On understanding of the Hofmeister effect: how addition of salt alters the stability of temperature responsive polymers in aqueous solutions
Esben
Thormann
*
KTH Royal Institute of Technology, Department of Chemistry, Surface and Corrosion Science, SE-100 44, Stockholm, Sweden. E-mail: esben@kth.se; Fax: +46 8 208284; Tel: +46 8 7909920
First published on 10th July 2012
Abstract
In the present study, differential scanning calorimetry was employed to investigate the temperature induced phase separation process of poly(propylene oxide) in a pure aqueous solution and in the presence of five different potassium salts at three different concentrations. The different salts affected the phase separation temperature in accordance with the Hofmeister series with the three salts, KF, KCl and KBr, inducing a clear salting-out effect, one salt, KSCN, inducing a clear salting-in effect and one borderline salt, KI, showing a salting-in or a salting-out effect depending on the salt concentration. It was further observed that the phase separation enthalpy was almost unaffected by the presence of KF, KCl, KBr and KI, while the presence of KSCN led to a significant decrease in this quantity. This suggests that KF, KCl, KBr and KI have a very moderate influence on the PPO hydration, while KSCN appears to decrease the hydrophobic hydration of the PPO chains. The order of how the salts affect the phase separation temperature is in agreement with data for the partition coefficients of the anions between bulk water and at the air–water interface, but only partially in agreement with data related to ion hydration and water structuring effects. These observations are discussed in relation to existing models of how the different nature of the ion and polymer hydration can lead to effective attractive and repulsive ion–polymer interactions depending of the exact chemistry of the ions and the polymer. It is suggested that the previous confusion about the Hofmeister effect is due to a misleading conceptual picture of how polymer hydration is affected by the presence of ions. It is concluded that the Hofmeister effects, in the present case, can be described by a balance between the effective interactions governed by the asymmetric hydration of ions and hydrophobic polymers.
Introduction
More than 100 years ago, Hofmeister showed that different salts could be used to increase or decrease the solubility of proteins in aqueous media in a very regular way depending on the type of salt and its concentration.1,2 Today, it is well-known that this effect, now known as the Hofmeister effect, is very general and can be used to alter the solubility of a wide range of polymers and smaller molecules, which are partially miscible with water.3–12 The typical order of how different anions change the stability of such solutions is found in the Hofmeister series:| SO42− > F− > Cl− > Br− > NO3− > I− > ClO42− > SCN−, | (1) |
Despite a very large number of studies on the Hofmeister effect, there is still confusion about the underlying mechanisms behind the effect. Originally, the ions in the Hofmeister series were divided into structure-breaking and structure-making ions depending on how they are affecting the bulk water structure and, thereby, how they also affect the solubility of hydrophobic molecules.5,13,14 Later, the importance of direct ion–solute interactions and ion polarizability was considered and, in particular, how ion–solute interactions affect the hydration of ions and solutes.3,8,9,17
In the present work, differential scanning calorimetry (DSC) was used to study how five different potassium salts affect the stability of aqueous solutions of poly(propylene oxide) (PPO). PPO is a polymer that is partially miscible with water, meaning that it is fully soluble in water at low concentrations and at low temperatures, while the system splits into a water rich and a polymer rich phase when the concentration and/or the temperature are increased. At a given fixed PPO concentration, DSC is thus a useful technique for studying the phase separation process occurring when scanning from low temperatures, where PPO is fully soluble, toward high temperatures, where the solution changes to a two-phase system. DSC thermograms provide information about the onset temperature and the width of the temperature range of the phase separation, as well as the change in enthalpy and heat capacity, due to the phase separation process. In the presence of different salts, DSC thermograms thus provide direct information about the shift in the phase separation temperatures (salting-in and salting-out effects) and the enthalpic changes, which are due to changes in PPO hydration. In the present study, the observed salting-in and salting-out effect and the hydration enthalpies have been combined with literature data for ion hydration and some existing models for ion–polymer interactions. This has been done with the purpose of finding a solution to the controversy about the water structuring effect versus ion–polymer interactions and to explain why a conceptual understanding of the Hofmeister effect has been hindered by a misleading classical picture of how ions affects polymer hydration.
