Nickel-complexes with a mixed-donor ligand for photocatalytic hydrogen evolution from aqueous solutions under visible light†
Jianyu
Han‡
ab,
Wei
Zhang‡
a,
Tianhua
Zhou
a,
Xin
Wang
a and
Rong
Xu
*a
aSchool of Chemical & Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459. E-mail: rxu@ntu.edu.sg; Fax: 65-67947553; Tel: 65-67906713
bEnergy Research Institute @ NTU, Nanyang Technological University, 50 Nanyang Drive, Singapore 637553
First published on 24th July 2012
Abstract
Increasing interest has been paid to the development of earth-abundant photocatalyst systems for production of hydrogen from water. In this work, we, for the first time, investigated the photocatalytic activities of complexes formed between β-mecaptoethylamine (L) and Ni(II), NiL2, and the trinuclear complex, Ni(NiL2)22+, with a mixed S and N donor environment. Both complexes were found to be active catalysts for hydrogen evolution in aqueous solutions. In particular, a high quantum efficiency of 30.9 % at 480 nm was obtained over NiL2 when Erythrosin yellowish was used as the photosensitizer.
Transition metal ions coordinated to various types of ligands are found in active sites of nature's most pervasive catalysts, hydrogenases.1 Over the past few decades, design of metal complex mimics has attracted intensive interest with the aim to provide mechanistic insights for the development of efficient chemical systems under non-biological settings.2 Though immensely inspired by nature, the research on artificial photosynthesis is still at an early stage and many technological challenges remain to be solved. In particular, finding inexpensive and efficient catalytic systems for hydrogen generation by photocatalytic water splitting is an important task. A few recent reviews on catalysts made of earth-abundant elements for water splitting have been published.3 Several promising examples for photocatalytic hydrogen evolution using Fe, Co and Ni complex catalysts and various types of photosensitizer (PS) have been found in the literature including the report by our group.4 Most of these catalysts can be sensitized by nonprecious metal-containing PS, such as xanthene dyes, Zn-containing porphyrin, etc.
In our previous work, a Ni-thiolate complex formed between Ni(II) ions and 2-mecaptoethanol (Ni-ME) was discovered as an efficient catalyst for photocatalytic hydrogen evolution by water splitting. A high quantum efficiency (QE) of 24.5 % was attained at 460 nm.4h In light of the knowledge that some enzymes have been shown to contain Ni in the active sites with a variety of coordination types,5 model complexes involving mixed donors are worth studying. In this work, a simple ligand with N and S donors, β-mecaptoethylamine (L), was chosen to form a mononuclear complex NiL2, which could be further transformed to trinuclear complex Ni(NiL2)2 reacting with Ni(II) (Fig. 1A). The activity of photocatalytic hydrogen evolution over both complexes was investigated. The facile preparation method of NiL2 crystal was described by Jicha and Busch (see Supporting Information†).6 The configuration around Ni(II) atom is square planar and the crystal structure is made up of trans NiL2 according to a single crystal X-ray diffraction study, although trans–cis isomerization could occur in the solution.7 The obtained green solid was characterized by XRD, FTIR, UV–vis and CHNS elemental analyses. All the analytic results consistently showed the formation of NiL2 complex (Fig. S1–S3). Although NiL2 is only slightly soluble in aqueous solution, upon raising the temperature to around 60 °C, the solid can be completely dissolved at a concentration of 1.6 mM (the optimized concentration for photoreaction) or higher and the solution exhibits a light orange color (Fig. 1B). The UV-vis spectrum (Fig. S3) shows the characteristic ligand-to-metal charge transfer (262 nm), 1A1g–1B2g transition (300 nm) and 1A1g–1A2g transition (473 nm) of NiN2S2 chromophore.8 In aqueous solution, two NiL2 complexes can further act as ligands to coordinate to the third Ni(II) ion via both thiolate sulfur donors to form a trinuclear cluster which is positively charged and soluble in water, Ni(NiL2)22+ (Fig. 1C and 1D).6 Similar characterization techniques were applied to Ni(NiL2)2Cl2 and the results in Fig. S4–S6 are also in good agreement with the literature data on the formation of the trinuclear complex. The additional absorption band appeared at 362 nm in Fig. S6 and can be assigned to 1A1g–1A2g transition of NiS4 chromophore. In addition, the broad and asymmetric bands indicate an overlap of transitions contributed by both terminal NiN2S2 and central NiS4.8,9 Although the properties of these two complexes have been studied to a certain extent, their catalytic activities in photocatalytic water splitting reactions have not been explored so far.
