Fluorescence properties of polyaryletherketone rare earth-coordinated complexes in near-infrared region†
Dan
Liu
ab and
Zhonggang
Wang
*a
aState Key Laboratory of Fine Chemicals, Department of Polymer Science and Materials, School of Chemical Engineering. Dalian University of Technology, Dalian 116024, China. E-mail: zgwang@dlut.edu.cn
bSchool of Biochemistry and Chemical Engineering, Jiaxing University, Jiaxing, China
First published on 29th August 2012
Abstract
Novel rare earth-coordinated polymers were prepared using carboxyl-containing polyaryletherketone (PEK) as the macromolecular ligand and 8-hydroxyquinoline (HQ) as the co-ligand to coordinate with erbium (Er3+), neodymium (Nd3+) and ytterbium (Yb3+), respectively. The FTIR, elemental analysis, inductively coupled plasma, and UV-vis results confirm that the rare earth ions have been attached to the polymer backbone via a coordination bond. The complexes can emit the characteristic near-infrared fluorescence of the corresponding rare earth ions. Moreover, it is found that the synergistic effect of PEK and HQ can significantly enhance the emission intensities. In particular, for PEK-Er3+-HQ, the full width at half maximum (FWHM) of the 4I13/2→4I15/2 transition is up to 78 nm. The combination of strong fluorescence emission in the near-infrared range, high glass transition temperature and wide gain bandwidth provides promising potential for optical amplification applications.
Introduction
Rare earth polymeric complexes are of great value in the materials science field because of their high luminous intensity, long luminous lifetime and extremely sharp emission bands arising from the 4f electronic transitions of rare earth ions. In the past decades, however, studies in this area were mainly focused on the visible range (400–800 nm) for optical display devices, whereas less attention has been paid to the fluorescence materials in the near-infrared range (800–2000 nm).1–5 Only recently, the importance of rare earth complexes containing erbium (Er3+), neodymium (Nd3+) and ytterbium (Yb3+) ions is being recognized, owing to their increasing potentials in optical amplification (1500 nm emission of Er3+ complex), laser systems (1060 nm emission of Nd3+ complex), and medical diagnosis (980 nm emission of Yb3+ complex).6–13 A review of the literature reveals that most of the above materials are prepared by doping the rare earth ions into inorganic matrixes such as sol–gel glass,14–18 but organic polymeric rare earth complexes with fluorescence emitting in the near-infrared region are seldom reported.Compared to inorganic materials, organic polymers have some intrinsic advantages, such as flexibility, excellent processability and convenient control of polymer properties by varying the molecular structures. Nevertheless, rare earth-doped polymers prepared by the simple mixing of rare earth complexes with a polymer matrix often give rise to serious fluorescence quenching due to the ionic aggregates, even at low rare earth ion content.19,25 It is desirable that rare earth ions are coordinated with some polymer functional groups. Thus, rare earth ions attached to the polymer backbone via a coordination bond can be well dispersed within the polymer matrix at the molecular level.
Polyaryletherketone (PEK), simultaneously bearing carboxyl and alkyl substitutent groups on the phenylene ring, is a new polymer recently developed in our group.26 The aromatic backbone and bulky alkyl substituents on the phenylene rings result in the high glass transition temperatures (over 210 °C), excellent heat-resistance and high mechanical strength. Meanwhile, the carboxyl groups in the PEK chain provide the sites to coordinate with the rare earth ions, which make it very suitable for utilization as a macromolecular ligand. In the present work, using PEK as a macromolecular ligand and HQ as a synergistic ligand, rare earth coordination polymers (PEK-RE3+-HQs, RE3+ = Er3+, Nd3+, Yb3+) are prepared and characterized. Their near-infrared fluorescence properties in the near-infrared region are investigated in detail.
Experimental
Materials
Rare earth nitrate hexahydrates (RE(NO3)3·6H2O, RE3+ = Er3+, Nd3+, Yb3+) and 8-hydroxyquinoline (HQ) were purchased from Shandong Qingda Fine Chemical Factory and Shenyang Chemical Reagent Factory, respectively, and used without further purification. N,N-Dimethylformamide (DMF) was dried and distilled under reduced pressure just before use. Other chemical reagents were of analytical reagent grade and used as received.The film samples of PEK-RE3+-HQs for FTIR, elemental analysis, XRD and TGA measurements were obtained from their DMF solutions, which were cast onto a clean glass plate at 60 °C. After 8 h, the films were transferred to vacuum oven and dried further at 150 °C for 24 h to constant weight.
The powder samples of PEK-RE3+-HQs and PEK-RE3+s for the UV-Vis absorption measurements were also prepared from their DMF solutions, which were slowly poured into ethanol under constant stirring. After filtration, the powder products were dried at 80 °C under vacuum to constant weight before measurements. The powder samples of RE3+-HQs for the FTIR and XRD measurements were also obtained from their DMF solutions by the above method.
Instruments
Fourier transform infrared spectra (FTIR) were recorded on a Nicolet 20XB FT-IR spectrophotometer using KBr pellets at room temperature.Elemental analyses were performed on an Elementar Vario EL III elemental analyzer. The rare earth ions content was determined by an optimer 2000dv inductively coupled plasma (ICP).
The ultraviolet-visible (UV-Vis) absorption spectra were measured by a UV-550 spectrophotometer at room temperature.
X-ray diffraction (XRD) measurements were recorded on a D/Max-2400 X-ray diffractometer with Cu-Kα1 radiation (λ = 1.54 Å) over the 2θ range 4 to 60°.
Differential scanning calorimetries (DSC) were run on a NETZSCH DSC204 in the temperature range 50–350 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere. The glass transition temperatures (Tg) were read at the middle of the change in heat capacity.
The fluorescence excitation and emission spectra of rare earth coordination polymers were measured on a FS 920 steady-state fluorescence spectrofluorometer with a scanning rate of 300 nm min−1 at room temperature. The slit widths for both excitation and emission were set as 15 nm, and the concentrations of DMF solutions of rare earth coordination polymers were 1 × 10−3 mol L−1.
Results and discussion
Synthesis and characterization of rare earth-coordinated polymers
As shown in Scheme 1, a series of novel rare earth-coordinated polymers (PEK-RE3+-HQs, RE3+ = Er3+, Nd3+, Yb3+) were synthesized in DMF solution through a co-coordination reaction with carboxyl-containing polyaryletherketone (PEK) and 8-hydroxyquinoline (HQ). Their structures were characterized by the FTIR spectra, elemental analysis, inductively coupled plasma, UV-Vis absorption spectra and XRD methods.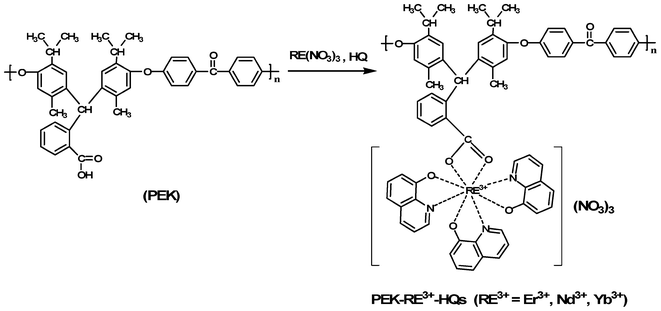 | ||
| Scheme 1 Synthesis of rare earth coordination polymers (PEK-RE3+-HQs). | ||
The FTIR spectra of HQ, Er3+-HQ, PEK and PEK-Er3+-HQ are illustrated in Fig. 1. For HQ, the characteristic absorption corresponding to the C–O stretching vibration is located at 1093 cm−1. After coordination, the spectra of Er3+-HQ and PEK-Er3+-HQ show that the band at 1093 cm−1 shifts to 1107 cm−1, and the absorptions due to the Er3+-O and Er3+-N stretching vibrations appear at 490 cm−1 and 818 cm−1, respectively, suggesting that HQ is coordinated to the Er3+ ion.27,28 Moreover, the comparison between PEK and PEK-Er3+-HQ reveals that the characteristic carboxyl absorption for PEK at 1725 and 1696 cm−1 completely disappears in the spectrum of PEK-Er3+-HQ. Instead, a new peak at about 1384 cm−1 due to the symmetric stretching vibration (COO−) appears, indicative of the formation of a coordination bond between the Er3+ ion and the carboxyl group in the PEK backbone. In addition, in the FTIR spectra of PEK-Er3+-HQ and Er3+-HQ, no adsorption at 460 cm−1 is observed, suggesting that the solvent DMF molecules do not participate in coordination with the Er3+ ion.29 The reason can be attributed to the fact that the carboxyl groups of PEK and HQ have strong coordination ability, which can fully exclude coordination of the solvent molecules (DMF) to the Er3+ ion. The results of elemental analysis of PEK-Er3+-HQ agree well with the calculated ones (Table 1).
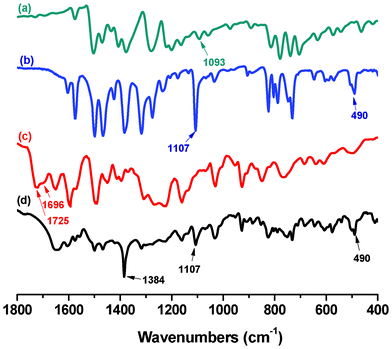 | ||
| Fig. 1 FTIR spectra of (a) HQ, (b) Er3+-HQ, (c) PEK, (d) PEK-Er3+-HQ. | ||
| Element | Found (%) | Calc. (%) |
|---|---|---|
| C | 58.10 | 58.17 |
| H | 4.63 | 4.01 |
| N | 5.86 | 6.07 |
| Er | 12.18 | 12.09 |
PEK can dissolve easily in N,N-dimethylformamide (DMF), N,N-dimethlyacetamide (DMAc), dimethyl sulfoxide (DMSO), N-methylpyrrolidone (NMP), tetrahydrofuran (THF) and acetone at room temperature. Relative to the pure PEK, the three rare earth complexes exhibit obviously decreased solubility due to enhancement of the interaction between the polymer chain and the rare earth ion, which makes the diffusion of solvent molecules between the polymer chains more difficult. Nevertheless, PEK-RE3+-HQs can still be soluble in DMF, DMAc, DMSO and NMP at room temperature. More importantly, the films of PEK-RE3+-HQs can be obtained from their solutions and the thickness of the films can be controlled by changing the concentration of the solutions to meet the requirement of practical applications.
The XRD patterns of PEK-Er3+-HQ, Er3+-HQ and PEK are compared in Fig. 2. Er3+-HQ displays some sharp crystalline peaks, while PEK exhibits a wide hump, indicative of its amorphous structure. However, PEK-Er3+-HQ only shows an amorphous hump similar to PEK, and the sharp peaks corresponding to Er3+-HQ cannot be detected, suggesting that, for PEK-Er3+-HQ, the rare earth ions are well dispersed in the polymer matrix. The other two polymer complexes also give similar results.
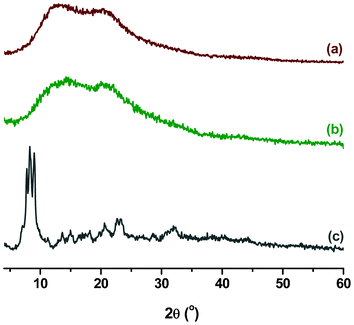 | ||
| Fig. 2 XRD patterns of (a) PEK, (b) PEK-Er3+-HQ, and (c) Er3+-HQ. | ||
The DSC measurements for both PEK and PEK-Er3+-HQ were conducted at a heating rate of 10 °C min−1 in a N2 atmosphere. As illustrated in Fig. 1S (ESI†), PEK shows quite a high glass transition temperature (Tg) at 230 °C because of its rigid aromatic backbone. After coordination with the rare earth ion, for PEK-3-Er3+-HQ, the former glass transition disappears, and no obvious Tg can be detected in the whole measurement temperature range. The possible reason may be due to the enhanced interchain interaction caused by the introduction of rare earth ions, leading to a significantly increased Tg beyond the measurement range. The high glass transition temperature is especially favorable for optical amplification application of PEK-Er3+-HQ.