Materials and method
Materials
Poly(propylene oxide) (Mw ≈ 2000 g mol−1, PPO) and homopolymers with an average composition of (C3H6O)39 were purchased from Waters Corporation and used as received. The salts, which were of an analytical grade or better, were purchased from Merck. Aqueous solutions containing the salts and 0.5 wt% PPO were prepared by continuous slow rotation of the solution-flasks at a low temperature (5 °C) for at least 24 h.Differential scanning calorimetry (DSC)
Thermograms showing relative heat capacity versus temperature in the interval between approximately 10 and 50 °C were obtained using a MC-2 differential scanning calorimeter (MicroCal VP-DSC, Northhampton, MA) combined with an external temperature regulated water bath. To avoid bubble formation during the temperature scan, the samples were degassed immediately prior to the measurements and the solution in the measuring cell was kept at a pressure of approximately 2 atm. The thermograms were obtained at a scan rate of 20 K h−1. In the case of PPO in pure water, three consecutive thermograms were obtained by reloading the measuring cell with fresh solution and the minor differences between these repeated measurements were used to calculate the standard derivations of the quantities deduced from the thermograms. It is assumed that these values also approximately represent the uncertainties of the measurements performed on the different PPO/salt solutions.In the DSC system, the heat capacity of a constant volume of the sample solution was measured relative to a reference solution containing the same salt concentration as the polymer solution. This means that the measured signal is an excess heat capacity, which is related to changes in the heat capacity of the polymers, as well as the change in the heat capacity of the solvent caused by the presence of the polymers. In a system operating with a constant volume, the absolute mass of the polymer in the measuring cell and, thus, the absolute signal intensity will depend on the density of the solution. Since the density slightly varies for solutions with different salt species and for different salt concentrations, the specific excess heat capacity is normalized by the polymer concentration, the cell volume and the density of the solution to account for this:
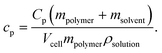 | (2) |
Results and discussion
Thermograms for poly(propylene oxide)
Fig. 1 shows a thermogram for PPO in pure water. Here cp only displays very small relative changes up to approximately 19 °C, while a strong endothermic peak is observed between approximately 19 to 35 °C. This peak is a result of the phase separation and the system in this temperature interval thus consists of polymers in aqueous solution in equilibrium with polymers in a polymer rich phase. Although the phase separation takes place over a broad temperature range, for simplicity, in the following we refer to the temperature with the highest value of cp (the peak point) as the phase separation temperature, TPS. The observed endothermic process shows that the phase separation is entropically driven, which is in agreement with the common understanding that the solubility of a range of polymers decreases with increasing temperature due to entropically unfavorable water structures in the hydration zone of the polymer chains.18–21 At higher temperatures, where most of the PPO chains have entered the polymer rich phase, cp returns to an approximately constant level, which is lower than the constant level before the phase separation process started. This change in cp over the phase separation is also a common observation in the dehydration of hydrophobic interfaces and is normally explained by the lower heat capacity of the solvent after “melting” the well-organized water structures in the hydration zone.22–26 An estimate of the magnitude of the change in water structure can be calculated from the change in enthalpy, ΔH, found by integrating the endothermal peak. If it is assumed that the enthalpic contribution of approximately 150 J g−1 of PPO (corresponding to 7.5 kJ mol−1 of PO units) is mainly used to break down the strongly hydrogen bound water structures in the hydration zone of the PPO chains, then ΔH provides information about how many hydrogen bonds which exist between the water molecules that are present in the PPO solution relative to the number in bulk water. If the strength of a hydrogen bond is assumed to be in the order of 15 kJ mol−1, as suggested in the literature,27–31 the number of hydrogen bonds, NHB, which are broken during the phase separation will be given by: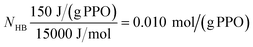 | (3) |
| NHB = 0.010 mol/(g PPO) × 2000(g PPO)/mol = 20 | (4) |
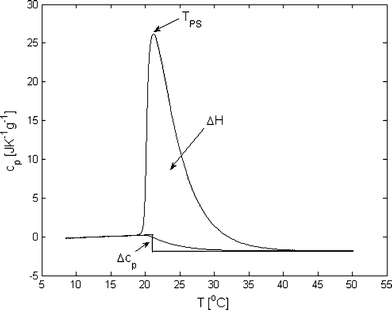 | ||
| Fig. 1 Thermogram of 0.5 wt% poly(propylene oxide) in water showing the phase separation process that occurs with increasing temperature. From the thermogram, one can obtain the phase separation temperature, TPS, the change in heat capacity, Δcp, and the change in enthalpy, ΔH. | ||
This estimate thus shows that the hydration of a PPO chain gives rise to approximately twenty extra hydrogen bonds compared to bulk water, which corresponds to half an extra hydrogen bond per PO unit. If it is further assumed that each water molecule in bulk water is, on average, involved in approximately 3.5 hydrogen bonds to other water molecules,30,32–35 the relative increase in the number of hydrogen bonds due to the hydration of the PPO chains is given by:
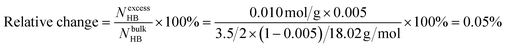 | (5) |
Although the real number might be slightly smaller or larger since the contribution from water–PPO interactions was ignored in this calculation, this relatively small increase in the apparent number of hydrogen bonds indicates that the hydration and dehydration of a PPO chain only has a very local effect on the water structure and does not significantly alter the bulk behavior of water. The magnitude of ΔH can also be used to estimate the entropy change ΔShyd = ΔH/TPS ≈ 0.51 J g−1 K−1 related to the hydration of the PPO chains (corresponding to 25.5 J mol−1 of PO units K−1). These numbers will be relevant in the forthcoming comparison between the hydration of PPO and different ions.