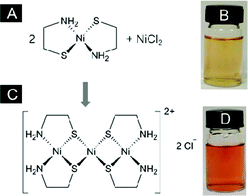 | ||
| Fig. 1 Molecular structure of the complexes, (A) NiL2 and (C) Ni(NiL2)2Cl2, and photos of aqueous solutions of (B) NiL2 (60 °C, 1.6 mM) and (D) Ni(NiL2)2Cl2 (room temperature, 0.8 mM). | ||
The photocatalytic hydrogen evolution reactions were carried out with NiL2 and Ni(NiL2)22+ catalysts dispersed and dissolved in aqueous solutions, respectively. Erythrosin yellowish (ErY) and triethanolamine (TEOA) were used as the PS and sacrificial reagent, respectively. The time course of hydrogen evolution is shown in Fig. 2. Control experiments showed that no hydrogen was evolved in the absence of any of the three components, PS, catalyst, and TEOA. It can be seen that both the mono- and trinuclear complexes are active catalysts under visible light irradiation for hydrogen generation. Under the optimized conditions for NiL2, the amount of hydrogen produced reached 3.8 mmol and the reaction ceased at above 20 h (Fig. 2B). At a fixed ErY concentration of 3.2 mM, a slight decrease in the amount of hydrogen generation resulted when the concentration of NiL2 was lowered to 0.8 mM (Fig. 2A) or increased to 2.4 mM (Fig. 2C). As the catalyst was not dissolved in the solution, the photocatalytic reaction rate was linearly dependent on the catalyst concentration. When the concentration of solid catalyst exceeds the optimal value, light filtering by solid particles can lead to inefficient light absorption and hence lower activity. Other conditions including the type of xanthene dyes and the pH value of the reaction solution were also optimized. The optimum pH value was found as 8.5. In the current system, a basic condition was necessary to keep the dyes in the deprotonated anion form which is dissolvable in water for efficient light absorption and electron transfer to the catalyst. In addition, it has been shown that protonated TEOA under acidic condition is a less effective donor.4c Under the optimized conditions, the activity of the water-soluble trinuclear complex is slightly lower, as 3.2 mmol of hydrogen was produced (Fig. 2D). In all these cases, an induction period of about 1 h was found before substantial amounts of hydrogen evolved. A similar observation was reported by Eisenberg and co-workers on cobaloxime based catalyst.3c The exact reason for this needs further investigation.
![Time course of hydrogen evolution using Ni(ii)- mecaptoethylamine complexes, (A) [NiL2] = 0.8 mM, (B) [NiL2] = 1.6 mM, (C) [NiL2] = 2.4 mM, and (D) [Ni(NiL2)2]2+ = 0.8 mM (100 mL solution, 15 vol% TEOA, [ErY] = 3.2 mM, pH = 8.5, light source: 300 W Xe lamp, λ > 420 nm).](/image/article/2012/RA/c2ra21422a/c2ra21422a-f2.gif) | ||
| Fig. 2 Time course of hydrogen evolution using Ni(II)- mecaptoethylamine complexes, (A) [NiL2] = 0.8 mM, (B) [NiL2] = 1.6 mM, (C) [NiL2] = 2.4 mM, and (D) [Ni(NiL2)2]2+ = 0.8 mM (100 mL solution, 15 vol% TEOA, [ErY] = 3.2 mM, pH = 8.5, light source: 300 W Xe lamp, λ > 420 nm). | ||
Compared to inorganic semiconductor photocatalysts with a narrow light absorption range in the visible light region, the advantage of dye sensitized photocatalyst systems lies on the extended visible light absorption to longer wavelengths. As displayed in Fig. 3, when the cut-off filter of longer wavelengths up to 520 nm was used, the total amount of hydrogen producible from both systems did not change much, although the hydrogen production rate was lowered mainly due to the reduction in light intensity. More remarkably, the quantum efficiency (QE) measured for the system containing NiL2 complex is as high as 30.9 % at 480 nm and still remains at 21.8 % at 520 nm (Fig. 3B). Such features are desirable toward more efficient utilization of visible light (400–700 nm). Compared to NiL2 system, the trinuclear complex Ni(NiL2)22+ gave rise to lower QEs with the highest value of 19.3 % obtained at 520 nm.
