Stereoselective synthesis of C-2-methylene and C-2-methyl-C-glycosides by Claisen rearrangement of 2-vinyloxymethyl glycals†
Perali Ramu
Sridhar
* and
Chalapala
Sudharani
School of Chemistry, University of Hyderabad, Hyderabad-500 046, India. E-mail: prssc@uohyd.ernet.in; Fax: +91 4023012460; Tel: +91 4066794823
First published on 30th July 2012
Abstract
An efficient protocol for the stereoselective synthesis of C-2-methylene-α- and -β-C-glycosides by a Claisen rearrangement of 2-vinyloxymethyl glycal derivatives is reported. A plausible mechanism for the formation of α-selective-C-glycoside was proposed. The methodology was further extended to the high diastereoselective synthesis of C-2-methyl-C-glycosides.
Carbon branched sugars, in which the hydroxyl groups on pyranose/furanose ring is replaced with carbon are widely present in a number of natural products. Notably, C-2-methyl analogues of nucleosides have shown potential inhibitory activity against hepatitis C viral RNA replication.1 On the other hand, C-2-methylene- and C-2-methyl-C-glycoside derivatives exist ubiquitously in nature as subunits of several highly bioactive natural products such as spliceostatins, spongistatins, phorboxazoles, brevenal, brevetoxin, gambieric acids and halichondrins etc..2 One of the widely used approaches for stereoselective construction of C-glycosides is a Claisen or Ireland-Claisen rearrangement3 of glucal derived allylvinyl ethers that has been discussed extensively in the literature.4,5 Due to the intriguing features of the Claisen-[3,3]-sigmatropic rearrangement reaction, it has been utilized to provide key intermediates for the total synthesis of a number of natural products possessing C-2,6 C-3,7 C-48 and C-59 carbon branched carbohydrate frameworks.
Surprisingly, all the reported Claisen rearrangement (CR) reactions involving carbohydrate moieties with endo-cyclic allyl alcohols include at least one chiral center in the rearrangement sequence, which directs the stereochemical course of the newly generated stereocenters. In our interest towards the stereoselective synthesis of C-branched sugars,10 we envisaged that CR of glycal derived allylvinyl moieties might provide a straightforward access to the formation of C-2-methylene-C-glycosides and intern offer access to the preparation of 2-C-branched-C-glycosides. Even though, no chiral center is involved in the rearrangement sequence, we anticipated the reaction to proceed in a stereoselective fashion due to the concerted nature, highly organized transition state in CR reactions and the influence of additional stereocenters in the molecule. Chemoselective hydrogenation of the C-2-methylene group further provides an efficient route for the diastereoselective preparation of C-2-methyl-C-glycosides.
Thus, mercuric acetate-catalyzed trans vinylation of alcohol 1 with ethylvinyl ether provided the required 2-vinyloxymethyl-3,4,6-tri-O-benzyl-D-glucal 2 in 65% yield. When compound 2 was heated in a sealed tube at 180 °C for 6 h in toluene the reaction produced the expected C-2-methylene-C-glycosides 3α and 3β in 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 ratio, respectively.11,12 Interestingly, the α-anomer 3α was found to be very unstable and converted to the β-anomer 3β13 in the purification process using silicagel column chromatography, probably via ring opening to form α,β-unsaturated aldehyde 4 followed by intramolecular oxa-Michael addition reaction. Whereas, direct reduction of the crude aldehyde mixture (3α and 3β) with NaBH4/EtOH at −10 °C produced the corresponding alcohols 5α and 5β in a 84:16 ratio respectively (Scheme 1).
:16 ratio, respectively.11,12 Interestingly, the α-anomer 3α was found to be very unstable and converted to the β-anomer 3β13 in the purification process using silicagel column chromatography, probably via ring opening to form α,β-unsaturated aldehyde 4 followed by intramolecular oxa-Michael addition reaction. Whereas, direct reduction of the crude aldehyde mixture (3α and 3β) with NaBH4/EtOH at −10 °C produced the corresponding alcohols 5α and 5β in a 84:16 ratio respectively (Scheme 1).
 | ||
| Scheme 1 Claisen-rearrangement of 2-vinyloxymethyl glucal derivative. | ||
The formation of the major α-C-glycoside can be explained by considering the following facts. (a) Due to the vinylogous anomeric effect,14 an oxocarbenium ion formation can be visualized during the course of the rearrangement. (b) The electrostatic stabilization of oxocarbenium ion by C-3 and C-4 alkoxy groups assume pseudo axial positions in their half chair conformation.15 (c) The nucleophilic attack on the six membered ring oxocarbenium ion occur through a chair like transition state preferably along the axial trajectory.16 Thus, for both the possible conformations, 5H42a and 4H52b, the CR will have an early transition state with bond breaking well in advance with respect to bond making that may lead to the formation of ionic resonance structures 6a and 6b. For transition state 6a the approach of nucleophilic carbon on to the oxocarbenium ion along the stereochemically preferred axial trajectory ensures syn-pentane interaction17 that builds up between the nucleophile and substituent on C-5 as well as smaller syn-butanol18 interaction with the substituent on C-3. Due to these interactions, the transition state 6avia5H4 is destabilized compared to the transition state 6bvia4H5 in the reaction coordinate. As a result, the formation of C-2-methylene-α-C-glycoside 3α is major through the lowest-energy transition sate, namely via4H5.
However, compound 3α suffers with 1,3-diaxial interactions, that drives the formation of α,β-unsaturated aldehyde derivative 4 which might further undergo intramolecular oxa-Michael addition reaction to give stable C-2-methylene-β-C-glycoside 3β during the purification process (Fig. 1). On the other hand, the direct reduction of the crude aldehyde mixture provides the corresponding C-2-methylene-C-glycosides 5α and 5β in 84:16 ratio respectively, reflecting the ratio in the crude aldehyde mixture.
 | ||
| Fig. 1 Proposed mechanism for the Claisen rearrangement of 2-vinyloxymethyl glucal derivative. | ||
Interestingly, galactose derived vinyl ether 7 upon CR provided a mixture of aldehyde 8α as a single diastereomer along with unexpected aldehyde 9 in a ratio of 76:24 respectively. However, column chromatography of this mixture over silicagel provided only the ring opened α,β-unsaturated aldehyde derivative 10.19 A one-pot CR followed by reduction of the crude aldehyde provided alcohols 11 and 12. Due to the difficulty in separation of this alcoholic mixture the crude product was acetylated to provide galactose derived C-2-methylene-C-glycoside 13 as a single diastereomer and C-2 alkylated galactal derivative 14 (Scheme 2).
 | ||
| Scheme 2 Claisen-rearrangement of 2-vinyloxymethyl galactal derivative. | ||
The observation of unexpected aldehyde 9 supports the formation of fully separated ionic resonance structures in the proposed mechanism. As can be expected, the transition state 7a with 5H4 conformation will lead to the ionic resonance structure 15a. The nucleophilic attack at the anomeric position (path a) via15a suffers with higher steric strain that prohibits the formation of C-2-methylene-β-C-glycoside 8β. On the other hand, nucleophilic attack at the less hindered site through a (1,3)-sigmatropic rearrangement (path b) leads to the formation of aldehyde 9 (Fig. 2).
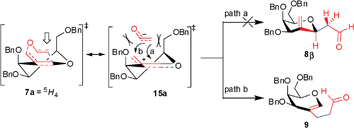 | ||
| Fig. 2 Proposed mechanism for the unexpected formation of C-2-alkyl galactal derivative 9 under Claisen rearrangement reaction conditions. | ||
Encouraged with the above results we further applied this methodology to various 2-vinyloxymethyl-glycal derivatives. Thus, rhamnal derived allylvinyl ether 16 upon CR provided the corresponding C-2-methylene-C-glycosides 17α and 17β in 87:13 ratio, respectively in good yield (Table 1, entry 1). Again, a similar kind of anomerization mentioned in the case of 3α above was observed while carrying out the purification over silicagel leading to the formation of 17β as a single diastereomer.
| Entry | Vinyl ether | C-2 Methylene C-glycoside (%)a |
α:β Ratio |
|---|---|---|---|
| a Yield represents pure and isolated products. b The mixture upon column chromatography provided only 17β as a single diastereomer. c The vinyl ether was subjected to CR and the obtained crude product was directly reduced with NaBH4/EtOH. | |||
| 1 |

|
 b
b
|
87:13 |
| 2 | 16 |
 c
c
|
87:13 |
| 3 |

|
|
50:50 |
| 4 |
|

|
50:50 |
| 5 |

|
|
50:50 |
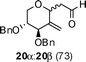
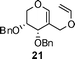
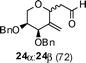
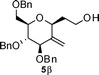
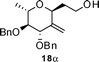
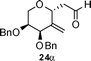
Nevertheless, direct reduction of the crude aldehyde mixture, 17α and 17β, provided C-2-methylene-C-glycosides 18α and 18β in 87:13 ratio respectively in 85% yield (Table 1, entry 2). However, pentose derived allylvinyl ethers 19, 21 and 23 upon thermal CR provided a 50:50 mixture of 20α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :20β, 22α:22β and 24α:24β respectively in good yield (Table 1, entries 3, 4 and 5). These observations clearly indicate the significance of the C-5-substituent in directing the stereochemical outcome of the rearrangement reaction.
:20β, 22α:22β and 24α:24β respectively in good yield (Table 1, entries 3, 4 and 5). These observations clearly indicate the significance of the C-5-substituent in directing the stereochemical outcome of the rearrangement reaction.
The importance of the methodology was further enhanced by applying it to synthesize a series of C-2-methyl-C-glycosides. Thus, C-2-methylene-β-C-glycoside 5β was hydrogenated with 10% Pd/C, H2 in MeOH in the presence of Na2CO320 to provide C-2-methyl-β-C-glycoside 25 as a single diastereomer in excellent yield, 93% (Scheme 3).
 | ||
| Scheme 3 Synthesis of glucose derived C-2-methyl-β-C-glycoside. | ||
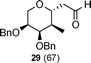
Further, application of the selective hydrogenation protocol to other C-2-methylene-C-glycosides 5β, 18α, 22α and 24α also provided C-2-methyl-C-glycosides 26–29 in excellent yields with very high diastereoselectivity (Table 2).




In conclusion, an efficient methodology for the stereoselective synthesis of C-2-methylene-C-glycosides as well as C-2-methyl-C-glycosides was developed. Importantly, the method is applicable to synthesize α- as well as β-C-glycosides in a stereoselective fashion. This novel method may provide an easy access to the synthesis of natural products possessing carbon branched sugar subunits.
We thank University Grant Commission (UGC), New Delhi, Grant number 40-56/2011 (SR).
Note added after first publication
This article replaces the version published on 16th August 2012, which contained errors in Table 1.References
- A. B. Eldrup, M. Prhavc, J. Brooks, B. Bhat, T. P. Prakash, Q. Song, S. Bera, N. Bhat, P. Dande, P. D. Cook, C. F. Bennett, S. S. Carroll, R. G. Ball, M. Bosserman, C. Burlein, L. F. Colwell, J. F. Fay, O. A. Flores, K. Getty, R. L. LaFemina, J. Leone, M. MacCoss, D. R. McMasters, J. E. Tomassini, D. V. Langen, B. Wolanski and D. B. Olsen, J. Med. Chem., 2004, 47, 5284–5297 CrossRef CAS.
- Please see the supporting information.
- (a) L. Claisen, Ber. Dtsch. Chem. Ges., 1912, 45, 3157–3166 CrossRef CAS; (b) R. E. Ireland and R. H. Mueller, J. Am. Chem. Soc., 1972, 94, 5897–5898 CrossRef CAS.
- For a review see, B. Werschkun and J. Thiem, Top. Curr. Chem., 2001, 215, 293–325 CrossRef CAS.
- (a) R. E. Ireland, C. S. Wilcox, S. Thaisrivongs and N. R. Vanier, Can. J. Chem., 1979, 57, 1743–1745 CrossRef CAS; (b) B. Fraser-Reid, R. D. Dawe and D. B. Tulshian, Can. J. Chem., 1979, 57, 1746–1749 CrossRef CAS; (c) R. E. Ireland, S. Thaisrivongs and C. S. Wilcox, J. Am. Chem. Soc., 1980, 102, 1155–1157 CrossRef CAS; (d) M. P. Edwards, S. V. Ley, S. G. Lister and B. D. Palmer, J. Chem. Soc., Chem. Commun., 1983, 630–633 RSC; (e) R. E. Ireland, P. G. M. Wuts and B. Ernst, J. Am. Chem. Soc., 1981, 103, 3205–3207 CrossRef CAS; (f) R. E. Ireland and M. G. Smith, J. Am. Chem. Soc., 1988, 110, 854–860 CrossRef CAS; (g) H. Y. Godage and A. J. Fairbanks, Tetrahedron Lett., 2000, 41, 7589–7593 CrossRef CAS; (h) H. Y. Godage, D. J. Chambers, G. R. Evans and A. J. Fairbanks, Org. Biomol. Chem., 2003, 1, 3772–3786 RSC.
- (a) R. J. Ferrier and N. Vethaviyasar, J. Chem. Soc., Perkin Trans. 1, 1973, 1791–1793 RSC; (b) D. P. Curran, P. B. Jacobs, R. L. Elliott and B. H. Kim, J. Am. Chem. Soc., 1987, 109, 5280–5282 CrossRef CAS.
- (a) K. Heyns and R. Hohlweg, Chem. Ber., 1978, 111, 1632–1645 CrossRef CAS; (b) L. Cottier, G. Remy and G. Descotes, Synthesis, 1979, 711–712 CrossRef CAS; (c) A. de Raadt and R. J. Ferrier, Carbohydr. Res., 1991, 216, 93–107 CrossRef CAS.
- (a) E. J. Corey, M. Shibasaki and J. Knolle, Tetrahedron Lett., 1977, 18, 1625–1626 CrossRef; (b) O. Hernandez, Tetrahedron Lett., 1978, 19, 219–222 CrossRef; (c) C. Kuroda, P. Theramongkol, J. R. Engebrecht and J. D. White, J. Org. Chem., 1986, 51, 956–958 CrossRef CAS.
- J.-M. Vatele, Tetrahedron, 1986, 42, 4443–4450 CrossRef CAS.
- (a) P. R. Sridhar, P. V. Kumar, K. Seshadri and R. Satyavathi, Chem.–Eur. J., 2009, 15, 7526–7529 CrossRef CAS; (b) P. R. Sridhar, K. Seshadri and G. M. Reddy, Chem. Commun., 2012, 48, 756–758 RSC.
- A one-pot ortho ester-Claisen (Johnson-Claisen) rearrangement of 1 was unsuccessful and provided a complex mixture of products.
- The ratio was calculated by taking the crude 1H NMR and integrating the aldehyde peaks. The stereochemistry at the anomeric center was established by considering the product ratio in the later stages.
- The configuration at the anomeric center was assigned by NOE experiments.
- (a) D. P. Curran and Y.-G. Suh, J. Am. Chem. Soc., 1984, 106, 5002–5004 CrossRef CAS; (b) D. P. Curran and Y.-G. Suh, Carbohydr. Res., 1987, 171, 161–191 CrossRef CAS; (c) R. M. Coates, B. D. Rogers, S. J. Hobbs, D. R. Peck and D. P. Curran, J. Am. Chem. Soc., 1987, 109, 1160–1170 CrossRef CAS.
- (a) J. A. C. Romero, S. A. Tabacco and K. A. Woerpel, J. Am. Chem. Soc., 2000, 122, 168–169 CrossRef CAS; (b) L. Ayala, C. G. Lucero, J. A. C. Romero, S. A. Tabacco and K. A. Woerpel, J. Am. Chem. Soc., 2003, 125, 15521–15528 CrossRef CAS.
- (a) R. V. Stevens and A. W. M. Lee, J. Am. Chem. Soc., 1979, 101, 7032–7035 CrossRef CAS; (b) P. Deslongchamps, Stereoelectronic Effects in Organic Chemistry, Pergamon, New York, 1983 Search PubMed.
- (a) K. B. Wiberg and M. A. Murcko, J. Am. Chem. Soc., 1988, 110, 8029–8038 CrossRef CAS; (b) R. W. Hoffmann, M. Stahl, U. Schopfer and G. Frenking, Chem.–Eur. J., 1998, 4, 559–566 CrossRef CAS.
- K. Ohno, H. Yoshida, H. Watanabe, T. Fujita and H. Matsuura, J. Phys. Chem., 1994, 98, 6924–6930 CrossRef CAS.
- No trace amount of cyclic product was able to be isolated. Probably compound 9 might have decomposed during the column chromatography.
- G. R. Cook, L. G. Beholz and J. R. Stille, J. Org. Chem., 1994, 59, 3575–3584 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: See DOI: 10.1039/c2ra21505e/ |
| This journal is © The Royal Society of Chemistry 2012 |