UV-Vis absorptions and near-infrared emissions of rare earth-coordinated polymers
The UV-Vis absorption spectra of PEK and PEK-Er3+-HQ are presented in Fig. 3. Relative to PEK, the absorption bands of PEK-Er3+-HQ are apparently red-shifted, which is caused by the decrease in electron density around PEK due to the coordination bond between the Er3+ ion and the oxygen atom of the carboxyl group in PEK. The weak peaks at 488, 522 and 653 nm are attributed to the 4I15/2→4F7/2, 4I15/2→2H11/2 and 4I15/2→4F9/2 transitions of Er3+ ions, respectively.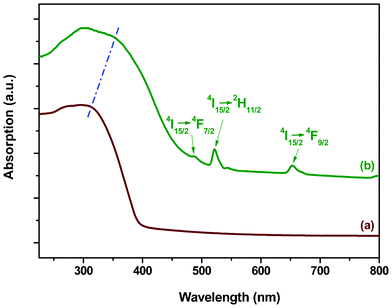 | ||
| Fig. 3 UV-Vis absorption spectra of (a) PEK and (b) PEK-Er3+-HQ. | ||
The emission spectrum of PEK-Er3+-HQ is shown in Fig. 4. Under excitation at 392 nm, a broad emission band at 1527 nm, attributed to the typical 4I13/2→4I15/2 transition of the Er3+ ion appears, which is just located in the desired wavelength range for optical-fiber telecommunication. Moreover, the full width at half maximum (FWHM) of PEK-Er3+-HQ is up to 78 nm, much wider than other Er3+-containing materials, such as Er3+-doped phosphate glass (40 nm).30 The above reason can be attributed to the structure of PEK, which simultaneously contains multiple alkyl substituents and a bulky side group. As a result, the resultant PEK-Er3+-HQ backbone becomes seriously twisted, and the chain packing is very disordered. The previous study has revealed that the electron transition of a rare earth ion is very sensitive to the coordination environment.31 The disordered coordination environment of the Er3+ ion can disturb the energy transfer of the ligands, and thus lead to the broadened FWHM of PEK-Er3+-HQ. The wide gain bandwidth is desirable for optical amplification applications.
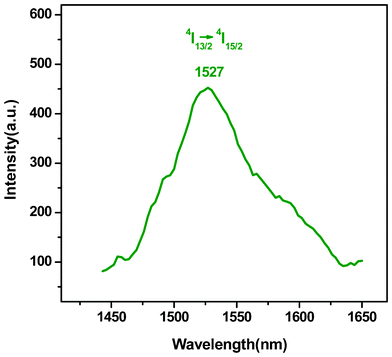 | ||
| Fig. 4 Emission spectrum of PEK-Er3+-HQ. | ||
The replacement of Er3+ with Nd3+ (Fig. 5) makes the emission bands of PEK-Nd3+-HQ shift toward the shorter wavelengths appearing at 898, 1060 and 1328 nm, corresponding to the 4F3/2→4I9/2, 4F3/2→4I11/2 and 4F3/2→4I13/2 transitions, respectively. Among them, the emission intensity at 1060 nm due to the 4F3/2→4I11/2 transition is the strongest, suggesting that PEK-Nd3+-HQ is suitable for the laser materials corresponding to the 1060 nm wavelength.6–13
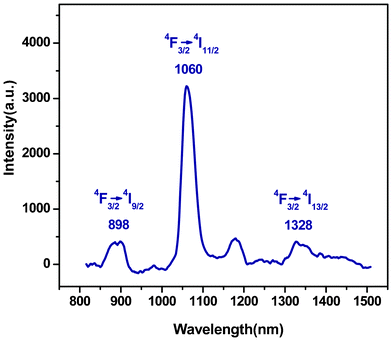 | ||
| Fig. 5 Emission spectrum of PEK-Nd3+-HQ. | ||
The excitation spectra of PEK-Yb3+-HQ, PEK-Yb3+ and Yb3+-HQ, by monitoring the emission of the Yb3+ ions at 980 nm, are shown in Fig. 6. The inset is the enlarged view of the excitation spectrum of PEK-Yb3+, in which the broad band centered at 350 nm was attributed to the π→π* electronic transition of PEK, and the sharp peak at 490 nm was associated to the 4f–4f transitions of the Yb3+ ion. The excitation intensity of the former is higher than that of the latter, implying that the fluorescence emission of the PEK-Yb3+ is mainly sensitized by the absorption of the organic ligand rather than that of Yb3+ ion. This deduction can be further proved by the excitation spectra of PEK-Yb3+-HQ and Yb3+-HQ (Fig. 6a and Fig. 6b), in which the excitation peaks corresponding to the 4f–4f transitions of Yb3+ ion can hardly be observed. The maximum excitation wavelengths of PEK-Yb3+-HQ and Yb3+-HQ are located at 392 and 388 nm, respectively. On the other hand, PEK-Yb3+-HQ displays a stronger and broader excitation band than PEK-Yb3+ and Yb3+-HQ. The reason can be due to the fact that, relative to PEK-Yb3+ and Yb3+-HQ, PEK-Yb3+-HQ has the greater π-electronic system, owing to the simultaneous introduction of macromolecular ligand PEK and small molecule HQ, which is favorable for the absorption of the ultraviolet light.