Salt effects on the phase separation
In Fig. 2, thermograms of PPO dissolved in water and in 0.1 M, 0.5 M and 1 M solutions of KSCN, KI, KBr, KCl and KF are presented. It is here observed that the presence of salt has a clear effect on the phase separation temperatures and on the shape of the peaks. In Fig. 3, TPS, ΔH and Δcp are plotted as a function of the salt concentration for the five different salts (in the case of 1 M KF, ΔH and Δcp are not reported since the baseline at a low temperature could not be accurately determined). It is here seen that the addition of KF, KCl and KBr decreases the phase separation temperature, TPS, with KF being the salt that has the largest effect and KBr the salt with the smallest effect. The effect is, for these three salts, further observed to be approximately proportional to the salt concentration. The effect of adding KI is small at all salt concentrations. However, while the phase separation temperature is slightly increased at a KI concentration of 0.1 and 0.5 M, it is slightly decreased at 1 M. The addition of KSCN does, in contrast to the other salts, increase the phase separation temperature relative to the salt-free system at all three measured concentrations but has a nonlinear incremental increase with an increasing salt concentration.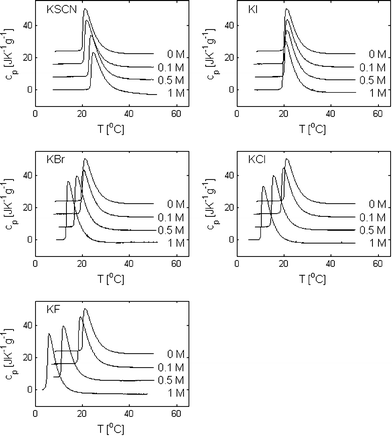 | ||
| Fig. 2 Thermograms of 0.5 wt% poly(propylene oxide) in aqueous solutions containing 0, 0.1, 0.5 and 1 M KSCN, KI, KBr, KCl and KF. For clarity, the thermograms obtained at different salt concentrations have been shifted vertically. | ||
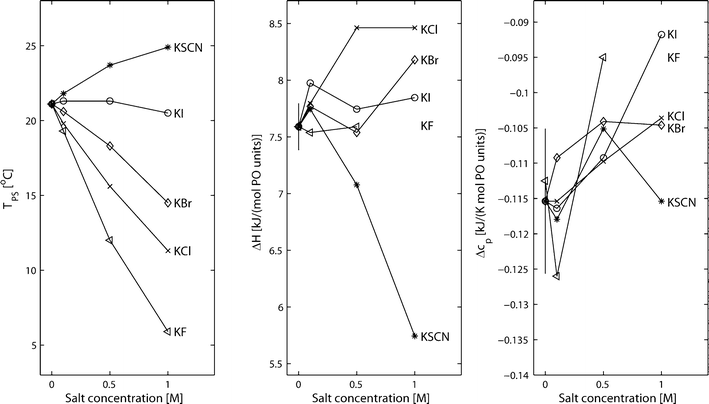 | ||
| Fig. 3 Phase separation temperatures, TPS, ΔH values and Δcp values for 0.5 wt% poly(propylene oxide) in aqueous solutions containing 0, 0.1, 0.5 and 1 M KSCN, KI, KBr, KCl and KF. The error bars for the system in pure water correspond to twice the standard derivation calculated from the results from three repeated measurements. In the case of TPS, the error bar is 0.1 °C and, due to this small value, is not visible in the figure. | ||
The values of Δcp only show minor and unsystematic variations for the different salts. There might be a weak trend towards numerically lower Δcp values with an increasing salt concentration but the variations are in the order of the magnitude of the standard deviation.
In the case of KI, KBr, KCl and KF, the ΔH values also do not vary significantly between the different salts and the different salt concentrations. However, in the case of KSCN, the numerical value of ΔH is significantly reduced with an increasing salt concentration. This indicates that the hydration of PPO is almost unaffected by the addition of salt in the case KI, KBr, KCl and KF, while the addition of KSCN appears to decrease the hydrophobic hydration.