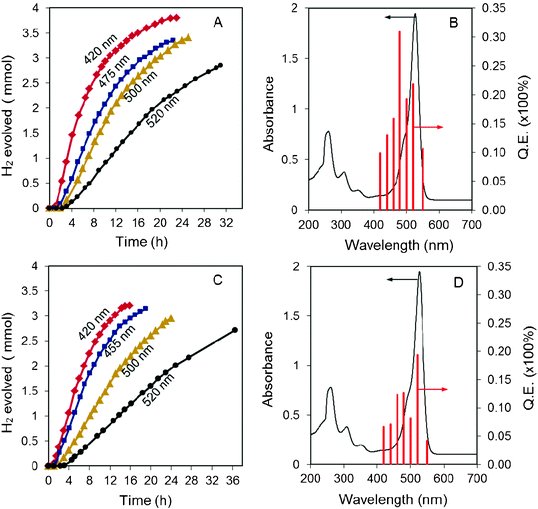 | ||
| Fig. 3 Time course of hydrogen evolution using (A) NiL2 (1.6 mM) and (C) Ni(NiL2)22+ (0.8mM) with a 300 W Xe lamp equipped with different long-pass cut-off filters, and the QEs under photons with different wavelengths over (B) NiL2 and (D) Ni(NiL2)22+ complexes. Other reaction conditions were the same as those in Fig. 2. | ||
The stability of the PS was studied by monitoring the UV–vis absorption spectrum of the reaction mixture at different reaction times. Heavy halogen-substituted xanthene dyes are known to have high intersystem crossing yield favourable for the formation of long-lived 3ππ* triplet state which injects electron to the catalysts. Nevertheless, the halogen groups can be easily photobleached through reductive quenching.10 The xanthene ring of ErY is substituted with two iodide atoms with its halogen-free analogue as fluorescein (FL). The maximum absorption of ErY and FL in aqueous TEOA solution (pH = 8.5) is at around 520 nm and 488 nm, respectively. As shown in Fig. 4, the PS in the reaction mixture in the presence of NiL2 is much more stable (Fig. 4A) compared to that without the catalyst but in the presence of TEOA electron donor (Fig. 4B). At 2 h of photoreaction, the peak position was shifted to 500 nm due to the cleavage of one C–I bond. The peak intensity was also slightly lowered. At longer reaction time, the peak position progressively shifted to 488 nm, corresponding to the loss of the remaining iodide leading to FL. Compared to the previous studies on Ni-thiolate systems involving xanthene dyes,[4g,h] a better stability of PS in this system was found since there was no significant change in peak intensity in the period of 2–24 h of reaction. Similar observations were made when trinuclear Ni(NiL2)22+ complex was used as the catalyst (Fig. S7). In contrast, photobleaching of ErY occurred rapidly in the absence of the catalyst. Fig. 4B shows that the peak intensity was already greatly reduced at 0.5 h of irradiation. Based on the above results, it can be concluded that the favourable electron transfer from the excited state of PS to the catalyst plays an imperative role in improving the stability of PS.
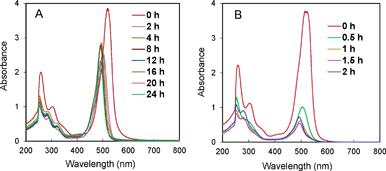 | ||
| Fig. 4 UV–vis spectra of (A) the reaction mixture of NiL2 system at different reaction times (the reaction mixture was filtered before analysis), and (B) the mixture without NiL2 catalyst while keeping other conditions the same. All solutions were diluted 10 times before analysis. The reaction conditions were the same as those in Fig. 2. | ||
Electrocatalytic properties of the complexes were investigated in order to provide insight to the reaction mechanism. The black cyclic voltammogram (CV) curve in Fig. 5A for NiL2 in the absence of acid exhibits no reduction waves. In the presence of dilute acetic acid (HAc), the colored curves show that there is an obvious increase in the current. At an HAc concentration of 0.1 mM, a reduction wave can be observed at around −0.5 V (green curve) for Ni(II/I). Hence, it is suggested that the protonated form of NiL2 can accept electrons. A similar observation was reported by Eisenberg and co-workers for [Ni(pyS)3]− complex in which Ni(II) is in a mixed S and N donor environment.4g Further increase of acid concentration led to better developed reduction waves. At higher acid concentration of 0.5–0.7 mM, the waves were shifted to more positive positions at about −0.4 V, indicating that protonation of the Ni(I) centre takes place at a rate faster than the overall catalytic rate.11 Under this condition, the half wave potential of Ni(II/I) is determined by the protonation rate while the current is determined by the overall catalytic rate. The CV curves shown in Fig. 5B present different behaviour of Ni(NiL2)22+ complex. A broad reduction wave around −0.4 V was observed in the absence of acid, consistent with accepting of electrons by the NiS4 chromophore as shown in our earlier work.4h However, the current did not increase significantly when the acid concentration was increased.
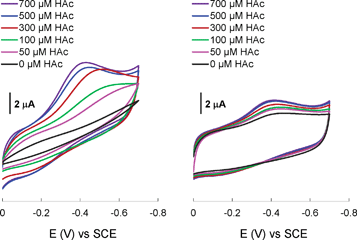 | ||
| Fig. 5 Cyclic voltammograms of (A) NiL2 (1.6 mM, 60 °C) and (B) Ni(NiL2)22+ (0.8 mM) in water using a glassy carbon working electrode with a scan rate of 50 mV s−1. | ||
In light of the results and phenomena obtained so far, a photocatalytic hydrogen evolution mechanism over NiL2 complex is proposed in Scheme 1. The complex is first protonated by adopting a proton from water to the amine species which could cause the breaking of the N chelating to the Ni(II) centre. The resultant protonated complex then electrostatically interacts with deprotonated PS under basic conditions and further accepts an electron from the excited triplet state of the PS (3*ErY) leading to reduction of Ni(II) to Ni(I) species. Both theoretical studies and experimental works have suggested that the base sites like N atoms near the metal centres could act as an intramolecular proton transfer relay for more efficient hydrogen processing.11,12 In line with this, it is proposed that the proton from the N atom is subsequently transferred to the Ni centre and becomes a hydride intermediate, in conjunction with the acceptance of another electron from the excited PS. In the following step, the complex adopts another proton from water to the same N atom for charge neutralization. Finally, the proton (H+) and hydride (H−) attached to N and Ni atoms, respectively, form a hydrogen molecule as the expected product. After releasing of molecular hydrogen, the complex is restored for the next cycle of hydrogen evolution reaction. Simultaneously, the oxidized PS is reduced by TEOA electron donor. The oxidative quenching pathway was confirmed by the CV studies of the redox potentials of ErY and TEOA.
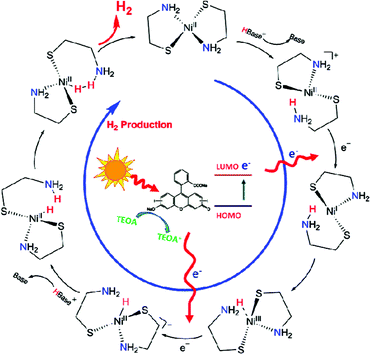 | ||
| Scheme 1 Proposed mechanism of photocatalytic hydrogen evolution over NiL2 complex using ErY and TEOA as the PS and sacrificial electron donor, respectively. | ||
The present work has shown that the complexes formed between Ni(II) and β-mecaptoethylamine with a mixed S and N donor environment are active and inexpensive catalysts for photocatalytic hydrogen evolution by water reduction. An excellent quantum efficiency of 30.9 % was obtained at 480 nm. A tentative reaction mechanism involving intramolecular proton transfer from N atom to Ni centre was proposed. This work provides us with useful clues to further explore noble-metal free complexes with a relevant environment to natural enzymes toward development of earth-abundant photocatalyst systems for solar hydrogen production.
Acknowledgements
This work was supported by AcRF grants: ARC25/08 from Ministry of Education, Singapore. J. Han acknowledges the research scholarship from Energy Research Institute @ NTU.References
- (a) Y. Nicolet, B. J. Lemon, J. C. Fontecilla-Camps and J. W. Peters, Trends Biochem. Sci., 2000, 25, 138 CrossRef CAS; (b) D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268 RSC; (c) A. L. d. Lacey, V. M. Fernández and M. Rousset, Coord. Chem. Rev., 2005, 249, 1596 CrossRef.
- (a) E. Bouwman and J. Reedijk, Coord. Chem. Rev., 2005, 249, 1555 CrossRef CAS; (b) C. d. Tard and C. J. Pickett, Chem. Rev. (Washington, DC, U. S.), 2009, 109, 2245 Search PubMed; (c) A. C. Marr, D. J. E. Spencer and M. Schröder, Coord. Chem. Rev., 2001, 219–221, 1055 CrossRef CAS; (d) S. Tschierlei, S. Ott and R. Lomoth, Energy Environ. Sci., 2011, 4, 2340 RSC.
- (a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew Chem, Int Ed, 2011, 50, 7238 CrossRef CAS; (b) Y. Lin, G. Yuan, S. Sheehan, S. Zhou and D. Wang, Energy Environ. Sci., 2011, 4, 4862 RSC; (c) P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012 RSC.
- (a) A. M. Kluwer, R. Kapre, F. Hartl, M. Lutz, A. L. Spek, A. M. Brouwer, P. W. N. M. van Leeuwen and J. N. H. Reek, PNAS, 2009, 106, 10460 CAS; (b) P. Zhang, M. Wang, C. Li, X. Li, J. Dong and L. Sun, Chem. Commun., 2010, 46, 8806 RSC; (c) T. Lazarides, T. McCormick, P. W. Du, G. G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192 CrossRef CAS; (d) W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368 CrossRef CAS; (e) T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480 CrossRef CAS; (f) M. P. McLaughlin, T. M. McCormick, R. Eisenberg and P. L. Holland, Chem. Commun., 2011, 47, 7989 RSC; (g) Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem. Int. Ed., 2012, 51, 1667 CrossRef CAS; (h) W. Zhang, J. Hong, J. Zheng, Z. Huang, J. Zhou and R. Xu, J. Am. Chem. Soc., 2011, 133, 20680 CrossRef CAS.
- K. H. Chan, K. M. Lee and K. B. Wong, PLoS One, 2012, 7, e32592 CAS.
- D. C. Jicha and D. H. Busch, Inorg. Chem., 1962, 1, 878 CrossRef CAS.
- J. Suades, X. Solans and M. Font-Altaba, Polyhedron, 1984, 3, 1227 CrossRef CAS.
- M. A. Turner, W. L. Driessen and J. Reedijk, Inorg. Chem., 1990, 29, 3331 CrossRef CAS.
- A. P. Arnold, R. Bhula, X. Chen, R. J. Geue and W. G. Jackson, Inorg. Chem., 1999, 38, 1966 CrossRef CAS.
- T. Shimidzu, T. Iyoda and Y. Koide, J. Am. Chem. Soc., 1985, 107, 35 CrossRef CAS.
- U. J. Kilgore, M. P. Stewart, M. L. Helm, W. G. Dougherty, W. S. Kassel, M. R. DuBois, D. L. DuBois and R. M. Bullock, Inorg. Chem., 2011, 50, 10908 CrossRef CAS.
- (a) A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, J. Am. Chem. Soc., 2008, 130, 4533 CrossRef CAS; (b) M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62 RSC; (c) M. H. Chiang, Y. C. Liu, S. T. Yang and G. H. Lee, Inorg. Chem., 2009, 48, 7604 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: experimental methods, XRD, FTIR and UV–vis analysis results of metal complexes, and UV–vis spectra of the reaction mixture of Ni(NiL2)22+ system at different reaction time. See DOI: 10.1039/c2ra21422a/ |
| ‡ J. Han and W. Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |