Maltose-based gelators having azobenzene as light-sensitive unit†
María J.
Clemente
a,
Rosa M.
Tejedor
b,
Pilar
Romero
a,
Juliette
Fitremann
*c and
Luis
Oriol
*a
aInstituto de Ciencia de Materiales de Aragón (ICMA), Universidad de Zaragoza-CSIC, Dpt. Química Orgánica, Facultad de Ciencias, Pedro Cerbuna 12, Zaragoza, 50009, Spain. E-mail: loriol@unizar.es; Fax: +34 976 761209; Tel: +34 976 762276
bCentro Universitario de la Defensa, Academia General Militar, Carretera de Huesca s/n, 50090, Zaragoza, Spain. E-mail: rtejedor@unizar.es
cLaboratoire des IMRCP, UMR CNRS 5623, Bât. 2r1, Université Paul Sabatier, 118 route de Narbonne, F-31062, Toulouse Cedex 9, France. E-mail: fitremann@chimie.ups-tlse.fr; Fax: 05 61 55 81 55; Tel: 05 61 55 62 72
First published on 18th October 2012
Abstract
Three light-sensitive amphiphiles, based on azobenzene, have been synthesized as supramolecular gelators. A hydrophobic chain with an azobenzene group incorporated at different positions was click coupled to a maltose polar head by a copper(I)-catalysed azide–alkyne [3 + 2] cycloaddition. The liquid crystalline and gel properties of these azo-amphiphilic materials have been studied. Two of these azo-gelators containing maltose give rise to stable gels in water or in a mixture of water and DMSO at room temperature. The chiral supramolecular assemblies of these gelators have been characterised by NMR, electron microscopy and circular dichroism (CD). The light-response of azo-amphiphiles in supramolecular gels has been studied. Also azo-gels, which contained mixtures of the azo-amphiphilic compounds and a similar structural hydrogelator, have been investigated.
Introduction
Research on supramolecular gels has increased in recent years due to their potential applications1 as biomaterials, smart materials and electronic devices.2 These types of gels are formed by self-aggregation of low molecular weight gelator molecules, a process that gives rise to a supramolecular structure that can trap organic or aqueous solvents by means of a combination of non-covalent interactions, like H-bonding, π–π stacking, donor–acceptor interactions, solvophobic forces and van der Waals interactions.3 Taking into account the reversibility of these interactions, supramolecular gels are responsive to external stimuli. The type of response depends on the applied stimulus and can affect the supramolecular structure at different hierarchical levels. For instance, by triggering adequate modifications, these materials can be cycled between free-flowing liquids and non-flowing materials. However, other modifications can also be promoted, for example, in the chemical or physical properties, such as colour or conductivity or swelling and shrinking by extension or contraction of the network.4Changes in these supramolecular materials can be triggered by chemical or physical stimuli yielding smart materials. Different supramolecular gels have been reported to be chemo-responsive by host–guest complexation,5 a metal-ion interaction6 and pH changes.7 Apart from the response to temperature and mechanical stress, among the different physical stimuli, light is attractive because it is a remote stimulus that can promote spatially controlled changes. As photoactive gels, there are several examples of luminescent gels8 and phototunable gels. For this last kind of material, there is a structure transformation through a photochemical process of a photochromic unit. The photochromic unit can be a gelator itself or be added as a co-gelator. The gel response is a consequence of the ability of the photochromic unit to alternate between two different chemical forms with light, the two forms displaying different absorption spectra. Most often, the mechanisms involved at the molecular level are trans–cis isomerisation, tautomerisation and electrocycling ring closures and openings. The photoinduced response in gels can be irreversible or reversible.9 Azo dye systems are used in supramolecular assemblies to trigger reversible environmental changes, due to the reversible trans–cis photochemical isomerisation experienced by azobenzenes, where the cis isomer can promote a structural change and disruption.10
Examples of trans-isomer organogels giving rise to a photo-stationary state of trans–cis mixtures that provide a sol state, have been described on aza-crown-appended cholesterol derivatives11 and recently on hydrazine12 and lipid derivatives.13 By contrast, in organogels derived from bis-ureido-azobenzene derivatives, the trans–cis isomerisation is blocked in the gel state.4,14 The co-assembly of an azobenzene derivative with gels having a chiral nanotube structure has also been studied, in which reversible changes were regulated by light switching.15 Most reported examples are related to organogels, due to the difficulty of incorporating a hydrophobic photoresponsive part; however, a few photoresponsive hydrogels based on peptide derivatives have also been reported.16
Carbohydrates provide a water soluble hydrophilic building block and they can act as candidates in the preparation of hydrogelators based on the H-bonding of hydroxyl groups.3c Azobenzene chromophores can be incorporated into the glyco-amphiphilic structure.17 Reported azo-glycolipids can be classified into conventional single-head amphiphiles18 and bolaamphiphiles.19 Changes under irradiation in their structure,20 or gel–sol transition,21 have been described for sugar based azo-gelators.
Apart from their ability to self-assemble in solvents, glycolipids may exhibit liquid crystal (LC) phases due to the polar asymmetry of these compounds. The head groups are capable of H-bonding, while the alkyl chains self-aggregate into microsegregated hydrophobic regions.22 LC phases have also been observed in azo-glycoamphiphiles.23
We have recently studied a series of disaccharide-based hydrogelators.24 Some of them are efficiently synthesised by the click chemistry of a hydrophilic maltose head and a hydrophobic palmitic chain. These structures can promote supramolecular interactions to establish a self-assembled 3D network. The triazole ring gives additional π–π stacking and its dipolar moment increases the hydrophilicity of the amphiphile, contributing to the formation of the hydrogels.3c,25
We report here gels of new maltose-based photoactive gelators in water and DMSO/water mixtures prepared by a copper(I)-catalysed azide–alkyne [3 + 2] cycloaddition, with an azobenzene group at different positions on the hydrophobic chain (Fig. 1). The liquid crystalline behaviour, chiral supramolecular structures and light-response capability of these gels have been explored. Furthermore, a hydrogel was formed by a mixture of gelators, whose chiral arrangement can be easily modulated by light as a remote stimulus without changing the 3D gel structure. These reversible modifications found in a mixture of glycoamphiphiles open up the possibility to use them for rewritable or switchable soft materials.
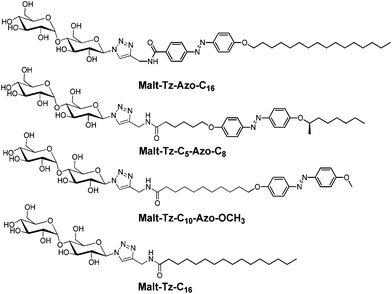 | ||
| Fig. 1 Chemical structure of the synthesized azo-glycolipids with the azobenzene group at different positions and the Malt–Tz–C16 hydrogelator. | ||
Results and discussion
Synthesis of materials
The glyco azo-amphiphiles were synthesised by click coupling maltosylazide and azo-contanining derivatives with a N-propargylamide end group using a copper(I)-catalysed azide–alkyne [3 + 2] cycloaddition. The resulting triazole ring connects the maltose polar head with the hydrophobic part. The hydrophobic part consists of an azobenzene group and an alkyl chain with the azobenzene at different positions: directly linked to the triazole (Malt–Tz–Azo–C16), in the middle of the chain (Malt–Tz–C5–Azo–C8) or at the end of the chain (Malt–Tz–C10–Azo–OCH3) (see Fig. 1).The synthetic pathway of the azo-glycoamphiphiles is shown in Scheme 1. Peracetylated maltose and the azobenzene-containing carboxylic acids, shown in Fig. 2, were used as starting materials. Compounds HOOC–Azo–C16, HOOC–C5–Azo–C8, and HOOC–C10–Azo–OCH3 were prepared by an azo coupling reaction26 by reacting sodium phenoxide and ethyl p-aminobenzoate for HOOC–Azo–C16, (2R)-octyloxyaniline for HOOC–C5–Azo–C8 and p-methoxyaniline for HOOC–C10–Azo–OCH3, respectively. Compound (2R)-octyloxyaniline was prepared from 4-nitrophenol by etherification with (2R)-octanol and further reduction of the nitro group. The carboxylic aliphatic chains were introduced by a Williamson reaction, followed by the hydrolysis of the ester group to yield the desired acid.
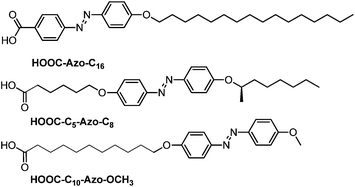 | ||
| Fig. 2 Chemical structure and nomenclature of the azobenzene acids. | ||
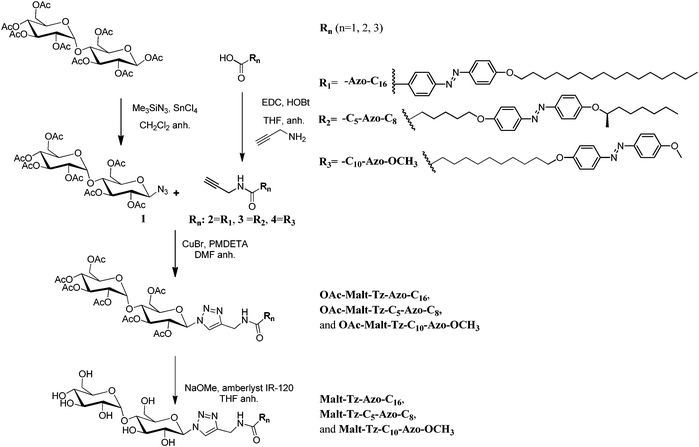 | ||
| Scheme 1 Synthesis of Malt–Tz–Azo–C16, Malt–Tz–C5–Azo–C8, and Malt–Tz–C10–Azo–OCH3 by a click reaction. | ||
Maltosylazide 1 (see Scheme 1) was synthesised in a stereoselective manner by treating maltose octa-acetate with trimethylsilyl azide and tin tetrachloride, as a Lewis acid catalyst, employing the general procedure described by Paulsen.27 This compound can react through a 1,3-dipolar cycloaddition with propargylamides of the azobenzene acids.
