by our research group has been applied for the synthesis of NiO nanopowders by thermal decomposition of ion-exchanged nickel alginate. Three dimensional structures of the nickel alginate have been non-destructively analyzed before and after calcination, at 700 °C for 1 h, using X-ray Micro-Tomography (XMT) in order to obtain an insight into the pore structure of the freeze dried nickel alginate beads. The morphology of the nanoparticles and their crystal structure are characterized by Transmission Electron Microscopy (TEM) and X-ray diffraction (XRD), respectively. Details of the research work and its findings are described in the following sections.
Experimental
Commercial nickel nitrate hexahydrate Ni(NO3)2·6H2O (98+% purity) and sodium alginate powders were purchased from Fisher Scientific Ltd., UK. Nickel alginate beads were prepared by sol–gel method using an ion-exchange process between Ni2+ containing aqueous salt solution and sodium alginate gel. Nickel alginate beads were produced by dropping Na-ALG solution (4 wt%, 200 mL) into nickel salt solution (30 g L−1, 200 mL) through a 16 gauge (1.194 mm inner diameter) stainless steel needle attached to a 20 cm3 hypodermic syringe. The formed jelly beads were maintained in the gelling medium for 2 h under gentle magnetic stirring for the ion-exchange reaction to take place between Na+ and Ni2+. During the ion-exchange process the clear beads turned into uniformly light green color beads due to the Ni2+ ions replacing Na+ ions in the alginate structure. The nickel alginate beads were dried using a freeze dryer (Scanvac Cool Safe 55–9, Denmark). Freeze-drying was achieved in a two step process. The first step was to freeze the samples in a −80 °C freezer. The pre-frozen sample was then placed in the equipment at −52 °C for a period of 48 h until they were completely dried. The freeze dried beads were calcined at 700 °C for 1 h in ambient atmosphere to produce nanoparticles of NiO. The calcined beads tend to retain their original bead like physical shape and structure even after calcination at elevated temperature.The physical structure of nickel alginate beads before and after calcination at 700 °C for 1 h was non-destructively determined using X-ray Micro-Tomography (XMT). A Phoenix X-ray Nanotom 160NF (GE Phoenix X-ray, Germany) was used for the X-ray attenuation scan. A constant cone beam of X-ray was generated by a tungsten filament at ∼80 kV and 80 μA. The scan was carried out with a rotational sample stage, located between the X-ray source and a detector (2304 × 2304 pixel), at resolutions of 1∼3 micron per pixel and a rotation step of 0.25 degree. Projective attenuation images of 1440 were thus obtained for each scan and reconstructed using the reconstruction software of Phoenix for digital volume structures. The volume data were visualized and analyzed using VGStudio (v2.1.1).
The calcined beads were broken into fine powders and, the particle size and morphological features of the NiO nanopowders were observed by Transmission Electron Microscopy (FEI Tecnai TF20 FEG-TEM). Finally, the crystal structure and phase form of the nanopowder NiO sample was analyzed using X-ray diffraction (XRD, P'Analytical X'Pert MPD, Netherlands) employing Cu-Kα radiation. A Rietveld size/strain structural refinement was performed using the resultant XRD data (X'Pert HighScore Plus, P'Analytical, Netherlands) to determine the crystallite size, lattice parameter and micro-strain in the NiO nanopowders. Details of the Rietveld refinement method using this software package can be found elsewhere.14–19
Results and discussion
Fig. 1 shows ion-exchanged nickel alginate beads in the wet state and after freeze drying. The wet beads appear as spherical light green in color having a diameter of ∼5 mm. It can be seen that the beads changed their color to bright green, characteristic color of nickel compounds, and nearly maintained their shape and size after freeze drying. The freeze dried nickel alginate beads were then calcined at 700 °C for 1 h in ambient air giving rise to black spherical beads with sizes in the range of ∼2 mm. The calcined samples are very fragile due to high degree of porosity. | ||
| Fig. 1 Nickel alginate wet beads (left) and freeze dried beads (right). | ||
Freeze dried nickel alginate beads and calcined samples were individually scanned using XMT. Fig. 2 and 3 show their reconstructed 3D images from outside, inside, transverse and cross sections, respectively. It can be seen that both the freeze dried and calcined samples have foam-like structures composed of a dominant proportion of voids and thin films as the wall of the foam structure. The outside walls have out-pointed wrinkles, that have very similar thickness to the adjacent wall. These wrinkle structures formed during freeze drying remained after calcinations (comparing Fig. 2a with Fig. 3a and 3e). The inside walls in the freeze dried sample are relatively thinner than its outside wall, in the range of ∼20 μm and ∼30 μm, respectively, and occasionally shows smaller wrinkles at the side towards the outside. Both the outside and inside films showed good continuity at the scale examined.