Although the addition of salt does not significantly change the integral of the peak (except in the case of KSCN), the height-to-width ratio of the peaks is altered. This change is related to the nature of the phase separation process. By the onset of the peak in the thermogram, PPO starts to precipitate and an equilibrium between the polymers in the aqueous phase and the polymer rich phase is established. As the temperature is further increased, more and more PPO leaves the aqueous phase until a temperature where most of the polymers have precipitated and the value of the heat capacity reaches a new baseline level. When the solvent quality is changed by the addition of salt, both the onset of the phase separation process, as well as the temperature range where PPO is found in equilibrium between the two phases, are affected. It is evident from the data in Fig. 2 that the salts with a salting-in effect (KF, KCl and KBr) both lower the temperature for the onset of the phase separation and decrease the temperature range where PPO can coexist in the two phases. The opposite is the case for KSCN, which has a salting-out effect. These observations are in agreement with a study of the effect of NaCl and NaI on the phase behavior of heptaethylene glycol dodecyl ether in aqueous solution, where the range of the two-phase region was observed to decrease by addition of NaCl and increase by addition of NaI.36
Models for the Hofmeister effect
When a salt dissolves in water it is known to have a significant influence on the water structure and dynamics.39–42 The electric field from an ion has a tendency to orient the water molecules and thereby change the hydrogen bonding network compared to bulk water. Close to the ion, the water molecules are oriented with the oxygen atom pointing towards the cation and one of the hydrogen atoms pointing towards the anion. Due to the high dielectric constant of water, the electric field from the ion is effectively shielded and, at a distance of 10–20 Å43 from the ion, the tetrahedral hydrogen bonding network is re-established. However, in the intermediate layer, the water molecules will be somewhat frustrated since they both try to align the dipole moment with the electric field and fit into the bulk network. This is normally described in terms of different zones: the primary hydration zone, where the water molecules are aligned with the electric field; the secondary hydration zone, where the water structure is partly broken due to frustration and, finally, the bulk water, where the molecules are unaffected by the ion.44
The hydration of an ion can be described from the hypothetical process of transferring an ion from a vacuum to an aqueous solution and the change in free energy, ΔGhyd, is then related to the direct ion–water interaction and the indirect change in the intermolecular interactions between the water molecules. This change can be described in terms of a change in enthalpy, ΔHhyd, given by changes in intermolecular interactions and a change in entropy, ΔShyd, given by changes in degrees of freedom. The magnitudes of ΔGhyd, ΔHhyd and ΔShyd thus contain important information about how the ion influences the water structure. Values of these variables for the ions used in this study can be found in the literature34,37,45–47 and are reported in Table 1. Both ΔHhyd and ΔShyd are, in all cases, observed to be negative and the values become numerical smaller with an increasing size of the ion. The sign of ΔHhyd is mainly the result of the energetically favorable direct interactions between the ion and the water molecules, which also explains why the effect is larger for small compact ions, where the electric field close to the ion is stronger. The interpretation of the sign of ΔShyd is less obvious. However, the entropy change related to ion hydration is a sum of different contributions, such as structural changes in the network of the water molecules, electrostatic contributions, the effects of volume changes and contributions from the limited translational freedom of the water molecules in the ions’ primary hydration zone.14,34 Since the discussion refers to the ions’ structure-breaking or structure-making effect, it is of interest to be able to distinguish the part of ΔShyd that comes from structural changes with other contributions. Based on experimental data, Marcus et al. have calculated this structural contribution, ΔSstr, by subtracting the other contributions.34,37,38,47 For the ions used in this study, these values are also reported in Table 1. As seen in this table, only hydration of F− has a negative change in Sstr and thus an apparent structure-making effect, while the cation K+ and the anions Cl−, Br−, I− and SCN− all have positive changes in Sstr and thus an apparent structure-breaking effect.
| Ion | ΔGhyd [kJ mol−1] | ΔHhyd [kJ mol−1] | ΔShyd [kJ mol−1 K−1] | ΔSstr [kJ mol−1 K−1] | ΔGHB | Partition coefficient |
|---|---|---|---|---|---|---|
| K+ | −300a | −321a | −70a | 47d | −0.7e | 0.12f |
| −324b | ||||||
| −304b | −321c | −80b | ||||
| F− | −459a | −504a | −150a | −27d | 0.1e | 0.53f |
| −519b | ||||||
| −472b | −503c | −150b | ||||
| Cl− | −334a | −361a | −90a | 58d | −0.7e | 0.69f |
| −376b | ||||||
| −347b | −369c | −87b | ||||
| Br− | −309a | −330a | −70a | 81d | −0.95e | 0.86f |
| −345b | ||||||
| −321b | −336c | −70b | ||||
| I− | −270a | −285a | −50a | 117d | −1.25e | 1.18f |
| −300b | ||||||
| −283b | −298c | −47b | ||||
| SCN− | −287b | −307b | −66b | 83d | −0.9e | 1.64f |
Ben-Naim and Marcus have also developed a methodology to describe how the hydration of an ion influences the water structure with respect to the number of hydrogen bonds.37,38,48 They introduced a geometric factor, GHB, which describes whether a hydrogen bond exists or not. The quantity of ΔGHB then describes the change in the average geometrical factors from all water molecules by the hydration of a given ion, and a positive value of ΔGHB means that the hydration leads to an increase in the number of hydrogen bonds, while a negative value means a decrease in the number of hydrogen bonds. As seen in Table 1, there exists a strong correlation between the change in the structural entropy and the change in hydrogen bonding.