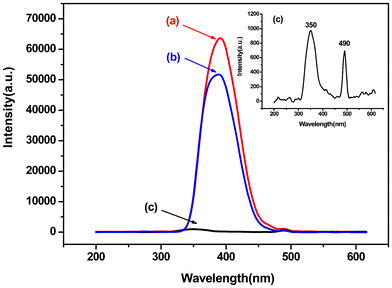 | ||
| Fig. 6 Excitation spectra of (a) PEK-Yb3+-HQ, (b) Yb3+-HQ, and (c) PEK-Yb3+. | ||
In order to further investigate the synergistic effect of PEK and HQ on the fluorescence properties of polymeric rare earth complexes, the comparative emission spectra of PEK-Yb3+-HQ, PEK-Yb3+ and Yb3+-HQ are measured and illustrated in Fig. 7. The inset presents the enlarged emission spectrum of PEK-Yb3+. It can be seen that all the complexes emit the characteristic fluorescence of the Yb3+ ion at about 980 nm, corresponding to the 4F5/2→2F7/2 transition, but their emission intensities are different. Among them, the emission intensity of PEK-Yb3+ is the lowest, due to the lack of effective energy transfer within the complex system. In contrast, the simultaneous coordination of PEK and HQ to the Yb3+ ion leads to the PEK-Yb3+-HQ sample having the strongest fluorescence. The reason is attributed to the fact that, firstly, the coordination numbers of the Yb3+ ions in the PEK-Yb3+-HQ system can be much more easily satisfied than in either the PEK-Yb3+ or the Yb3+-HQ system and, secondly, the macromolecular ligand PEK is quite rigid, which makes the coordination structure of PEK-Yb3+-HQ more stable, thereby effectively inhibiting the non-radiative transition of rare earth ions.
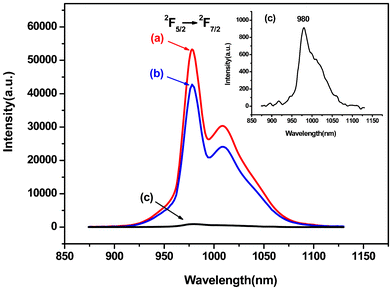 | ||
| Fig. 7 Emission spectra of (a) PEK-Yb3+-HQ, (b) Yb3+-HQ, and (c) PEK-Yb3+. | ||
Conclusions
A series of novel rare earth coordination polymers (PEK-RE3+-HQs, RE3+ = Er3+, Nd3+, Yb3+) with near-infrared fluorescence emission were successfully prepared, in which the RE3+ ions were attached onto the polymer backbone via coordination bonds, and surrounded by both PEK and low molecular weight ligand HQ, and thus distributed homogeneously in the polymer matrix. The rare earth coordination polymers could dissolve in the common solvents at room temperature and were easily cast into uniform and smooth films, which is important in future practical applications. PEK-Er3+-HQ, PEK-Nd3+-HQ and PEK-Yb3+-HQ emit intense fluorescence in the near-infrared region and the strongest emission peaks are located at 1527, 1060 and 978 nm, respectively. The synergistic effects of PEK and HQ could significantly improve their emission intensities. These rare earth coordination polymers with excellent near-infrared fluorescence properties and good film-forming properties showed potential applications in the optical communication, laser and bio-analysis fields.Acknowledgements
The Program for New Century Excellent Talents in University of China (Grant No. NCET-06-0280), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (Grant No. 2005-546), and the Supporting Program for Optoelectronic Materials of Dalian University of Technology is gratefully acknowledged. We are grateful to Prof. Chunying Duan for the fluorescence measurements.References
- M. Seitz, E. G. Moore, A. J. Ingram, G. Muller and K. N. Raymond, J. Am. Chem. Soc., 2007, 29, 15468 CrossRef.
- E. Stathatos, P. Lianos, E. Evgeniou and A. D. Keramidas, Synth. Met., 2003, 139, 433 CrossRef CAS.
- I. Georgieva, N. Trendafilova, A. J. A. Aquino and H. Lischka, Inorg. Chem., 2007, 46, 10926 CrossRef CAS.
- Y. S. Song, B. Yan and Z. X. Chen, J. Solid State Chem., 2004, 177, 3805 CrossRef CAS.
- S. V. Eliseeva, O. V. Kotova, F. Gumy, S. N. Semenov, V. G. Kessler, L. S. Lepnev, J. C. G. Bunzli and N. P. Kuzmina, J. Phys. Chem. A, 2008, 112, 3614 CrossRef CAS.
- L. N. Sun, W. P. Mai, S. Dang, Y. N. Qiu, W. Deng, L. Y. Shi, W. Yan and H. J. Zhang, J. Mater. Chem., 2012, 22, 5121 RSC.
- L. N. Sun, S. Dang, J. B. Yu, J. Feng, L. Y. Shi and H. J. Zhang, J. Phys. Chem. B, 2010, 114, 16393 CrossRef CAS.
- A. F. Silva, D. S. Moura, A. S. Gouveia-Neto, E.A. Silva, L. A. Bueno, E. B. Costa and E. N. Azevedo, Opt. Commun., 2011, 284, 4501 CrossRef.
- G. S. Yi, Y. F. Peng and Z. Q. Gao, Chem. Mater., 2011, 23, 2729 CrossRef CAS.
- L. Bertolo, S. Tamburini, P. A. Vigato, W. Porzio, G. Macchi and F. Meinardi, Eur. J. Inorg. Chem., 2006, 12, 2370 CrossRef.
- C. Gao, K. Cui, J. B. She, C. Q. Hou, H. Y. Guo and W. Zhao, Inorg. Chim. Acta, 2009, 362, 2001 CrossRef CAS.
- S. Comby, D. Imbert, A. S. Chauvin and J. C. G. Bü1nzli, Inorg. Chem., 2006, 45, 732 CrossRef CAS.
- G. A. Kumar, R.E. Riman, L. A. D. Torres, O. B. Garcia, S. Banerjee, A. Kornienko and J. G. Brennan, Chem. Mater., 2005, 17, 5130 CrossRef CAS.
- B. H. Tong, S. J. Wang, Y. Z. Meng and B. Wang B, Photochem. Photobiol. Sci., 2007, 6, 519 CAS.
- P. Lenaerts, E. Ryckebosch, K. Driesen, V. R. Deun, P. Nockemann P and C. Görller-Walrand, J. Lumin., 2005, 114, 77 CrossRef CAS.
- L. N. Sun, H. J. Zhang, J. B. Yu, S. Y. Yu, C. Y. Peng and S. Dang, Langmuir, 2008, 24, 5500 CrossRef CAS.
- V. Prajzler, I. Hüttel, O. Lyutakov, J. Oswald, V. Machovic and V. Jerabek, Polym. Eng. Sci., 2009, 49, 1814 CAS.
- B. Chen, J. Xu, N. Dong, H. Liang, Q. J. Zhang and M. Yin, Spectrochim. Acta, Part A, 2004, 60, 3113 CrossRef.
- N. V. Petrochenkova, A. G. Mirochnik and V. E. Karasev, Koord. Khim., 1991, 17, 1567 CAS.
- A. G. Mirochnik, N. V. Petrochenkova and V. E. Karasev, Russ. Chem. Bull., 1997, 46, 2135 CrossRef CAS.
- P. Lenaerts, K. Driesen, R. Van Deun and K. Binnemans, Chem. Mater., 2005, 17, 2148 CrossRef CAS.
- L. H. Slooff, A. Polman and F. Caciallib, Appl. Phys. Lett., 2001, 78, 2122 CrossRef CAS.
- L. H. Slooff, A. Blaaderen and A. Polman, Appl. Phys. Lett., 2000, 77, 1253 CrossRef.
- M. J. A. Dood, L. H. Slooffa and A. Polman, Appl. Phys. Lett., 2001, 79, 3585 CrossRef.
- R. Pogreb, O. Popov, V. Lirtsman, O. Pyshkin and A. Kazachkov, Polym. Adv. Technol., 2006, 17, 20 CrossRef CAS.
- D. Liu and Z. G. Wang, Polymer, 2008, 49, 4960 CrossRef CAS.
- Q. G. Li, X. Li, Y. Huang, S. X. Xiao, D. J. Yang, L. J. Ye, D. L. Wei and Y. Liu, Thermochim. Acta, 2006, 441, 195 CrossRef CAS.
- J. T. Wang and X. X. Zhou, Chin. J. Inorg. Chem., 1989, 5, 102 CAS.
- Q. L. Suo, F. Lu, J. W. Shi, H. L. Hong and J. P. Luo, J. Rare Earths, 2009, 27, 28 CrossRef.
- Q. Y. Jiang, M. J. Chen, Y. M. Hu, J. Shen and G. Q. Zhong, Chin. Chem. Res. Appl., 2008, 20, 58 CAS.
- M. Yu, J. Lin, Z. Wang, J. Fu, S. Wang, H. J. Zhang and Y. C. Han, Chem. Mater., 2002, 14, 2224 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21336b |
| This journal is © The Royal Society of Chemistry 2012 |