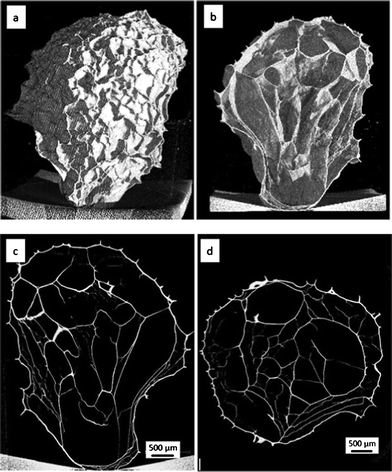 | ||
| Fig. 2 XMT images of the freeze dried nickel alginate bead: (a and b) 3D structure from outside and inside, (c and d) transverse and cross sections, respectively. | ||
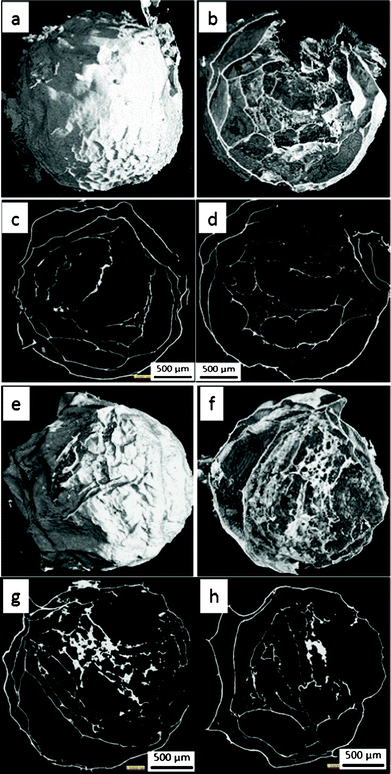 | ||
| Fig. 3 XMT images of two calcined beads: (a, b, e and f) 3D structure from outside and inside, (c, d, g and h) transverse and cross sections, respectively. | ||
The calcined bead samples show similar structures to their precursors, although they are obviously smaller in size (being shrunk from ∼5 mm to ∼2 mm). Their outside walls are ∼20 μm thick, generally thinner than that of the freeze dried sample, while their inside walls vary from less than 5 μm to more than 25 μm in thickness. The formation of the thicker inside wall could suggest a certain degree of aggregation during the fast calcination process due to the coalition of pores formed in the film (Fig. 3b and 3f). The aggregation and pore formation results in a lower continuity of the calcined inside film than the frozen analogue.
Fig. 4 presents the volume distribution of the freeze dried and calcined samples at different linear attenuation coefficients. The X-ray attenuation is a function of product of the linear attenuation coefficient μ cm−1 of the compound and the voxel length (x), following the Beer's law (eqn 1).
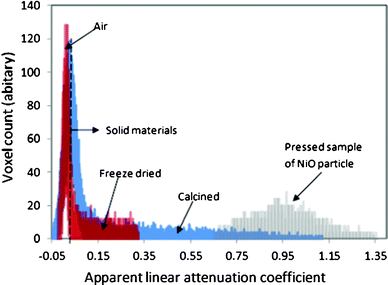 | ||
| Fig. 4 Comparison of the grey scale histogram of X-ray attenuation of the freeze dried and calcined samples. | ||
| I = I0e−μx | (1) |
For the highly porous materials such as samples studied here, a low value close to zero refers to empty space with air, and a higher apparent linear attenuation coefficient refers to the wall materials composed of components with solid materials, a larger population of nickel atoms or a larger proportion of the voxel packed by the nickel compounds.
Fig. 4 also shows that the freeze dried and calcined structures are mostly composed of air, and only a small fraction of the total volume are composed of solid wall materials. The apparent linear attenuation coefficient spreads in the range of 0.05–0.43 for the freeze dried sample and 0.05–1.27 for the calcined sample. The figure also gives the apparent linear attenuation coefficient of a sample pressed from pure NiO nanopowder. It is in the range of 0.65–1.43 with a peak at 0.95.