To introduce the alkyne, the propargylamide derivatives 2, 3 and 4, were synthesised by EDC coupling reagent with hydroxybenzotriazole. A click reaction was carried out in DMF using CuBr and N-pentamethyldiethylenetriamine (PMDETA) to give the azo-glycosyl products with an 80–90% yield. All of the protected derivatives were deacetylated at room temperature with MeONa and Amberlyst IR120 in anhydrous THF to give the final product at a yield of 75–90%.
Compounds were characterised by 1H NMR, 13C NMR, IR and mass spectrometry (see experimental section and for characterisation of intermediate compounds see SI1, ESI†). Peracetylated compounds (OAc–Malt–Tz–Azo–C16, OAc–Malt–Tz–C5–Azo–C8 and OAc–Malt–Tz–C10–Azo–OCH3) and azo-glycoamphiphilic compounds (Malt–Tz–Azo–C16, Malt–Tz–C5–Azo–C8 and Malt–Tz–C10–Azo–OCH3) were characterised by additional 2D NMR experiments (COSY, TOCSY, NOESY, HSQC and HMBC) to corroborate their chemical structure. As an example of the characterisation studies, Fig. 3 shows 1H NMR experiments on OAc–Malt–Tz–C10–Azo–OCH3 and Malt–Tz–C10–Azo–OCH3, and Fig. 4 shows a 2D TOCSY experiment on OAc–Malt–Tz–C10–Azo–OCH3, where all the sugar ring protons can be assigned. The exact masses of the azo-glycoamphiphiles were determined by mass spectrometry (see SI2, ESI,† for MicroTOF Mass Spectrometry of Malt–Tz–C10–Azo–OCH3 as an example) and the results also confirmed the proposed structures of these materials.
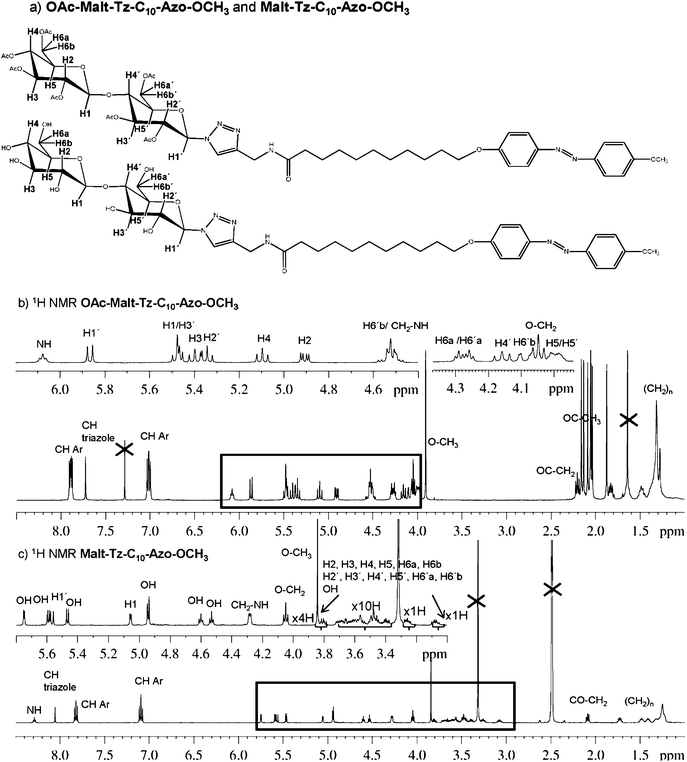 | ||
| Fig. 3 (a) OAc–Malt–Tz–C10–Azo–OCH3 and Malt–Tz–C10–Azo–OCH3 chemical structure and nomenclature, (b) 1H NMR spectrum of OAc–Malt–Tz–C10–Azo–OCH3 with CDCl3 as solvent at 25 °C, (c) 1H NMR spectrum of Malt–Tz–C10–Azo–OCH3 with DMSO as solvent at 25 °C. | ||
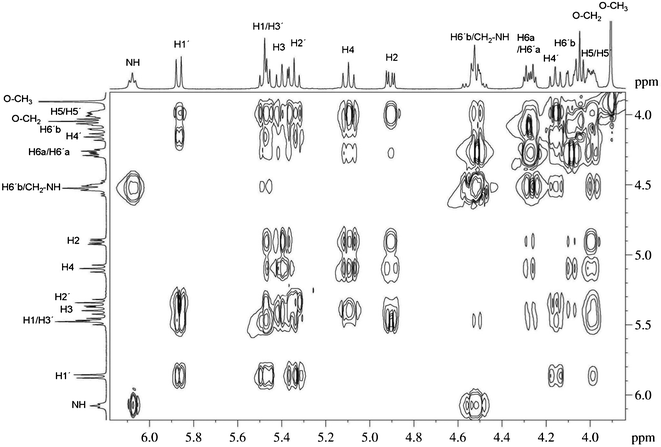 | ||
| Fig. 4 TOCSY experiment of OAc–Malt–Tz–C10–Azo–OCH3 with CDCl3 as solvent at 25 °C and 60 ms of mixing time. | ||
Thermal properties
It is known that carbohydrate amphiphiles usually have liquid crystal properties. These compounds have the ability to self-assemble and undergo microphase segregation due to hydrophobic interactions of aliphatic chains and the extensive hydrogen bonded network formed by the polar carbohydrate heads. Phase transition temperatures are dependent on the nature of the carbohydrate moiety, the length of the hydrophobic alkyl chain and the type of link between the two parts, as was previously reported for Malt–Tz–C16 and analogous compounds.24 The thermal properties of the synthesised azo-glycolipids were studied by thermogravimetric analysis (TGA), polarised light microscopy and differential scanning calorimetry (DSC). Compounds were observed after drying under vacuum.In peracetylated precursors, 5% weight losses were observed at temperatures close to 300 °C. However, thermogravimetric curves for deprotected azo-glycolipids display 5% weight losses at temperatures of 170–245 °C (in samples previously dried and immediately analysed). See SI3, ESI,† for the TGA analysis.
The peracetylated precursors were studied by polarised optical microscopy and DSC as a function of temperature (see Table 1). Mesomorphic behaviour was not observed in OAc–Malt–Tz–Azo–C16 and OAc–Malt–Tz–C10–Azo–OCH3, and the compounds melted directly from a crystalline state to an isotropic liquid. OAc–Malt–Tz–C5–Azo–C8 was an amorphous solid having a glass transition at around 57 °C (DSC).
| Compound | Thermal transition (°C) [ΔH (kJ mol−1)]a |
|---|---|
| a DSC thermal cycles were carried out in a nitrogen atmosphere (10 °C min−1). b The heating cycles were carried out up to 200 °C. Data corresponding to the second heating scan. c The first and second heating cycles were carried out up to 120 °C. d Data from polarised light microscopy. Cr = crystal, I = isotropic liquid, g = glassy state, Sm = smectic phase. | |
| OAc–Malt–Tz–Azo–C16b | Cr 171 [48.3] I |
| OAc–Malt–Tz–C5–Azo–C8b | g 57 I |
| OAc–Malt–Tz–C10–Azo–OCH3b | Cr 132[38.4] I |
| Malt–Tz–Azo–C16c | Cr 58 [5,2] Cr′ 79 [3.3] Cr′′ 140d Sm |
| Malt–Tz–C5–Azo–C8c | g 78 Sm |
| Malt–Tz–C10–Azo–OCH3c | g 67 Sm + Dec. |
Decomposition of azo-glycolipids was observed by optical microscopy at temperatures above 170 °C and the sample became brown, most probably due to decomposition of the sugar units.28 An experiment performed up to 200 °C showed decomposition at around 170–175 °C, which was confirmed by DSC for Malt–Tz–Azo–C16 and Malt–Tz–C5–Azo–C8; while for Malt–Tz–C10–Azo–OCH3, the decomposition started at 120 °C and a decrease of the baseline was observed, according to TGA experiments. DSC studies of the azo-glycolipids were then performed by heating the compounds to 120 °C (maximum) to prevent the thermal decomposition of the samples. Under these conditions, the second and successive scans were reproducible. From the DSC measurements, Malt–Tz–Azo–C16 has a thermal transition at around 80 °C; however, above this temperature, the sample is highly viscous and difficult to characterise by optical microscopy. Nevertheless, using optical microscopy, the sample becomes more fluid at temperatures above approximately 140 °C, and it can be characterised as a liquid crystal according to the optical observations. Malt–Tz–C5–Azo–C8 has a glass transition, observed by DSC measurement, at around 78 °C and also exhibits a birefringent texture corresponding to a highly viscous mesophase (see SI4, ESI†). In Malt–Tz–C10–Azo–OCH3, a glass transition was observed at 67 °C for the second and third cycles.
In an effort to identify the mesophase by XRD, attempts were made to obtain oriented fibres of the compounds. A fibre of Malt–Tz–C5–Azo–C8 compound was obtained around 160 °C; at lower temperatures any fibre can be drawn. Oriented fibres of Malt–Tz–Azo–C16 and Malt–Tz–C10–Azo–OCH3 could not be obtained. X-ray patterns of Malt–Tz–C5–Azo–C8 fibre were recorded at room temperature for 15 h. Bragg reflections were found in the low-angle region corresponding to the second, third and fifth lamellar order. The lamellar spacing is close to 61.6 Å, which is larger than the length of one molecule (44.1 Å), but smaller than twice the extended molecular length. This result indicates that an interdigitated bilayer is probably formed. In the high angle region a diffuse, broad maximum was found.
Gel properties
The solubility and gelation abilities of the azo-glycolipids were examined in different solvents by dissolving the compound in the corresponding solvent with a concentration in the range 0.5–5% by weight. Malt–Tz–Azo–C16 and Malt–Tz–C10–Azo–OCH3 azo-glycoamphiphiles are not soluble at room temperature (RT) in any of the selected solvents, except DMSO. However, the Malt–Tz–C5–Azo–C8 compound has a different behaviour: it is soluble at RT in solvents such as chloroform, THF, DMF, DMSO, methanol and the DMSO/water mixture. It is not soluble in water at RT, unless the water is heated and then cooled to RT.The results for the three compounds in the selected solvents are summarised in Table 2. If the compound was not soluble at RT, the mixtures were first dissolved by heating and then cooling to RT, with a solution, precipitate or gel being observed depending on the solvent. Malt–Tz–Azo–C16 gelates in water at a minimum gelation concentration of 5 wt% to form a gel in the absence of other organic solvents. This gel is opaque, but becomes a little transparent in a DMSO/water mixture, as can be seen in Fig. 5. The minimum gelation concentration decreases to 1.5 wt% for the mixture of the two solvents (DMSO/water, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w). Malt–Tz–C10–Azo–OCH3 also formed a gel in a DMSO/water mixture (1:1 w/w) at 2 wt% as a minimum gelation concentration; this gel is also opaque. These molecules have a highly hydrophobic tail and the presence of an organic co-solvent such as DMSO, in addition to water, is required for complete solubilisation on cooling and subsequent gel formation. In all the cases, the gels are stable at RT and thermoreversible. Sol states were reached by heating the septum-capped test tube in a block heater, for Malt–Tz–Azo–C16 (5.0 wt% water) at 90 °C, Malt–Tz–Azo–C16 (1.5 wt% DMSO/water 1:1 w/w) at 85 °C and for Malt–Tz–C10–Azo–OCH3 (2.0 wt% DMSO/water 1:1 w/w) at 100 °C. However, Malt–Tz–C5–Azo–C8 cannot form a gel even in mixtures of solvents.
:1 w/w). Malt–Tz–C10–Azo–OCH3 also formed a gel in a DMSO/water mixture (1:1 w/w) at 2 wt% as a minimum gelation concentration; this gel is also opaque. These molecules have a highly hydrophobic tail and the presence of an organic co-solvent such as DMSO, in addition to water, is required for complete solubilisation on cooling and subsequent gel formation. In all the cases, the gels are stable at RT and thermoreversible. Sol states were reached by heating the septum-capped test tube in a block heater, for Malt–Tz–Azo–C16 (5.0 wt% water) at 90 °C, Malt–Tz–Azo–C16 (1.5 wt% DMSO/water 1:1 w/w) at 85 °C and for Malt–Tz–C10–Azo–OCH3 (2.0 wt% DMSO/water 1:1 w/w) at 100 °C. However, Malt–Tz–C5–Azo–C8 cannot form a gel even in mixtures of solvents.
 | ||
| Fig. 5 Left: gels of Malt–Tz–Azo–C16 5 wt% water and 1.5 wt% DMSO/water. Right: gel of Malt–Tz–C10–Azo–OCH3, 2 wt% DMSO/water. | ||
| Solvent | Malt–Tz–Azo–C16 | Malt–Tz–C5–Azo–C8 | Malt–Tz–C10–Azo–OCH3 | |
|---|---|---|---|---|
| I = insoluble, P = precipitate, S = solution, NT = not tested, G = gel (minimum gelation concentration).a Soluble on heating for 2.5 wt%. | ||||
| Toluene | I | I | I | |
| Chloroform | I | S | I | |
| THF | P | S | P | |
| Dodecanol | I | I | I | |
| Acetone | I | I | I | |
| DMF | P | S | P | |
| DMSO | S | S | S | |
| Methanol | I | S | I | |
| Water | G (5 wt%) | Sa | I | |
| DMSO/water | G (1.5 wt%, 1:1 w/w) |
S | G (2 wt% 1:1 w/w) |
|
To corroborate the presence of π–π stacking and H-bonding interactions, 1H-NMR experiments were performed to study the groups involved in the self-assembly. An experiment was carried out starting from a DMSO solution of our amphiphile and subsequent addition of water according to the experiment described by Suzuki and coworkers.29 For example, Fig. 6a shows the 1H NMR spectra of the Malt–Tz–C10–Azo–OCH3 compound (2.9 mg) in 0.40 mL DMSO-d6 at RT from 8.50 to 6.90 ppm (see SI5, ESI,† for spectra from 3.00 to 6.00 ppm). The spectra were recorded upon successive additions of 0.04 mL water to the DMSO-d6 solution. The signals corresponding to CH or CH2 of the carbohydrate remain unchanged. Meanwhile a displacement of the rest of the signals was observed up to the addition of 0.16 mL water (1:0.4 DMSO/water). A shielding was found for the H signal of the triazole ring (Δδ(HTz) = 0.072 ppm) and for the H signals of the azobenzene aromatic ring (Δδ(Harom) = 0.036 ppm in both signals). These azobenzene hydrogens have a similar shift in DMSO-d6 solution, but become unequally shifted as the aggregation takes place, due to parallel displacement of the rings, minimising the unfavourable electrostatic effects in the π–π stacked arrangements.30 This supports the contribution of π–π stacking to the aggregation of the amphiphiles promoted by water addition. A simultaneous deshielding for the NH signal is detected (Δδ(HNH) = 0.075 ppm) as well as in the OH signals, which can be assigned to H-bonding interactions. These facts are in accordance with self-assembly upon addition of water, which finally led to a swollen gel aggregate macroscopically detected in the NMR tube.
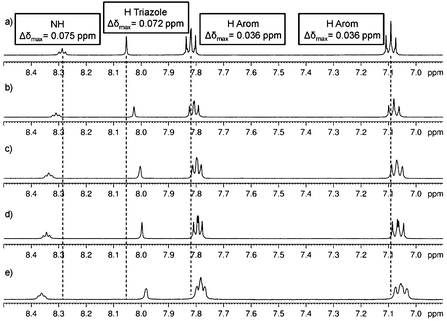 | ||
| Fig. 6
1H NMR experiments in DMSO-d6 increasing water concentration. Initial solution (a) 2.9 mg Malt–Tz–C10–Azo–OCH3 in 0.40 mL DMSO-d6. Addition of water to the initial solution: (b) 0.04 mL H2O (1:0.1 DMSO/water), (c) 0.08 mL H2O (1:0.2 DMSO/water), (d) 0.12 mL H2O (1:0.3 DMSO/water) and (e) 0.16 mL H2O (1:0.4 DMSO/water). | ||
Different solubilities and gelation abilities were observed in the synthesised compounds, related to the position of the azobenzene inside the hydrophobic chain. When the azobenzene group is located in the middle (Malt–Tz–C5–Azo–C8), the compound is soluble in several organic solvents and surprisingly soluble in water, while the azobenzene being located at the edge means a gel can form. If it is directly linked to the sugar polar head, the compound (Malt–Tz–Azo–C16) gelates in water (and water/DMSO), but when the azobenzene is at the end of the hydrophobic chain (Malt–Tz–C10–Azo–OCH3), the gel had to be formed in a mixture of a solubilising solvent, DMSO, and a non-solubilising solvent, water.
The self-assembled structures of the gels derived from Malt–Tz–Azo–C16 and Malt–Tz–C10–Azo–OCH3 were studied by electron microscopy (SEM and TEM), which revealed a characteristic fibrillar network of supramolecular gels.3,4,9
SEM measurements of the xerogels show bundles of fibres, which form the 3D network responsible for the gel structure. The fibres of the Malt–Tz–Azo–C16 gel, either in water or in a mixture of 1:1 DMSO/water (w/w), show diameters of around 40–60 nm. However, fibres from gels in water seem to be shorter than for the mixture of solvents, where they are several μm in length. Moreover, some wider fibres of around 100 nm in diameter can also be measured in the gel from the mixture of solvents (see SI6, ESI†). Malt–Tz–C10–Azo–OCH3 xerogel images show fibre diameters mainly around 200–250 nm, although narrower fibres of around 80 nm can also be observed (see Fig. 7a).
 | ||
| Fig. 7 (a) SEM image Malt–Tz–C10–Azo–OCH3 xerogel (2.0 wt% DMSO/water 1:1 w/w), (b) SEM image of a single twisted ribbon. | ||
Fibrillar structures were also observed by TEM. However, in this technique, a dried dilute solution of the gel must be used (0.1 wt% in water or DMSO/water) and negative staining with uranyl acetate was done. The observed fibres were over 10–20 nm in diameter for the Malt–Tz–Azo–C16 and Malt–Tz–C10–Azo–OCH3 samples, see SI6, ESI.†
Torsion was detected on the microphotographs, specifically with Malt–Tz–Azo–C16 (see TEM microphotograph in SI6, ESI†) and Malt–Tz–C10–Azo–OCH3 DMSO/water gels; Fig. 7b shows a single Malt–Tz–C10–Azo–OCH3 twisted ribbon observed by SEM. This torsion could be related to the molecular chirality of the molecules, which is traduced into the chiral helicity of the supramolecular arrangement.
To obtain hydrogels with a lower ratio of azo-derivatives, mixtures were prepared with an azo-glycolipid (Malt–Tz–C5–Azo–C8 or Malt–Tz–Azo–C16) and a previously known hydrogelator Malt–Tz–C16 (see Fig. 1). Malt–Tz–C5–Azo–C8 and Malt–Tz–Azo–C16 azo-glycolipids were selected because of their structural similarity to the hydrogelator and their solubility or ability to gelify in water. Malt–Tz–C10–Azo–OCH3 was not used because it is not soluble in water. In these cases, CD and absorption spectra were recorded from 200 to 600 nm to investigate both regions corresponding to the triazole and azobenzene groups; in contrast to the gels made from DMSO/water mixtures, where only the azobenzene group region can be observed. Gel I was formed by Malt–Tz–C5–Azo–C8 and Malt–Tz–C16 (1:10 molar ratio), at 1 wt% of Malt–Tz–C16 hydrogelator in water. In this mixture, the azo-amphiphile Malt–Tz–C5–Azo–C8 is able to gelate. In other mixtures with an increasing amount of the azo-compound, no gel formation was observed. To compare the influence of the azoamphiphile, Gel II was formed by Malt–Tz–Azo–C16 and Malt–Tz–C16, in the same ratio (1:10 molar ratio), at 1 wt% of the Malt–Tz–C16 hydrogelator in water. Microscopy observations show a similar fibrillar structure forming in these mixtures, when compared to the Malt–Tz–C16 compound alone.24
Study and control of the chiral supramolecular arrangement
The presence in the supramolecular gels of photoactive units as the azobenzene moieties allows the control of their nanostructure and subsequently of the gel properties. If a chiral organisation is expected in the gel, CD spectroscopy is of great interest to characterise the chiral assemblies and potential changes in the organisation. For this purpose, UV-vis and CD spectra of the azo-gelators were registered simultaneously in DMSO solutions as well as in gel states.In DMSO solutions with similar absorbance values to the gel states (5 × 10−5 M), the λmax in the UV absorption spectrum appears at 362 nm for all the compounds of Malt–Tz–Azo–C16, Malt–Tz–C5–Azo–C8 and Malt–Tz–C10–Azo–OCH3 (see SI7, ESI†), while the solutions were CD silent. These spectroscopic results point to the azobenzene chromophores being isolated in the DMSO solutions, with no aggregation being detected. However, a Malt–Tz–C5–Azo–C8 water solution, with a similar absorption to Gel I (4.5 × 10−5 M), exhibits a maximum absorption of the π–π* band of the azobenzene groups at 343 nm, i.e. the maximum absorption of the isolated azobenzene groups is blue-shifted. Moreover, CD was observed in the water solution of Malt–Tz–C5–Azo–C8. These results indicate azobenzene chromophores are H-aggregated in the water solution (see SI8, ESI†).
The absorption spectrum of Malt–Tz–C10–Azo–OCH3 gel (2 wt% DMSO/water, 1:1), see Fig. 8, exhibits a λmax at 335 nm, which is blue shifted with respect to the λmax of the free azobenzene units in DMSO solutions. This points to the azobenzene moieties being associated in the H-aggregates. Moreover, a negative Cotton effect is detected in the CD spectrum, corresponding with the absorption band of the azobenzene units. The UV-vis and CD spectra of the Malt–Tz–Azo–C16 gel (1.5 wt% DMSO/water, 1:1 w/w) could not be performed due to the sample crystallising inside the silica sandwich.
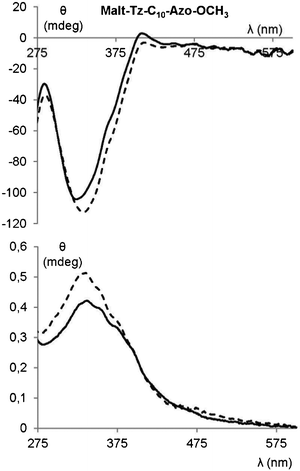 | ||
| Fig. 8 CD spectrum (top) and absorption spectrum (bottom) of Malt–Tz–C10–Azo–OCH3 gel (2 wt% DMSO/water 1:1) without irradiation (solid line), and later 2 h and 30 min of irradiation at 365 nm (dashed line). | ||
The presence of azobenzene groups can be used to induce modifications of the supramolecular structure of the gel by the irradiation of UV light. However, after UV irradiation at 365 nm for 150 min, no changes were detected in the UV-vis and CD spectra of Malt–Tz–C10–Azo–OCH3 gel (2 wt% DMSO/water 1:1) (see Fig. 8). The gel is stable under these conditions, indicating that trans–cis isomerisation of the azobenzene groups does not occur, probably because the dense packing of the azobenzene groups prevents the trans–cis reactivity.31
Multi-component supramolecular gels32 have been previously studied because the mixture of components33 allows tailoring of the properties of the resulting gel. If two components, which are themselves gelators are used, either co-gels or self-sorting gels can be formed. Azo mixtures have been studied by the potential light control of the gel structure through azo isomerisation.34 Furthermore, mixtures of amphiphiles based on sugars have also been described as self-sorted gels.35 In our study, sugar and azo based gelators of similar structure are combined in Gel I and Gel II in order to achieve a co-assembled photoresponsive structure.
The π–π* bands of the azobenzene units in the UV-vis spectra of Gel I (Malt–Tz–C5–Azo–C8 as photoactive compound) and Gel II (Malt–Tz–Azo–C16 as photoactive compound) show a hypsochromic shift in relation to the band of free azobenzene chromophores, suggesting H-aggregation. Both gels show CD signals due to the triazole and azobenzene groups (see Fig. 9), however there are some notable differences. At a shorter wavelength, Gel I exhibits a Cotton effect around 240 nm due to the absorption band of the triazole group, which is directly bonded to the chiral group (maltose unit). At a longer wavelength, a negative Cotton effect associated with the π–π* absorption band of the azobenzene moiety is detected. Moreover, the CD spectrum of the fresh Gel II seems to show two positive couplets, corresponding to triazole groups (shorter wavelength) and azobenzene units (longer wavelength). Surprisingly, after storing Gel II for 6 h, the ellipticity values of the negative bands increase and a new CD band is detected at 365 nm. A reorganisation of the sample probably occurs13 in the sandwich formed under measurement conditions.
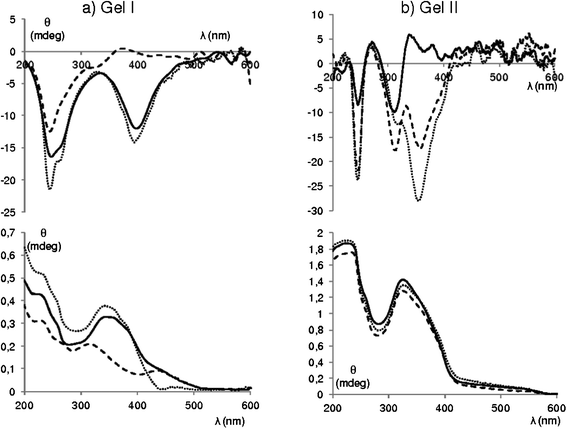 | ||
| Fig. 9 (a) CD (top) and absorption (bottom) spectrum of fresh Gel I, solid line; after 2 min of irradiation with UV light at 365 nm, dashed line; stored after 24 h irradiation, dotted line. (b) CD (top) and absorption (bottom) spectrum of fresh Gel II, solid line; later 6 h in the sandwich, dotted line, later 150 min of irradiation with UV light at 365 nm, dashed line. | ||
Both Gel I and Gel II were irradiated with UV light at 365 nm to evaluate the effect on the supramolecular organisation of the trans–cis isomerisation of the azobenzene groups. The UV-vis spectrum of Gel I (see Fig. 9) after 365 nm irradiation, clearly shows the presence of a cis-azoisomer: the absorption decreases at 365 nm and increases at 450 nm. The photostationary state is reached after irradiation for 2 min. The trans–cis isomerisation reduces the trans-azoisomer concentration; thus the negative CD signal related to the azobenzene group disappears, while the negative signal corresponding to the triazole only decreases. It can be deduced that gel–sol transition does not occur as the signals were not fully extinct. This was macroscopically corroborated by gel irradiation on a tube: gel disruption was not detected and only a colour change from red to yellow was obtained, probably due to partial disruption of the aggregates.13 When the sample was kept for 24 h in the dark, a total recovery of the initial UV-vis and CD spectra was achieved. After irradiation for 150 min, Gel II showed no evidence of trans–cis isomerisation, but the CD band at 365 nm was seen to increase (see Fig. 9). This band is probably caused by a photoinduced reorganisation of the chiral supramolecular structure, or a simple reorganisation over time involving the azobenzene groups. By storing the sample for 24 h in the dark, the CD and absorption signals were not modified.
Gel I works as a photoresponsive mixture, while Gel II has no response to light. This may be due to the presence of the azobenzene in the middle of the hydrophobic chain, in contrast to the azobenzene derivative in Gel II, which is directly linked to the sugar polar head (see SI9, ESI,† for a schematic representation of this effect).
Experimental section
Characterization data (elemental analysis, 1H and 13C NMR, FTIR and MS) for the intermediate compounds (1–4) are collected in the ESI,† see SI1. Only data corresponding to the azobenzene acid precursors, peracetylated and final azo-amphiphilic glycolipids are included in this section.HOOC–Azo–C16 (C29H42N2O3)
1H NMR (400 MHz, DMSO-d6, 70 °C, δ (ppm)): 0.85 (t, 3H, J = 6.8 Hz, –(CH2)12–CH3), 1.17–1.52 (m, 24H, –(CH2)12–CH3), 1.69–1.80 (m, 2H, –O–CH2–CH2–), 4.10 (t, 2H, J = 6.6 Hz, –O–CH2–CH2–), 7.10–7.14 (m, 2H, HAr), 7.86–7.92 (m, 4H, HAr), 8.09–8.12 (m, 2H, HAr).13C NMR (100 MHz, DMSO-d6, 70 °C, δ (ppm)): 14.2 –(CH2)12–CH3, 22.4, 25.8, 29.0, 29.0, 29.3, 29.3, 29.3, 31.6 –(CH2)15–, 68.7 –O–CH2, 115.4, 122.5, 125.3, 130.9 CHAr, 132.8, 146.8, 155.2, 162.6 CAr, 167.1 COOH. ESI: 467.2 [M + H]+. IR (KBr, cm−1): 2953, 2919, 2868, 2850, 1680, 1601, 1582, 1501, 1470, 1418, 1404, 1303, 1290, 1248, 1143, 1107, 1026, 941, 864, 838, 809, 775, 721, 544.HOOC–C5–Azo–C8 (C26H36N2O4)
1H NMR (400 MHz, DMSO-d6, 50 °C, δ (ppm)): 0.85 (t, 3H, J = 6.8 Hz, –(CH2)5–CH3), 1.26 (d, 3H, J = 6.4 Hz, –CH–CH3), 1.20–1.80 (m, 16H, –CH2–), 2.24 (t, 2H, J = 7.3 Hz, –CH2–COOH), 4.05 (t, 2H, J = 6.4 Hz, –O–CH2–CH2–), 4.55 (m, 1H, –O–CH–CH3), 7.07–7.10 (m, 4H, HAr), 7.79–7.82 (m, 4H, HAr).13C NMR (100 MHz, DMSO-d6, 50 °C, δ (ppm)): 13.9 –CH2–CH3, 19.4 CH3–O–, 21.9, 24.2, 24.7, 25.0, 28.3, 28.6, 31.2, 33.5, 35.7 –CH2–, CO–CH2–CH2–, 67.7 O–CH2–CH2, 73.4 –O–CH–CH3, 114.9, 115.7, 124.0, 124.1 CHAr × 8, 145.8, 146.0, 160.0, 160.8 CAr × 4, 174.4 NH–CO–CAr. ESI: 441.1 [M + H]+, 463.1 [M + Na]+. IR (KBr, cm−1): 2936, 2859, 2868, 1706, 1600, 1578, 1498, 1473, 1464, 1314, 1257, 1241, 1206, 1146, 1111, 1059, 1042, 1006, 972, 961, 935, 854, 845, 553.HOOC–C10–Azo–OCH3 (C24H32N2O4)
1H NMR (400 MHz, DMSO-d6, 45 °C, δ (ppm)): 1.17–1.53 (m, 14H, –(CH2)7–), 1.68–1.77 (m, 2H, CH2–CH2–O), 2.15 (t, 2H, J = 7.6 Hz, –CH2–CO), 3.84 (s, 3H, O–CH3), 4.05 (t, 2H, J = 6.8 Hz, –O–CH2–CH2–), 7.03–7.12 (m, 4H, CHAr), 7.76–7.85 (m, 4H, CHAr). 13C NMR (100 MHz, DMSO-d6, 45 °C, δ (ppm)): 25.0, 25.9, 29.0, 29.1, 29.2, 29.3 –(CH2)8–, 34.4 CH2–CH2–CO, 56.0 –O–CH3, 68.5 –O–CH2–CH2–, 115.0, 115.5, 124.5, 124.5 CHAr, 146.7, 146.8, 161.4, 161.9 CAr, 174.9 CO–NH. ESI: 413.1 [M + H]+, 435.1 [M + Na]+. IR (KBr, cm−1): 2936, 2916, 2848, 1709, 1620, 1582, 1497, 1469, 1465, 1317, 1296, 1278, 1246, 1151, 1107, 1029, 1018, 843, 823, 558.Synthesis of hepta-O-acetyl-β-maltosyl azide 1
Trimethylsilyl azide (543 μl, 4.13 mmol) and tin tetrachloride (173 μl, 1.48 mmol) were added, at room temperature and under argon, to a solution of β-D-maltose octaacetate (2.00 g, 2.95 mmol) in dry CH2Cl2 (6 mL, 0.5 M). The reaction mixture was stirred at room temperature and the reaction was monitored by TLC (6:4 hexane/ethyl acetate). After 24 h, CH2Cl2 was added and the solution was washed with saturated Na2CO3 and twice with water. The organic layer was dried over MgSO4, filtered and evaporated under reduced pressure. The product was purified by flash chromatography using hexane/ethyl acetate 6:4. A white solid was obtained (1.59 g, 80%). For characterization data see ESI.†
Synthesis of propargylamide azo derivatives 2, 3 and 4
Azobenzene acids (1.05 g, 2.25 mmol) and hydroxybenzotriazole (0.35 g, 2.60 mmol) were dissolved in 20 mL anhydrous tetrahydrofuran. Propargylamine (0.16 mL, 2.50 mmol) was added. The solution was cooled to 0 °C. A solution of EDC (418 mg, 2.18 mmol) in 15 mL anhydrous THF was added. The reaction mixture was stirred for 2 days at room temperature or under gentle heating. The reaction was monitored by TLC with hexane/ethyl acetate 7:3 as the eluent. The mixture was filtered and the solvent was removed under reduced pressure. 250 mL dichloromethane was added and the organic phase was washed three times with 1 M KHSO4 solution, and three times with 1 M NaHCO3 solution. The organic layer was dried over anhydrous MgSO4. The solution was filtered and the solvent was removed under reduced pressure. The resulting white solid was purified by recrystallization or flash chromatography. A red solid was obtained (yield around 40–70% depending on the compound). For characterization data see ESI.†
Synthesis of acetylated maltose conjugates OAc–Malt–Tz–Azo–C16, OAc–Malt–Tz–C5–Azo–C8, and OAc–Malt–Tz–C10–Azo–OCH3
Propargyl derivatives (433 mg, 0.83 mmol), maltosylazide (562 mg, 0.85 mmol), copper(I) bromide (27.1 mg, 0.19 mol) and N-pentamethyldiethylenetriamine (PMDETA) (35 μL, 0.17 mmol) were dissolved in anhydrous dimethylformamide (6 mL) under an argon atmosphere. The mixture was stirred at room temperature for 2 days. The reaction was monitored by TLC with hexane/ethyl acetate 1:1 as the eluent. The catalyst was removed by filtration and the solvent was removed under reduced pressure. The reaction was poured into 150 mL water. The aqueous phase was extracted three times, each with 150 mL hexane/ethyl acetate 1:1. The organic phase was dried with anhydrous MgSO4. The solution was filtered and the solvent was removed under reduced pressure. The resulting solid was purified by flash chromatography using dichloromethane/ethyl acetate 6:4, initially, and then increasing the polarity. A red solid was obtained (819 mg, 82–90%).
OAc–Malt–Tz–Azo–C16 (C58H80N6O19)
1H (400 MHz, CDCl3, 25 °C, δ (ppm)): 0.87 (t, 3H, J = 6.7 Hz, –(CH2)11–CH3), 1.18–1.40 (m, 22H, –CH2–(CH2)11–CH3), 1.41–1.52 (m, 2H, –CH2–(CH2)11–CH3), 1.78–1.86 (m, 2H, –O–CH2–CH2–), 1.85 (s, 3H, CH3–CO–O–C2′), 2.01 (s, 3H), 2.03 (s, 3H), 2.06 (s, 3H), 2.10 (s, 3H), 2.12 (s, 3H), 2.17 (s, 3H), (CH3–CO–O–), 3.97–3.99 (m, 2H, H5′, H5), 4.05–4.10 (m, 4H, –O–CH2–CH2, H4′, H6b), 4.23–4.27 (m, 2H, H6a, H6′a), 4.49 (dd, 1H, J5′,6′b = 1.8 Hz, J6′a,6′b = 12.5 Hz, H6′b), 4.69–4.79 (m, 2H, NH–CH2–triazole), 4.88 (dd, 1H, J1,2 = 3.9 Hz, J2,3 = 10.5 Hz, H2), 5.07 (dd, 1H, J3,4 = 9.8 Hz, J4,5 = 9.8 Hz, H4), 5.34 (dd, 1H, J1′,2′ = 9.1 Hz, J2′,3′ = 9.3 Hz, H2′), 5.37 (dd, 1H, J2,3 = 10.5 Hz, J3,4 = 9.8 Hz, H3), 5.44 (d, 1H, J1,2 = 3.9 Hz, H1), 5.46 (dd, 1H, J2′,3′ = 9.3 Hz, J 3′,4′ = 9.1 Hz, H3′), 5.87 (d, 1H, J1′,2′ = 9.1 Hz, H1′), 6.91 (t, 1H, J = 5.5 Hz, –NH–CO), 6.99–7.01 (m, 2H, HAr), 7.81 (s, 1H, CH–triazole), 7.90–7.93 (m, 6H, HAr). 13C (100 MHz, CDCl3, 25 °C, δ (ppm)): 14.1 (CH2)14–CH3, 20.2, 20.6, 20.7, 20.8, 20.8 CH3–CO–O–, 22.7, 26.0, 29.2, 29.4, 29.5, 29.5, 29.6, 29.7, 30.9, 31.9 –(CH2)14–CH3, 35.4 triazole–CH2–NH, 61.5 C6, 62.4 C6′, 67.9 C4, 68.4 O–CH2–(CH2)14, 68.8 C5, 69.2 C3, 70.0 C2, 70.9 C2′, 72.4 C4′, 75.0 C3′, 75.4 C5′, 85.4 C1′, 95.9 C1, 114.8 CHAr, 121.2 CH–triazole, 122.6, 125.2, 127.9 CHAr, 134.9 CAr, 145.1 CHtriazole–Ctriazole–CH2, 146.8, 154.6, 162.3 CAr, 166.8 NH–CO–CAr, 169.1 CH3–CO–O–C2′, 169.4 CH3–CO–O–C4, 169.9, 169.9 CH3–CO–O–C3/C3′, 170.3, 170.5, 170.6 CH3–CO–O–C2/C6/C6′. MALDI-TOF (DCTB + NaTFA): 1187, 6 [M + Na]+. IR (KBr, cm−1): 3351, 2922, 2851, 1749, 1644, 1604, 1532, 1503, 1470, 1370, 1234, 1141, 1036, 859.OAc–Malt–Tz–C5–Azo–C8 (C55H74N6O20)
1H (400 MHz, CDCl3, 25 °C, δ (ppm)): 0.90 (t, 3H, J = 7.0 Hz, –(CH2)5–CH3), 1.27–1.89 (m, 16H, –CH2–), 1.35 (d, 3H, J = 6.2 Hz, CH3–CH–O), 1.87 (s, 3H), 2.03 (s, 3H), 2.04 (s, 3H), 2.05 (s, 3H), 2.08 (s, 3H), 2.13 (s, 3H), 2.15 (s, 3H) (CH3–CO–O–), 2.26 (t, 2H, J = 7.6 Hz, CO–CH2–(CH2)3), 3.96–4.02 (m, 2H, H5, H5′), 4.04 (t, 2H, J = 6.4 Hz, –O–CH2–CH2), 4.05–4.11 (m, 1H, H6b), 4.15 (dd, 1H, J3′,4′ = 8.8 Hz, J4′,5′ = 9.8 Hz, H4′), 4.25–4.30 (m, 2H, H6a, H6′a), 4.43–4.58 (m, 4H, H6′b, O–CH–CH3–, Ctriazole–CH2–NH–), 4.90 (dd, 1H, J1,2 = 3.9 Hz, J2,3 = 10.5 Hz, H2), 5.09 (dd, 1H, J3,4 = 9.9 Hz, J4,5 = 9.9 Hz, H4), 5.34 (dd, 1H, J1′,2′ = 9.3 Hz, J2′,3′ = 9.5 Hz, H2′), 5.39 (dd, 1H, J2,3 = 10.5 Hz, J3,4 = 9.9 Hz, H3), 5.46 (d, 1H, J1,2 = 3.9 Hz, H1), 5.47 (dd, 1H, J2′,3′ = 9.5 Hz, J3′,4′ = 8.8 Hz, H3′), 5.87 (d, 1H, J1′,2′ = 9.3 Hz, H1′), 6.11 (t, 1H, J = 5.7 Hz, CH2–NH–CO), 6.98–6.99 (m, 4H, HAr), 7.73 (s, 1H, CH–triazole), 7.85–7.80 (m, 4H, HAr). 13C (100 MHz, CDCl3, 25 °C, δ (ppm)): 14.1 (CH2)5–CH3, 19.7 CH–CH3, 20.2 CH3–CO–O–C2′, 20.6, 20.7, 20.8, 20.8 CH3–CO–O– 22.6, 25.2, 25.5, 25.7, 28.9, 29.2, 31.8 –(CH2)–, 34.9 Ctriazole–CH2–NH, 36.3 –CO–CH2–CH2–, 36.4 CH3–CH–CH2–, 61.4 C6, 62.4 C6′, 67.9 C4, O–CH2–CH2, 68.8 C5, 69.2 C3, 70.0 C2, 70.9 C2′, 72.4 C4′, 74.2 O–CH–CH3, 75.1 C3′, 75.4 C5′, 85.3 C1′, 95.9 C1, 114.8, 115.2 CHAr, 120.7 CHtriazole, 124.3, 124.3 CHAr, 145.2 CHtriazole–Ctriazole–CH2, 146.8, 147.0, 160.4, 160.9 CAr, 169.1 CH3–CO–O–C2′, 169.4 CH3–CO–O–C4, 169.9, 169.9 CH3–CO–O–C3/C3′, 170.3, 170.5, 170.6 CH3–CO–O–C2/C6/C6′, 172.8 NH–CO–CH2. MALDI-TOF (DCTB + NaTFA): 1161,5 [(M + Na]. IR (KBr, cm−1): 3326, 2935, 2858, 1748, 1599, 1499, 1373, 1234, 1147, 1036, 841.OAc–Malt–Tz–C10–Azo–OCH3 (C53H70N6O20)
1H (400 MHz, CDCl3, 25 °C, δ (ppm)): 1.31–1.72 (m, 14H, –(CH2)7–), 1.77–1.87 (m, 2H, –O–CH2–CH2–(CH2)–), 1.87 (s, 3H, CH3–CO–O–C2′), 2.03 (s, 3H), 2.05 (s, 6H), 2.09 (s, 3H), 2.01 (s, 3H), 2.16 (s, 3H) (CH3–CO–O–), 2.20 (t, 2H, J = 7.5 Hz, CO–CH2–CH2–), 3.91 (s, 3H, –O–CH3), 3.97–4.02 (m, 2H, H5, H5′), 4.05 (t, 2H, J = 6.8 Hz, –O–CH2–CH2), 4.06–4.12 (m, 1H, H6b), 4.16 (dd, 1H, J3′,4′ = 9.1 Hz, J4′,5′ = 9.1 Hz, H4′), 4.25–4.30 (m, 2H, H6a, H6′a), 4.47–4.58 (m, 3H, H6′b, Ctriazole–CH2–NH–), 4.91 (dd, 1H, J1,2 = 4.2 Hz, J2,3 = 10.6 Hz, H2), 5.10 (dd, 1H, J3,4 = 9.8 Hz, J4,5 = 9.8 Hz, H4), 5.34 (dd, 1H, J1′,2′ = 9.1 Hz, J2′,3′ = 9.1 Hz, H2′), 5.40 (dd, 1H, J2,3 = 9.8 Hz, J3,4 = 9.8 Hz, H3), 5.46–5.50 (m, 2H, H1, H3′), 5.86 (d, 1H, J1′,2′ = 9.1 Hz, H1′), 6.08 (t, 1H, J = 5.6 Hz, CH2–NH–CO), 6.99–7.03 (m, 4H, HAr), 7.72 (s, 1H, CH–triazole), 7.87–7.90 (m, 4H, HAr). 13C (100 MHz, CDCl3, 25 °C, δ (ppm)): 20.2 CH3–CO–O–C2′, 20.6, 20.7, 20.8, 20.8 CH3–CO–O–, 25.5, 26.0, 29.2, 29.3, 29.3, 29.3, 29.4, 29.5 –(CH2)–, 34.8 Ctriazole–CH2–NH, 36.5 –CO–CH2–CH2–, 55.6 –O–CH3, 61.4 C6, 62.4 C6′, 67.9 C4, 68.3 O–CH2–CH2, 68.8 C5, 69.2 C3, 70.0 C2, 71.0 C2′, 72.4 C4′, 75.1 C3′, 75.4 C5′, 85.3 C1′, 95.9 C1, 114.2, 115.6 CHAr, 120.8 CHtriazole, 124.3, 124.3 CHAr, 145.3 CHtriazole–Ctriazole–CH2, 146.9, 147.1 CAr, 161.2 CAr–O–CH2, 161.5 CAr–O–CH3, 169.1 CH3–CO–O–C2′, 169.4 CH3–CO–O–C4, 169.9, 169.9 CH3–CO–O–C3/C3′, 170.3, 170.5, 170.6 CH3–CO–O–C2/C6/C6′, 173.2 NH–CO–CH2. MALDI-TOF (DCTB + NaTFA): 1133,5 [M + Na]+. IR (KBr, cm−1): 3360, 2926, 2851, 1748, 1652, 1601, 1582, 1501, 1370, 1238, 1147, 1038, 840.Synthesis of azo-glycosyl conjugates
The protected triazole-disaccharide-heptaacetate derivatives (OAc–Malt–Tz–Azo–C16, OAc–Malt–Tz–C5–Azo–C8 and OAc–Malt–Tz–C10–Azo–OCH3) (197.7 mg, 0.17 mmol) were dissolved in 7.5 mL anhydrous THF. Sodium methoxide (81.0 mg, 1.50 mmol) was added. The solution was stirred at room temperature until the reaction was complete (TLC, dichloromethane/ethyl acetate 1:1). Amberlyst IR 120 (H+ form) was added to exchange sodium ions to reach pH = 6–7, the resin was filtered off and the solvent was evaporated in vacuo. The resulting solid was purified by flash chromatography using dichloromethane/methanol acetate 9:1, initially, and then increasing the polarity. A red solid was obtained (75–90%).
Malt–Tz–Azo–C16 (C44H66N6O12)
1H (400 MHz, DMSO-d6, 25 °C, δ (ppm)): 0.85 (t, 3H, J = 7.0 Hz, –(CH2)13–CH3), 1.20–1.40 (m, 26H, –CH2–(CH2)13–CH3), 1.73–1.76 (m, 2H, –CH2–(CH2)13–CH3), 3.04–3.87 (m, 12H, H2, H3, H4, H5, H6a, H6b, H2′, H3′, H4′, H5′, H6′a, H6′b), 4.09 (t, 2H, J = 6.4 Hz, O–CH2–CH2), 4.56 (d, 2H, J = 5.5 Hz, Ctriazole–CH2–NH), 5.04 (d, 1H, J1,2 = 3.6 Hz, H1), 5.56 (d, 1H, J 1′,2′ = 9.2 Hz, H1′), 7.12–7.15 (m, 2H, HAr), 7.68–7.93 (m, 4H, HAr), 8.08–8.09 (m, 2H, HAr), 8.18 (s, 1H, CH–triazole), 9.28 (t, 1H, J = 5.5 Hz, CH2–NH–CO). 13C (100 MHz, DMSO-d6, 25 °C, δ (ppm)): 14.4 (CH2)14–CH3, 22.5, 25.8, 28.9, 29.2, 29.4, 29.5, 31.7 –(CH2)14–CH3, 35.4 triazole–CH2–NH, 60.70, 61.3, 70.4, 71.5, 73.1, 73.7, 74.1, 77.1, 78.6, 79.7 C2, C3, C4, C5, C6, C3′, C4′, C5′, C6′, 68.5 O–CH2– (CH2)14, 71.5 C2′, 87.7 C1′, 101.7 C1, 115.6 CHAr, 122.4 CHtriazole, 125.3, 129.1 CHAr, 135.5 CAr, 145.4 CHtriazole–Ctriazole–CH2, 146.5, 153.9, 162.4 CAr, 165.4 NH–CO–Ar. MicrOTOF-Q: [M + Na]+ 893.461 calcd.: 893.463.IR (KBr, cm−1): 3320 (wide band), 2918, 2849, 1577, 1418, 1251, 1040, 850.Malt–Tz–C5–Azo–C8 (C41H60N6O13)
1H (400 MHz, MeOD, 25 °C, δ (ppm)): 0.92 (t, 3H, J = 6.8 Hz, –(CH2)5–CH3), 1.31–1.88 (m, 16H, –CH2–), 1.35 (d, 3H, J = 5.8 Hz, CH3–CH–O), 2.29 (t, 2H, J = 7.4 Hz, CO–CH2–(CH2)3–), 3.29–3.91 (m, 10H, H3, H4, H5, H6a, H6b, H3′, H4′, H5′, H6′a, H6′b), 3.48 (dd, 1H, J1,2 = 3.8 Hz, J2,3 = 9.7 Hz, H2), 3.95 (dd, 1H, J1′,2′ = 9.1 Hz, J2′,3′ = 9.1 Hz, H2′), 4.08 (t, 2H, J = 6.5 Hz, –O–CH2–), 4.47 (s, 2H, Ctriazole–CH2–NH–), 4.51–4.60 (m, 1H, O–CH–CH3), 5.24 (d, 1H, J1,2 = 3.8 Hz, H1), 5.62 (d, 1H, J1′,2′ = 9.1 Hz, H1′), 7.02–7.06 (m, 4H, HAr), 7.83–7.86 (m, 4H, HAr), 8.08 (s, 1H, CH–triazole).13C (100 MHz, MeOD, 25 °C, δ (ppm)): 13.0 CH2–CH3, 18.6 O–CH–CH3, 22.2, 25.1, 25.2, 25.3, 28.6, 29.0, 29.5, 31.6 –(CH2)–, 34.2 Ctriazole–CH2–NH, 35.4 –CO–CH2–CH2–, 36.2 CH3–CH–CH2–, 67.8 O–CH2–CH2, 72.2 C2′, 72.8 C2, 60.4, 61.3, 70.1, 73.5, 73.7, 73.8, 76.8, 78.2, 78.9 C3, C4, C5, C6, C3′, C4′, C5′, C6′ –O–CH–CH3, 88.0 C1′, 101.5 C1, 114.4, 115.4 CHAr, 122.0 CHtriazole, 123.9, 123.9 CHAr, 145.0 CHtriazole–Ctriazole–CH2, 146.6, 146.8, 160.5, 161.3 CAr, 174.7 NH–CO–CH2. MicrOTOF MS: 845.4309 [M + H]+, 867.4118 [M + Na]+. Calcd: 845.4218 [M + H]+, 867.4110 [M + Na]+. IR (KBr, cm−1): 3364, 2928, 2857, 1652, 1598, 1498, 1248, 1148, 1039, 840.Malt–Tz–C10–Azo–OCH3 (C39H56N6O13)
1H (400 MHz, DMSO-d6, 25 °C, δ (ppm)): 1.27–1.56 (m, 14H, –(CH2)7)–, 1.71–1.78 (m, 2H, –O–CH2–CH2–(CH2)–), 2.11 (t, 2H, J = 7.4 Hz, CO–CH2–CH2), 3.07–3.90 (m, 12H, H2, H3, H4, H5, H6a, H6b, H2′, H3′, H4′, H5′, H6′a, H6′b), 3.85 (s, 3H, Ar–O–CH3), 4.05 (t, 2H, J = 6.0 Hz, –O–CH2–CH2), 4.30 (d, 2H, J = 3.9 Hz, Ctriazole–CH2–NH–), 4.54–4.60 (m, 1H, OH), 4.60–4.67 (m, 1H, OH), 4.95–5.02 (m, 2H, OH), 5.07 (d, 1H, J1–2 = 2.6 Hz, H1), 5.50–5.51 (m, 1H, OH), 5.58–5.63 (m, 2H, H1′, OH), 5.79–5.80 (m, 1H, OH), 7.09–7.13 (m, 4H, HAr), 7.82–7.85 (m, 4H, HAr), 8.08 (s, 1H, CH–triazole), 8.33 (t, 1H, J = 5.6 Hz, CH2–NH–CO). 13C (100 MHz, DMSO-d6, 25 °C, δ (ppm)): 25.6, 25.9, 29.1, 29.2, 29.2, 29.3, 29.4, 29.5 –(CH2)–, 34.5 Ctriazole–CH2–NH, 35.7 –CO–CH2–CH2–, 56.1 –O–CH3, 68.4 O–CH2–CH2, 60.7, 61.2, 70.3, 72.0, 72.9, 73.7, 74.0, 77.2, 78.4, 79.6 C2, C3, C4, C5, C6, C2′, C3′, C4′, C5′, C6′, 87.6 C1′, 101.4 C1, 115.0, 115.4 CHAr, 122.3 CHtriazole, 124.6, 124.6 CHAr, 145.5 CHtriazole–Ctriazole–CH2, 146.5, 146.7 CAr, 161.4 CAr–O–CH2, 161.9 CAr–O–CH3, 172.6 NH–CO–CH2. MicrOTOF MS: 817.3968 [M + H]+, 839.3773 [M + Na]+. Calcd: 817.3978 [M + H]+, 839.3797 [M + Na]+. IR (KBr, cm−1): 3376 (wide band), 2920, 2850, 1642, 1582, 1602, 1253, 1148, 1025, 840.Characterization techniques
1H and 13C NMR spectra were recorded on a BRUKER AV-400 spectrometer. IR spectra were measured on a Thermo NICOLET Avatar 360 FT-IR spectrophotometer using KBr pellets. Mass Analysis was performed using a MALDI+/TOF Brüker Microflex system, with a different matrix depending on the compound (DCTB or DHB) and MicroTOF Brüker equipment for exact mass measurements. Elemental analysis was performed using a Perkin Elmer CHN2400 microanalyser.The mesogenic behaviour was studied by optical microscopy with an Olympus BH-2 polarising microscope equipped with a Linkam THMS hot-stage central processor and a CS196 cooling system. Differential scanning calorimetry (DSC) was performed using a DSC 2910 from TA Instruments, with samples sealed in aluminium pans and a scanning rate of 10 °C min−1 under a nitrogen atmosphere. Temperatures were read at the maximum of the transition peaks. Thermogravimetric analysis (TGA) was performed using a TGA Q5000IR from TA Instruments at a rate of 10 °C min−1 under a nitrogen atmosphere. XRD measurements of the oriented fibre samples were performed with an evacuated pinhole camera (Anton-Paar), operating with a point-focused Ni-filtered Cu-Kα beam. Samples were held in Lidemann glass capillaries and the patterns collected on flat photographic films perpendicular to the X-ray beam.
CD spectra were performed using a Jasco J-810 instrument. To check for the lack of contribution of linear dichroism in the circular dichroism, the CD spectra of the samples were performed by rotating the sandwich every 60 degrees around the light beam axis. Sample preparation for CD measurement: Malt–Tz–Azo–C16 and Malt–Tz–C10–Azo–OCH3 gels were heated and a drop of hot solution was placed over a UV fused silica sandwich with a 0.025 mm Teflon spacer and left to cool at room temperature for 10 min. Gel I and Gel II mixed hydrogels were placed onto the sandwich with a 0.025 mm spacer at room temperature.
SEM measurements were performed using a FESEM Carl Zeiss MERLIN at the Microscopy Service laboratory at Zaragoza University. The sample was fixed onto glass and coated with platinum. TEM measurements were performed using a TECNAI G2 20 (FEI COMPANY) at the Advanced Microscopy laboratory in the ‘Instituto de Nanociencia de Aragon’. For TEM sample preparation, a drop of the solution (0.1 wt% gel diluted) was placed on a copper grid and left to dry for 15 min. The copper grid was then placed again over a drop of 1% uranyl acetate solution as a negative stain for 30s and left to dry.
Gelation test: the gelator and the solvent were placed in a septum-capped test tube. The resulting mixture was heated until a clear solution was obtained. The solution was cooled to room temperature, and a gel was considered to have been formed if the solution did not flow after the test tube was turned upside down.
Conclusions
Three maltose-based azo-amphiphiles have been synthesised and gel-forming properties have been determined. The azobenzene chromophore was placed at different positions of the hydrophobic chain and a maltose sugar head was linked by a copper(I)-catalysed azide–alkyne [3 + 2] cycloaddition with a good yield. A mesomorphic fluid state was found for these compounds. While one of the three compounds synthesized was soluble in water, the two with the azobenzene close to the triazole group or at the end of the chain formed gels in pure water or a mixture of DMSO and water at different minimum concentrations from 1.5 to 5 wt%. Gels are stable at room temperature. Torsion on the resulting self-assembled fibrillar network was observed by different electronic microscopy and a chiral arrangement was confirmed by CD.The designed molecules have a photosensitive group and a modulation of the supramolecular arrangement under testing with light. No changes were obtained in the DMSO/water gels due to their highly ordered organisation. Moreover, the hydrogels formed by the combination of azobenzene compounds synthesised in this study and a previously prepared hydrogelator were also studied. The 3D network of the hydrogel was successfully doped with a similar structure that had a photoresponsive unit. Reversible modifications in the azobenzene structure under irradiation with UV light were performed when the water soluble azobenzene derivative was used. The CD signal related to the azobenzene group disappeared and a photostationary state was reached. It indicates that some structural modifications have been promoted, making this material capable of switching from one part of this structure while the gel structure remained unchanged. This mixture forms a rewritable supramolecular hydrogel to be applied in the field of soft materials.
Acknowledgements
This work was supported by MINECO, Spain, under the project MAT2011-27978-C02-01, Fondo Europeo de Desarrollo Regional (FEDER), with the Government of Aragón being gratefully acknowledged. The authors also acknowledge the Zaragoza University Microscopy Service for the SEM measurements and the Advanced Microscopy Laboratory (LMA) at the Instituto de Nanociencia de Aragón (INA) for the TEM measurements.References
- (a) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS; (b) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766–1776 CrossRef CAS.
- (a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef; (b) D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS; (c) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS; (d) D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC; (e) S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC; (f) J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC; (g) D. K. Smith, Tetrahedron, 2007, 63, 7283–7284 CrossRef CAS.
- J. J. De Jong, B. L. Feringa and J. Van Esch, Responsive Molecular gels, in Molecular gels, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006, 895-927 Search PubMed.
- (a) S. K. Chang and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 1318–1319 CrossRef CAS; (b) K. Hanabusa, T. Miki, Y. Taguchi, T. Koyama and H. Shirai, J. Chem. Soc., Chem. Commun., 1993, 18, 1382–1384 RSC; (c) K. Inoue, Y. Ono, Y. Kanekiyo, T. Ishi-i, K. Yoshihara and S. Shinkai, J. Org. Chem., 1999, 64, 2933–2937 CrossRef CAS.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS.
- (a) J. L. Pozzo, G. M. Clavier and J. P. Desvergne, J. Mater. Chem., 1998, 8, 2575–2577 RSC; (b) W. Deng and D. H. Thompson, Soft Matter, 2010, 6, 1884–1887 RSC.
- (a) H. Ihara, T. Sakurai, T. Yamada, T. Hashimoto, M. Takafuji, T. Sagawa and H. Hachisako, Langmuir, 2002, 18, 7120–7123 CrossRef CAS; (b) P. Terech and A. Coutin, J. Phys. Chem. B, 2001, 105, 5670–5676 CrossRef CAS.
- A. P. Del Guerzo, J. L. Pozzo, Photoresponsive Gels, in Molecular Gels, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006, pp. 817–855 Search PubMed.
- (a) J. Griffith, Chem. Soc. Rev., 1972, 1, 481–493 RSC; (b) S. Mahesh, A. Gopal, R. Thirumalai and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 7227–7230 Search PubMed.
- (a) J. H. Jung, Y. Ono and S. Shinkai, Tetrahedron Lett., 1999, 40, 8395–8399 CrossRef; (b) K. Murata, M. Aoki, T. Nishi, A. Ikeda and S. Shinkai, J. Chem. Soc., Chem. Commun., 1991, 24, 1715–1718 Search PubMed; (c) K. Murata, M. Aoki and S. Shinkai, Chem. Lett., 1992, 5, 739–742 Search PubMed; (d) K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
- X. Ran, H. T. Wang, P. Zhang, B. L. Bai, C. X. Zhao, Z. X. Yu and M. Li, Soft Matter, 2011, 7, 8561–8566 RSC.
- P. F. Duan, Y. G. Li, L. C. Li, J. G. Deng and M. H. Liu, J. Phys. Chem. B, 2011, 115, 3322–3329 CrossRef CAS.
- (a) M. de Loos, J. van Esch, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 613–616 CrossRef CAS; (b) S. van der Laan, B. L. Feringa, R. M. Kellogg and J. van Esch, Langmuir, 2002, 18, 7136–7140 CrossRef CAS.
- H. Cao, J. Jiang, X. F. Zhu, P. F. Duan and M. H. Liu, Soft Matter, 2011, 7, 4654–4660 RSC.
- Y. C. Huang, Z. J. Qiu, Y. M. Xu, J. F. Shi, H. K. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 RSC.
- (a) A. Srivastava, S. Ghorai, A. Bhattacharjya and S. Bhattacharya, J. Org. Chem., 2005, 70, 6574–6582 CrossRef; (b) K. Tiefenbacher, H. Dube, D. Ajami and J. Rebek, Chem. Commun., 2011, 47, 7341–7343 RSC.
- M. Amaike, H. Kobayashi and S. Shinkai, Chem. Lett., 2001, 7, 620–621 Search PubMed.
- G. Narayan, N. S. Kumar, S. Paul, O. Srinivas, N. Jayaraman and S. Das, J. Photochem. Photobiol., A, 2007, 189, 405–413 CrossRef CAS.
- H. Kobayashi, A. Friggeri, K. Koumoto, M. Amaike, S. Shinkai and D. N. Reinhoudt, Org. Lett., 2002, 4, 1423–1426 CrossRef CAS.
- (a) Y. Y. Lin, A. D. Wang, Y. Qiao, C. Gao, M. Drechsler, J. P. Ye, Y. Yan and J. B. Huang, Soft Matter, 2010, 6, 2031–2036 RSC; (b) R. Rajaganesh, A. Gopal, T. M. Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748–751 Search PubMed; (c) Y. Ogawa, C. Yoshiyama and T. Kitaoka, Langmuir, 2012, 28, 4404–4412 Search PubMed.
- J. W. Goodby, V. Gortz, S. J. Cowling, G. Mackenzie, P. Martin, D. Plusquellec, T. Benvegnu, P. Boullanger, D. Lafont, Y. Queneau, S. Chambert and J. Fitremann, Chem. Soc. Rev., 2007, 36, 1971–2032 RSC.
- (a) N. Laurent, D. Lafont, F. Dumoulin, P. Boullanger, G. Mackenzie, P. H. Kouwer and J. W. Goodby, J. Am. Chem. Soc., 2003, 125, 15499–15506 CrossRef CAS; (b) S. Abraham, S. Paul, G. Narayan, S. K. Prasad, D. S. Rao, N. Jayaraman and S. Das, Adv. Funct. Mater., 2005, 15, 1579–1584 CrossRef CAS.
- M. J. Clemente, J. Fitremann, M. Mauzac, J. L. Serrano and L. Oriol, Langmuir, 2011, 27, 15236–15247 CrossRef CAS.
- H. Juwarker, J. M. Lenhardt, J. C. Castillo, E. Zhao, S. Krishnamurthy, R. M. Jamiolkowski, K. H. Kim and S. L. Craig, J. Org. Chem., 2009, 74, 8924–8934 CrossRef CAS.
- (a) Y. F. Zhou, T. Yi, T. C. Li, Z. G. Zhou, F. Y. Li, W. Huang and C. H. Huang, Chem. Mater., 2006, 18, 2974–2981 CrossRef CAS; (b) F. Vera, R. Tejedor, P. Romero, J. Barbera, M. B. Ros, J. L. Serrano and T. Sierra, Angew. Chem., Int. Ed., 2007, 46, 1873–1877 CrossRef CAS; (c) M. Haro, J. del Barrio, A. Villares, L. Oriol, P. Cea and M. C. Lopez, Langmuir, 2008, 24, 10196–10203 Search PubMed.
- Z. Gyorgydeak, L. Szilagyi and H. Paulsen, J. Carbohydr. Chem., 1993, 12, 139–163 CrossRef CAS.
- V. Molinier, P. H. J. Kouwer, J. Fitremann, A. Bouchu, G. Mackenzie, Y. Queneau and J. W. Goodby, Chem.–Eur. J., 2006, 13, 3547–3557 Search PubMed.
- M. Suzuki, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Chem.–Eur. J., 2003, 9, 348–354 CrossRef CAS.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef.
- J. del Barrio, R. M. Tejedor, L. S. Chinelatto, C. Sánchez, M. Piñol and L. Oriol, Chem. Mater., 2010, 22, 1714–1723 CrossRef CAS.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- (a) Y. Jeong, A. Friggeri, I. Akiba, H. Mansunaga, K. Sakurai, S. Sakurai, S. Okamoto, K. Inoue and S. Shinkai, J. Colloid Interface Sci., 2005, 283, 113–112 CrossRef CAS; (b) N. E. Botterhuis, S. Karthikeyan, D. Veldman, S. C. J. Meskers and R. P. Sijbesma, Chem. Commun., 2008, 33, 3915–3917 Search PubMed; (c) J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 3, 316–318 Search PubMed; (d) D. García-Velázquez and R. Luque, Chem.–Eur. J., 2011, 17, 3847–3849 CrossRef CAS; (e) A. Das and S. Ghosh, Chem. Commun., 2011, 47, 8922–8924 RSC; (f) M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC; (g) K. Kohno, K. Morimoto, N. Manabe, T. Yajima, A. Yamagishi and H. Sato, Chem. Commun., 2012, 48, 3860–3862 RSC; (h) D. Li, J. Liu, L. Chu, J. Liu and Z. Yang, Chem. Commun., 2012, 48, 6175–6177 RSC; (i) G. O. Lloyd, M. O. M. Pipenbrock, J. A. Foster, N. Clarke and J. W. Steed, Soft Matter, 2012, 8, 204–216 RSC.
- (a) S. Yagai, T. Karatsu and A. Kitamura, Langmuir, 2005, 21, 11048–11052 CrossRef CAS; (b) S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134–11139 CrossRef CAS; (c) Y. F. Zhou, M. Xu, Y. Xi, S. Z. Xiao, Z. G. Zhou, F. Y. Li and C. H. Huang, Langmuir, 2007, 23, 202–208 CrossRef CAS; (d) X. J. Liao, G. S. Chen, X. X. Liu, W. X. Chen, F. Chen and M. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4409–4413 CrossRef CAS; (e) J. S. Park, S. Jeong, B. Ahn, M. Kim, W. Oh and J. Kim, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 79–86 Search PubMed.
- J. H. Fuhrhop and C. Boettcher, J. Am. Chem. Soc., 1990, 112, 1768–1776 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Characterization data of intermediate compounds (1–4). MicroTOF Mass Spectrometry of Malt–Tz–C10–Azo–OCH3. Thermogravimetric analysis of peracetylated precursors and azo-glycolipids. Microphotograph of Malt–Tz–C5–Azo–C8. 1H NMR experiments in DMSO-d6 increasing water concentration from 6.00 to 3.00 ppm. SEM and TEM images of the synthesized azo-glycolipids. Absorption spectra of Malt–Tz–Azo–C16, Malt–Tz–C5–Azo–C8 and Malt–Tz–C10–Azo–OCH3 in DMSO solution (5 × 10−5 M). CD and absorption spectra of Malt–Tz–C5–Azo–C8 in a 4.5 × 10−5 M water solution. See DOI: 10.1039/c2ra21506c |
| This journal is © The Royal Society of Chemistry 2012 |