In Fig. 4 A–E, the values of the shifts in the phase transition temperatures are plotted against the values of ΔGhyd, ΔHhyd, ΔShyd, ΔSstr and ΔGHB for the anions. It is here observed that the order of salting-in and salting-out effects of KF, KCl, KBr and KI correlate with the ΔGhyd, ΔHhyd, ΔShyd, ΔSstr and ΔGHB values. However, KSCN appears to be an exception to this trend. KSCN has a larger salting-in effect than KI even though the SCN− ion has a water breaking effect, which is found to be between the effect of the Br− ions and the effect of I− ions.
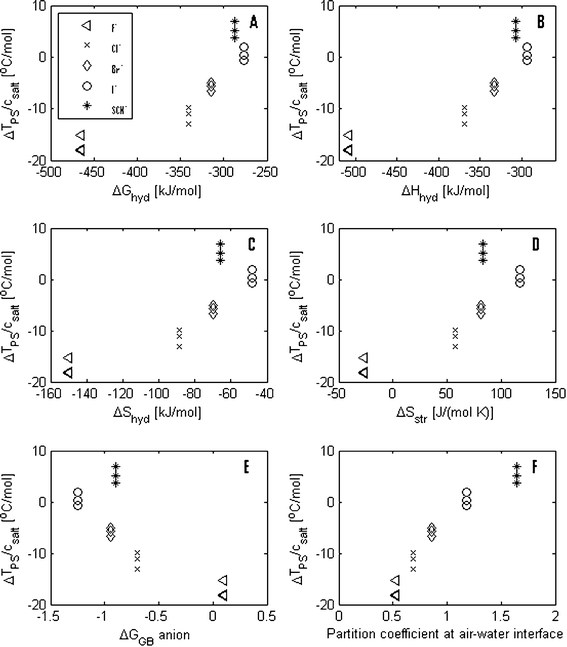 | ||
| Fig. 4 The salt induced changes in the phase separation temperature normalized by the salt concentration are plotted against (A) the change in hydration free energy, ΔGhyd, (B) the change in hydration enthalpy, ΔHhyd, (C) the change in hydration entropy, ΔShyd, (D) the change in structural entropy, ΔSstr, (E) the change in the geometrical factor, ΔGHB, giving the change in hydrogen bonding and (F) the partition coefficients between bulk water and at the air–water interface for the anions used in this study. | ||
Despite the fact that the order (F−, Cl−, Br− and I−) of the ions’ water structuring effect correlates qualitatively with the order of how the potassium salts containing these anions influence the phase separation of the PPO solutions, there does not seems be a simple quantitative correlation between the ions’ effect on ΔSstr and their salting-in and salting-out effect. This means that there is no simple correlation between which salts have a structure-breaking effect and a salting-in effect and, conversely, which salts have a structure-making effect and a salting-out effect. It is noteworthy that, of these four potassium halides, only the strong structure-breaking salt, KI, has a weak salting-in effect (and only at low salt concentrations), while the other structure-breaking salts, KBr and KCl together with KF (which has a structure-breaking and a structure-making effect), have a relatively large salting-out effect. In addition, KSCN has a structure-breaking effect, which corresponds in magnitude to the structure-breaking effect of KBr, but it also has a salting-in effect, which is stronger than the one of KI.
A mechanism where the Hofmeister effect is explained by the ions ability to adapt to the hydration zone of the polymers was actually suggested more than 25 years ago by Florin et al. in a comprehensive experimental and theoretical study of the influence of different salts on the phase separation temperature of aqueous solutions of high molecular weight poly(ethylene oxide) (PEO).3 In that work it was suggested that an overlap in the hydration zone of an ion and a polymer segment will lead to an effective force with a repulsive and an attractive component. As described in the section above (and shown in Table 1), hydration of an ion is a low free energy state and partial dehydration of the ion, due to overlap in the hydration zones, thus leads to an effective repulsive force between the ion and the polymer segment. In contrast, hydration of a (hydrophobic) polymer is a high free energy state and partial dehydration, due to overlap in the hydration zones, thus lead to an effective attractive force between the ion and the polymer segment. In accordance with such a description, the repulsive component will contribute to a salting-out effect, while the attractive component will contribute to a salting-in effect. However, the magnitude of the repulsive and attractive component will obviously depend on the type of ion and the type of polymer.
The suggested model by Florin is also in line with a more recent study by Pegram and Record Jr., where surface tension data were used to calculate the partition coefficient between different ions in bulk solution and at the air–water interface (see Table 1).16 Here, a value smaller than unity, as seen for K+, F−, Cl− and Br−, indicates that the ions are effectively repelled from the air–water interface, while a value larger than unity, as seen for I− and SCN−, indicates that the ions are effectively attracted to the interface. In Fig. 4F, the change in the phase separation temperature of the PPO solutions is plotted against these partition coefficients and it is seen that these numbers follow the same trend as the change in the phase separation temperature. Although the PPO–water interface cannot be directly compared to the air–water interface, it is also noticed that there is agreement between which anions are repelled from the air–water interface and which are leading to a salting-out of PPO. Oppositely, the anions which are attracted to the air–water interface are leading to salting-in of PPO. Thus, in contrast to the ΔGhyd, ΔHhyd, ΔShyd, ΔSstr and ΔGHB values, the partition coefficient seems to be a parameter that can describe the Hofmeister effect for all the anions in this study. Although the partition coefficient by itself is a phenomenological quantity and thus does not directly contain new knowledge, the direct correlation between this parameter and the Hofmeister effect works as a confirmation of the suggested mechanisms in Florin's model.
It should also be noted that the K+ cation, based on the partition coefficient, should have a much stronger salting-out effect than any of the anions, which is an observation that seems to contradict the general observation that anions are more important than cations. However, while the value suggests that K+ should be strongly repelled from the air–water interface, the calculations by Pegram and Record Jr. reveal almost identical partition coefficients for cations, such as Li+, Na+, K+ and Cs+. This could indicate that the cations indeed are important for the polymer/salt solution stability, but that they do not show significant variations as seen for the anions in the Hofmeister series.
In a more recent work, Zhang et al. adopted a similar description in order to explain the salt dependence of the phase separation of aqueous solutions of poly(N-isopropylacrylamide) (PNIPAM).8,9 Here, it was suggested that the Hofmeister effect can be explained by three mechanisms: (1) dehydration of the amide groups of PNIPAM due to competition with strongly hydrated ions; (2) interference with the hydration of the hydrophobic parts of the polymer due to surface tension effects and (3) direct ion binding to the polymer. The second mechanism is similar to the repulsive part of the ion–polymer interaction in Florin's model and is due to partial dehydration of the ion and thus is most significant for strongly hydrated ions, like F−. With respect to the third mechanism, direct ion binding between SCN− and PNIPAM has recently been reported from a study using isothermal calorimetry (ITC).49 In the work by Zhang et al., it is not specified if the third mechanism refers to a non-specific preferential association of the ion to the polymer segment, as described above, or due to a direct specific bond between the ion and a specific location on the polymer segment, as recently suggested in a study of ion association by NMR.17 However, in each of the cases, the effect will be the same and the different wording may simply reflect the uncertainty surrounding the nature of the attractive interaction. The first mechanism is slightly different from the descriptions in Florin's model since it involves dehydration of amide groups, which means removal of water molecules from a low free energy state and this process thus leads to an additional repulsive component to the force between the ion and the polymer segment. However, in the end, it all refers to the total free energy cost of an overlap in the hydration zones of the ion and the polymer segments.
In accordance with this understanding, the salting-out effect of KF, KCl and KBr found in the present study has to be an effect of the effective repulsion between the PPO chains and the F−, Cl− and Br− ions. However, whether the repulsion is purely due to the unfavorable dehydration of these ions, or also due to unfavorable dehydration of, for example, the oxide groups of PPO, cannot be distinguished here. The salting-in effect of KI (at 0.1 and 0.5 M) and, in particular, the salting-in effect of KSCN conversely has to be due to an effective attraction between the PPO chains and the I− and the SCN− ions. This interpretation is further supported by the ΔH values, which represent the enthalpies needed for dehydration of the PPO chains during the phase separation. As seen in Fig. 3, the ΔH values are almost unaffected by the presence of KF, KCl and KBr, which is in line with the model predicting that the F−, Cl− and Br− ions are repelled from the hydration zone of the PPO chains and, thus, consequently do not affect the hydrophobic hydration of PPO. KI is a borderline salt, which apparently does not have a measurable influence on the hydration of the PPO chains. However, in the case of KSCN, the numerical value of ΔH becomes significantly reduced with an increasing KSCN concentration, which is in line with the model predicting the SCN− ions are attracted to the PPO chains, which thus lowers the degree of hydrophobic hydration. The fact that the effects of KSCN and KI have different concentration dependence than the salt with a salting-out effect is also in line with this qualitative description. Due to the limited polymer–water interface, a limited number of ions can be attracted to the hydration zone of the polymers. It is thus in agreement with a saturation model that KSCN only has a weakly increasing salting-in effect with increasing salt concentration and that the effect of KI can change from salting-in to salting-out with the increasing salt concentration.