The distribution of the apparent linear attenuation coefficient demonstrates the effect of nickel concentration and spatial structure of the highly porous samples. The freeze dried sample contains ∼14% of nickel, while the calcined sample contains ∼78% of nickel. It explains that the highest apparent linear attenuation coefficient for the freeze dried sample is less than 0.34, because the linear attenuation coefficient of nickel is a few thousand times that of carbon, hydrogen and oxygen. The contribution of alginate to the apparent attenuation coefficient is relatively small. In contrast, the apparent linear attenuation coefficient of the calcined sample extends to 1.43.
The thin film structure results in the wider distribution of the histogram, because the voxel size (2 or 3 μm for the scan, 4 or 6 μm for the reconstructed structures is large compared to the thickness of the films (5–30 μm). It can be imagined that the large amount of voxels at the film surface are partially packed by the film and partially packed by the empty space, resulting in lower and scattered values of apparent linear attenuation coefficient. For the calcined sample, the apparent linear attenuation coefficient occurs in a wide range, but only a very small proportion reached the range of that for the compressed NiO sample (0.65–1.47), suggesting a possibility of the existence of free fissures between the nanoparticles in the size range of ∼50 nm (Fig. 5). The free space makes the calcined foam very fragile and easy to pulverize. Both structural and X-ray attenuation analysis clearly suggest that the foam-like structure and well separated thin film walls of the nickel alginate beads are beneficial for the formation of nickel oxide nanoparticles in a nanostructured dimension during the calcination stage.
 | ||
| Fig. 5 TEM image of NiO nanoparticles prepared by the calcinations of freeze dried nickel alginate beads at 700 °C. | ||
The morphology of the NiO nanopowders obtained by breaking the calcined sample has been investigated using TEM. As shown in Fig. 5, the nanoparticles have cubic morphology and are approximately ∼50 nm. This is in good agreement with that calculated using XRD Rietveld refinements shown in Fig. 6 and 7.
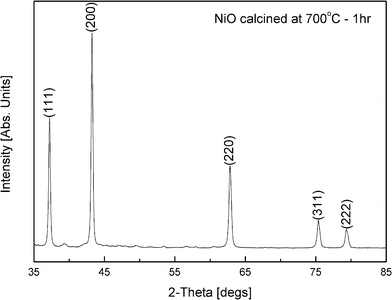 | ||
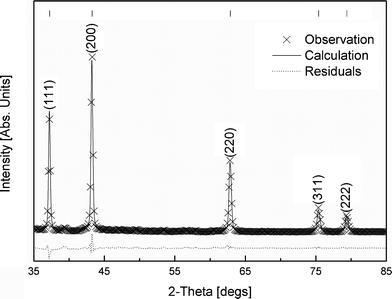
| Fig. 6 XRD of NiO nanopowders calcined at 700 °C for 1 h. The pattern is indexed using the (hkl) values from ICDD 04-002-0665 shown at the top of the peaks. | ||
 | ||
| Fig. 7 Size/strain Rietveld structural refinement analysis of NiO sample calcined at 700 °C–1 h. Tick marks for reference pattern of NiO, ICDD 04-002-0665, are shown at the top of the figure. | ||
Fig. 6 shows the XRD diffraction pattern of the NiO nanoparticles formed during the calcination of nickel alginate freeze dried beads at 700 °C for 1 h. It shows a perfect single phase cubic NiO powders. The XRD pattern could be entirely indexed with the reference pattern (ICDD 04-002-0665) available in the literature.
Fig. 7 shows a comparison between the calculated and the observed XRD patterns obtained experimentally of the nanopowders of NiO employing a Rietveld size/strain structural refinement. It is clearly seen from the figure that the agreement between the experimental data and the calculated XRD pattern is excellent (Rexp ≈ 0.50%, Rwp ≈ 2.18%) showing negligible residual pattern. This suggests that the nanoparticles of NiO are free of any strain. The mean crystallite size calculated from XRD Rietveld refinement is 49.1 ± 4 nm which is in excellent agreement with the average size of ∼50 nm obtained from TEM analysis (Fig. 5). The lattice parameter and d-spacing values for various crystal planes obtained from XRD Rietveld refinement analysis are compared with the ICDD 04-002-0665 reference data in Table 1. The results obtained in the present investigation are in good agreement with the reference data.
| h | k | l | Rietveld Refinement | ICDD 04-002-0665 |
|---|---|---|---|---|
| 1 | 1 | 1 | 2.42 | 2.41 |
| 0 | 0 | 2 | 2.09 | 2.09 |
| 0 | 2 | 2 | 1.48 | 1.48 |
| 1 | 1 | 3 | 1.26 | 1.26 |
| 2 | 2 | 2 | 1.21 | 1.21 |
| Lattice parameter a/Å | 4.1794 | 4.1780 | ||
Conclusions
In this paper, we have reported that it is possible to produce single cubic phase nickel oxide (NiO) nanoparticles using sodium alginate under relatively simple conditions. In this sol–gel production method, homogeneous distribution of metal ions and slow collapse of the carbohydrate structure during calcination prevents the rapid agglomeration of metal ions, which ensures small particle size of the product and high purity single phase material is formed at temperatures as low as 700 °C.The insight into the structures of nickel alginate freeze dried beads before and after calcination is obtained using XMT. The XRD data indicates that single phase cubic NiO is formed after being calcined at 700 °C for 1 h, confirming that the phase formation is complete. The crystalline size determined by Rietveld structural refinement is ∼50 nm which shows excellent agreement with data obtained from TEM images.
The ability of sodium alginate to absorb metal ions, such as Fe, Co, Ni and Cu, can be considered as a low cost, simple, environmentally friendly and non-toxic route for a large scale production of high purity single phase nanopowders at significantly low temperatures for other technical applications.
Acknowledgements
Zihua Wang wishes to thank IMR for partial financial aid. Authors wish to thank IMR, IPSE and SPEME for providing research facilities and infrastructure. The authors wish to thank Dr Ebun-Oluwa Oladele for assistance with the freeze drying experiment.References
- M. Atif, M. Nadeem, R. Grössinger and R. S. Turtelli, J. Alloys Compd., 2011, 509, 5720 CrossRef CAS
.
- N. V. Krstajić, Lj. Gajić-Krstajić, U. Lačnjevac, B. M. Jović, S. Mora and V. D. Jocić, Int. J. Hydrogen Energy, 2011, 36, 6441 CrossRef
- Z. W. Zhu and S. C. Deevi, Mater. Sci. Eng. A, 2003, 36, 228 CrossRef
- J. G. Cook and F. P. Koffyberg, Sol. Energy Mater., 1984, 10, 55 CrossRef CAS
- J. C. B. Nadesan and A. C. C. Tseung, J. Electrochem. Soc., 1985, 132, 2957 CrossRef CAS
- W. Z. Xiong and G. M. Kale, Anal. Chem., 2007, 79, 3561 CrossRef CAS PubMed
- S. Poomiapiradee, R. M. D. Brydson and G. M. Kale, Ceram. Trans., 2002, 130, 79 CAS
- C. R. H. Bahl and S. Morup, Nanotechnology, 2006, 17, 2835 CrossRef CAS
- S. Das, T. Ghoshal and P. M. G. Nambissan, Phys. Status Solidi C, 2009, 6, 2569 CrossRef CAS
- K. Hayat, M. A. Gondal, M. M. Khaled and S. Ahmed, J. Mol. Catal. A: Chem., 2011, 336, 64 CrossRef CAS
- H. Y. Lee and G. M. Kale, Int. J. Appl. Ceram. Technol., 2008, 5, 657 CrossRef CAS
- Q. Zhen, G. M. Kale, W. M. He and J. Q. Liu, Chem. Mater., 2007, 19, 203 CrossRef CAS
- Q. A. Zhen, G. M. Kale, G. Shi, R. Li, W. M. He and J. Q. Liu, Solid State Ionics, 2005, 176, 2727 CrossRef CAS
- W. R. Gombotz and S. F. Wee, Adv. Drug Delivery Rev., 1998, 31, 267 CrossRef CAS
- Z. H. Wang, T. P. Comyn, M. Ghadiri and G. M. Kale, J. Mater. Chem., 2011, 21, 16494 RSC
- Z. H. Wang, G. M. Kale and M. Ghadiri, J. Am. Ceram. Soc. DOI:10.1111/j.1551-2916.2012.05303
- Z. H. Wang, G. M. Kale and M. Ghadiri, Chem. Eng. J., 2012, 198–199, 149 CrossRef CAS
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS
-
R. A. Young, The Rietveld Method, Oxford University Press Inc., New York, 1993 Search PubMed
| This journal is © The Royal Society of Chemistry 2012 |