In light of the importance of ion hydration, it should no longer appear a mystery that the addition of salt to a polymer solution affects the solution stability and that different ions affect the stability in a different manner. One could also change their mind set and consider how addition of different polymers would change the stability of a salt solution at a given concentration. Such experiments have obviously been performed since the Hofmeister effect has been verified for a large range of molecules, polymers and proteins. These experiments have revealed that the Hofmeister effect is significantly different in the different cases. For example, in the present study, a 10 °C decrease in the phase separation temperature of PPO is observed in the case of 1 M KCl, while Florin et al. observed a 23 °C and 24 °C decrease in the phase separation temperature of PEO in the case of 1 M NaCl and KCl, respectively, and Zhang et al. observed a very moderate 1.5 °C decrease in the phase separation temperature of PNIPAM in the case of 1 M NaCl. Similar trends are observed for other salts and at other salt concentrations. This illustrates that, while it is the type of ion that determines the order of the salting-in and salting out effect (the Hofmeister series), it is the combination of the chemistry of both the ion and the polymer that determines the magnitude of the Hofmeister effect. It is also the chemistry of the polymer that determines whether a borderline salt, like KI, will lead to a salting-in or a salting-out effect.
Conclusions
In the present work, it was observed that five potassium salts, KF, KCl, KBr, KI and KSCN, had salting-in and salting-out effects on aqueous solutions of PPO in accordance with the Hofmeister series. It was, however, also observed that the phase separation enthalpy was almost unaffected by the presence of KF, KCl, KBr and KI, while the presence of KSCN led to a significant decrease in this quantity. Based on these observations and the discussion on water structuring effects versus ion–polymer interactions, it is suggested that the Hofmeister effect can be qualitatively explained in the following way: the interaction between an ion and a polymer segment contains a repulsive contribution originating from a partially dehydration of the ion and an attractive and a repulsive contribution originating from a partially dehydration of the polymer segment, where the first part is dependent on the nature of the ion and the last two are dependent of the nature of the polymer segment. It is the balance between these attractive and repulsive contributions that determines whether the addition of a given type of salt will lead to a salting-out effect or a salting-in effect. This also means that both the direction and the magnitude of the salt effect will depend on both the nature of the ion and the nature of the polymer.This explains the order of the ions in the Hofmeister series since this order can be related to the order of how the ions are partitioning between bulk water and a non-polar interface and it also explains the somehow counter intuitive result that KF, KCl, and KBr can have a large salting-out effect while, at the same time, barely affect the polymer hydration in terms of the ΔH values.
Although the, by now, better understanding of how hydrophobic molecules affect ion hydration leads to a better conceptual understanding of the Hofmeister effect, there are still some unanswered questions. Firstly, based on the partition coefficient data, it is still not clear how significantly the cations affect the solution stability compared to the anions. Secondly, the exact nature of the attractive ion–polymer interaction is still not fully understood. It is, however, likely that this is related to a complex interplay between the hydration and ion polarization, as suggested in a series of articles by Ninham and coworkers.15,50–52 To understand these issues is the next challenge in the story about the Hofmeister effect.
References
- F. Hofmeister, Naunyn-Schmiedebergs Arch. Pharmacol., 1888, 24, 247–260 Search PubMed.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS.
- E. Florin, R. Kjellander and J. C. Eriksson, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2889–2910 RSC.
- P. Bahadur, K. Pandya, M. Almgren, P. Li and P. Stilbs, Colloid Polym. Sci., 1993, 271, 657–667 CAS.
- R. L. Baldwin, Biophys. J., 1996, 71, 2056–2063 CrossRef CAS.
- V. I. Lozinsky, L. V. Domotenko, A. L. Zubov and I. A. Simenel, J. Appl. Polym. Sci., 1996, 61, 1991–1998 CrossRef CAS.
- P. Alexandridis and J. F. Holzwarth, Langmuir, 1997, 13, 6074–6082 CrossRef CAS.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS.
- Y. J. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- Y. K. Jhon, R. R. Bhat, C. Jeong, O. J. Rojas, I. Szleifer and J. Genzer, Macromol. Rapid Commun., 2006, 27, 697–701 CrossRef CAS.
- Y. Cho, Y. Zhang, T. Christensen, L. B. Sagle, A. Chilkoti and P. S. Cremer, J. Phys. Chem. B, 2008, 112, 13765–13771 CrossRef CAS.
- J. M. G. Swann, W. Bras, P. D. Topham, J. R. Howse and A. J. Ryan, Langmuir, 2010, 26, 10191–10197 CrossRef CAS.
- K. D. Collins and M. W. Washabaugh, Q. Rev. Biophys., 1985, 18, 323–422 CrossRef CAS.
- M. G. Cacace, E. M. Landau and J. J. Ramsden, Q. Rev. Biophys., 1997, 30, 241–277 CrossRef CAS.
- D. F. Parsons, M. Bostroem, P. Lo Nostro and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- L. M. Pegram and M. T. Record Jr., J. Phys. Chem. B, 2007, 111, 5411–5417 CrossRef CAS.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2011, 133, 7344–7347 CrossRef CAS.
- M. Malmsten, P. M. Claesson, E. Pezron and I. Pezron, Langmuir, 1990, 6, 1572–1578 CrossRef CAS.
- D. P. Chang, J. E. Dolbow and S. Zauscher, Langmuir, 2007, 23, 250–257 CrossRef CAS.
- A. Dedinaite, E. Thormann, G. Olanya, P. M. Claesson, B. Nystrom, A.-L. Kjoniksen and K. Zhu, Soft Matter, 2010, 6, 2489–2498 RSC.
- E. Thormann, P. M. Claesson and O. G. Mouritsen, Phys. Chem. Chem. Phys., 2010, 12, 10730–10735 RSC.
- A. Pertsemlidis, A. M. Saxena, A. K. Soper, T. HeadGordon and R. M. Glaeser, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10769–10774 CrossRef CAS.
- C. Tanford, The Hydrophobic Effect: Formation of Micelles and Biological Membranes, Wiley-Interscience, New York, 1973 Search PubMed.
- K. A. T. Silverstein, A. D. J. Haymet and K. A. Dill, J. Am. Chem. Soc., 2000, 122, 8037–8041 CrossRef CAS.
- N. Muller, Acc. Chem. Res., 1990, 23, 23–28 CrossRef CAS.
- T. B. Nielsen, S. Hvidt, S. R. Keiding, C. Petersen, P. Westh and K. Keiding, Phys. Chem. Chem. Phys., 2011, 13, 1182–1188 RSC.
- L. Grunberg and A. H. Nissan, Trans. Faraday Soc., 1949, 45, 125–137 RSC.
- G. Scatchard, G. M. Kavanagh and L. B. Ticknor, J. Am. Chem. Soc., 1952, 74, 3715–3720 CrossRef CAS.
- C. N. R. Rao, in Water - A Comprehensive Treatise, ed. F. Franks, Plenum Press, New York, 1972, vol. 1, pp. 93–113 Search PubMed.
- F. H. Stillinger, Science, 1980, 209, 451–457 CAS.
- C. Millot and A. J. Stone, Mol. Phys., 1992, 77, 439–462 CrossRef CAS.
- G. E. Walrafen, in Hydrogen-Bonded Solvent Systems, ed. A. K. Covington and P. Jones, Taylor & Francis Ltd., London, 1968, pp. 9–29 Search PubMed.
- A. Rahman and F. H. Stillinger, J. Am. Chem. Soc., 1973, 95, 7943–7948 CrossRef CAS.
- Y. Marcus and A. Ben-Naim, J. Chem. Phys., 1985, 83, 4744–4758 CrossRef CAS.
- K. A. T. Silverstein, A. D. J. Haymet and K. A. Dill, J. Am. Chem. Soc., 1998, 120, 3166–3175 CrossRef CAS.
- T. Inoue, Y. Yokoyama and L. Q. Zheng, J. Colloid Interface Sci., 2004, 274, 349–353 CrossRef CAS.
- Y. Marcus, Pure Appl. Chem., 2010, 82, 1889–1899 CrossRef CAS.
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef CAS.
- F. Rull and J. A. Desaja, J. Raman Spectrosc., 1986, 17, 167–172 CrossRef CAS.
- P. Terpstra, D. Combes and A. Zwick, J. Chem. Phys., 1990, 92, 65–70 CrossRef CAS.
- D. Bulone, V. Martorana, P. L. San Biagio and M. B. Palma-Vittorelli, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 6799–6809 CrossRef CAS.
- W. Wachter, W. Kunz, R. Buchner and G. Hefter, J. Phys. Chem. A, 2005, 109, 8675–8683 CrossRef CAS.
- K. Rechendorff, M. B. Hovgaard, M. Foss, V. P. Zhdanov and F. Besenbacher, Langmuir, 2006, 22, 10885–10888 CrossRef CAS.
- J. O. Bockris and A. K. Reddy, Modern electrochemistry, Plenum Press, New York, 1970 Search PubMed.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pretice Hall, Gosport, UK, 2001 Search PubMed.
- G. Pass, Ions in Solution 3, Oxford University Press, Bristol, UK, 1973 Search PubMed.
- Y. Marcus, J. Solution Chem., 1994, 23, 831–848 CrossRef CAS.
- A. Ben-Naim, J. Phys. Chem., 1975, 79, 1268–1274 CrossRef CAS.
- I. Shechter, O. Ramon, I. Portnaya, Y. Paz and Y. D. Livney, Macromolecules, 2010, 43, 480–487 CrossRef CAS.
- D. F. Parsons and B. W. Ninham, Langmuir, 2010, 26, 6430–6436 CrossRef CAS.
- D. F. Parsons, M. Bostrom, T. J. Maceina, A. Salis and B. W. Ninham, Langmuir, 2010, 26, 3323–3328 CrossRef CAS.
- M. Bostrom, W. Kunz and B. W. Ninham, Langmuir, 2005, 21, 2619–2623 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |