Absorption of SO2 from flue gas by aqueous fulvic acid solution†
Jitao
Yang
ab and
Guoxin
Hu
*a
aSchool of Mechanical and Power Engineering, Shanghai Jiaotong University, Shanghai 200240, China. E-mail: hugx@sjtu.edu.cn; Fax: +86-21-34206569
bSchool of Energy and Environment Engineering, Zhongyuan University of Technology, Zhengzhou 450007, China
First published on 25th September 2012
Abstract
A new regenerable flue gas desulfurization process was proposed, in which fulvic acid derived from biomass residues was used as an absorbent to absorb SO2 from flue gas, based on acid–base buffering capacity. Experiments have been carried out to examine the absorption, desorption and reabsorption performance of fulvic acid solution in a lab-scale reactor. The results show fulvic acid solution (0.04 g mL−1, pH 5.5) could excellently absorb SO2 with a maximum absorption efficiency of 97.5% (298 K, 2200 ppm SO2, 5% O2, 0.14 m3 h−1). In the process of SO2 absorption, chemical absorption is the predominant mechanism. The SO2-loaded solution is readily desorbed and regenerated under ambient pressure by heating at 343 K, and the regenerated fulvic acid solution still exhibits good absorption performance after seven absorption/desorption cycles. Trace metal ions binding to fulvic acid play a decisive role in the absorption process. Fulvic acid samples before and after absorbing SO2 were well characterized by Fourier transform infrared spectroscopy, near-edge X-ray absorption fine structure and X-ray photoelectron spectroscopy. These results demonstrate that no chemical change is found, except that carboxylate groups are protonated to carboxylic groups, indicating that fulvic acid is stable as a regenerable absorbent.
Introduction
The emissions of SO2 from the burning of fossil fuels have caused significant environmental and human health effects.1 To control SO2 emissions, the once-through non-regenerative wet flue gas desulfurization (WFGD) processes, based mainly on limestone scrubbing, are frequently used.2 Although these processes have a number of advantages, they also have some serious disadvantages such as the high capital and operating expenses, the large volume of water required, poor quality of the byproduct, and even causing secondary pollution.3 Hence, an environmentally friendly and cost-effective technology for removing SO2 from flue gas needs to be developed.Recently, researchers studied SO2 absorption in room-temperature ionic liquids (ILs), such as 1,1,3,3-tetramethylguanidinium lactate ([TMG]L),1 TMG-based ILs,4 hydroxyl ammonium ILs,5 and imidazole-based ILs.6 In 2010, Heldebrant et al. reported an emerging technology that can capture SO2 and regenerate it for reuse by the reversible zwitterionic SO2-binding organic liquid produced from the reaction of SO2 with N,N-dibutylundecanolamine.7 Although these technologies can effectively absorb SO2, the high production costs would limit their industrial applications in large-scale flue gas desulfurization. Humic substances have been widely utilized as environmentally friendly organic fertilizers because of their molecular structure.8 With the aim of a resourceful type of WFGD technology, Hu et al. proposed humate for the removal of SO2 from flue gas and the production of compound humic acid fertilizer,9 investigated the absorption of SO2 with sodium humate solution and elucidated the forming mechanism of desulfurization byproduction.10 Fulvic acid (FA) is the fraction of humic substances that is soluble under all pH conditions. One of the most important properties of FA is the high buffering capacity in a wide pH range, which essentially arises from the dissociation of acidic functional groups (e.g. carboxylic and phenolic groups, Fig. 1).11 FA can be extracted from sources such as peat, soil, water, lignite, agricultural and forest biomass residues etc.12 Now, FA extracted from biomass residues has been industrialized, owing to accessible raw materials and low cost. Although FA has good buffering capacity, to our knowledge there have been no reports related to removing SO2 from flue gas by its absorption. Thus we propose a novel regenerable flue gas desulfurization process in which SO2 is first absorbed by aqueous FA solution in a bubbling reactor and then desorbed from the loaded solution by heating; the regenerated FA solution is recovered and reused for absorbing SO2 from flue gas. Meanwhile, the desorbed SO2 can be recovered and used to produce sulfuric acid or liquid sulfur dioxide. Compared with conventional limestone wet scrubbing, the regenerative desulfurization process would neither require a large quantity of water nor cause the problem of disposal of large amounts of waste such as wastewater, CaSO4 and waste carbon dioxide.
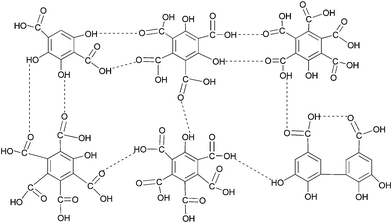 | ||
| Fig. 1 Representative structure of fulvic acid as proposed by Schnitzer and Khan (1972)11d. | ||
The aim of the present study is to investigate experimentally the feasibility and durability of FA as a regenerative flue gas desulfurization absorbent by the performances of absorption/desorption cycles in a lab-scale reactor; to obtain a more detailed understanding of the absorption mechanisms of SO2 with Fourier transform infrared spectroscopy (FTIR), near-edge X-ray absorption fine structure (NEXAFS) and X-ray photoelectron spectroscopy (XPS), etc., and demonstrate that FA is stable as an absorbent during SO2 absorption. Our findings signify the possibility that conventional limestone wet scrubbing could be replaced by regenerative absorption in FA solution.
Experimental
Sample preparation and treatment
In this work, powdered FA (≥95 wt%), extracted from crop straw, was provided by Shanghai Tongwei Biological and Technology Co., Ltd. (Shanghai, China). The FA sample was further purified using a DAX-8 resin (Supelco, Bellefonte, USA) column technique described by Thurman and Malcolm.13 The purified FA was converted to the protonated form by quickly passing it through a column of Amberlite IR-120 (H+ form) cation-exchange resin (Acros Organics, USA), and then freeze-dried for desulfurization experiment and chemical analysis. Deionized water (18 MΩ, Milli-Q, Millipore) was used for all experimental procedures.After the SO2 absorption test, the SO2-loaded solution was dried for approximately 72 h at 313 K under vacuum, and then the dried FA desulfurization products were obtained and provided for characterization analysis.
Absorption and desorption experiments
A schematic diagram of the experimental apparatus is shown in Fig. S1 (see ESI†). The experiments for SO2 absorption were conducted in a lab-scale bubbling reactor at ambient pressure and room temperature. A simulated flue gas consisted of 2200 ppm SO2, 5 vol% O2, and the balance was N2. The SO2 (99.95%), O2 (99.99%) and N2 (99.99%) gases were supplied from cylinders. The total flow rate of the simulated flue gas was controlled by rotameters. The SO2 absorption efficiency was defined in our previous literature.14 The FA solution (0.04 g mL−1, initial pH of 5.5) transformed into loaded solution after absorbing SO2 for some time; the SO2-loaded solution was also called FA desulfurization liquid. The desorption of the SO2-loaded solution was conducted in nitrogen gas by heating at 343 K under ambient pressure and FA solution was regenerated, from which the escaping steam–SO2 mixture was passed through a condenser for condensing water vapour and recovering the water into the regenerated FA solution that would be used to reabsorb SO2 gas. The desorption process was continued until the pH of the solution remained constant (pH 4.7). When the desorption was done, the first absorption/desorption cycle (Cycle 1) finished. The next reabsorption of SO2 in the regenerated FA solution was carried out, and so on. In the present study, we investigated eight absorption/desorption cycles. Furthermore, the two absorption experiments in purified FA solution and deionized water were also performed, which would be helpful to discussion of the role of trace metal ions binding to FA in the SO2 absorption. All absorption experiments were performed under the same conditions. In the absorption and desorption processes, the pH of the solution was monitored using a digital pH meter (PHB-5, Shanghai Weiye Instrument Plant, China).Analysis methods
The changes in SO2 concentration at the inlet and outlet of the reactor were monitored by a flue gas analyzer (Testo-350XL, Germany). The S-containing ion (SO42− and SO32−) was measured by modular ion chromatograph (Metrohm, Switzerland). Both FA and FA desulfurization products were analyzed for their elemental compositions. The ultimate analysis was performed on a CNS-Analyzer (Vario EL III, Elementar, Germany). The inorganic elements concentrations were analyzed by inductively coupled plasma atomic emission spectrometry (ICP-AES, IRIS Advantage 1000, THERMO, U.S.).The functional groups of FA and FA desulfurization products were identified by a Fourier transform infrared spectrometer (Equinox 55, Germany Bruker), in the range 4000–400 cm−1 with a resolution better than 1 cm−1. To record FTIR spectra, the KBr pressed disk technique was used with 1 mg of sample dispersed in 200 mg KBr. The spectra were processed by the OPUS 5.5 software.
XPS was performed with a Kratos Axis Ultra DLD X-ray photoelectron spectrometer, employing a monochromated Al-Kα X-ray source (hν = 1486.6 eV) at 150 W (15 kV, 10 mA), hybrid (magnetic/electrostatic) optics, a hemispherical analyzer, a multichannel plate and delay line detector (DLD). The powdered FA and FA desulfurization products were pressed onto a stainless-steel sample-holder using double-sided scotch tape. Spectrometer pass energies of 160 eV for the survey spectra and 40 eV for high resolution spectra were used for all elemental spectral regions. As charge effects were expected in the study of these poorly conducting samples, the binding energy of C1s from contamination at 284.7 eV was used as an internal reference to calibrate the spectra. The CasaXPS software (version 2.3.14) was used for all quantitative analysis and peak-fitting routines. The C1s, O1s and S2p envelopes were fitted using mixed Gaussian–Lorentzian component profiles after subtraction of a Shirley background.
The oxygen K-edge NEXAFS spectroscopy characterizations were carried out at the soft X-ray beamline BL08U of Shanghai Synchrotron Radiation Facility (SSRF) with a 150 MeV linac, a 3.5 GeV booster, a 3.5 GeV storage ring, and seven beamlines. The FA solution and FA desulfurization liquid were uniformly deposited on Au-coated silicon wafers, and then dried by gentle heating with a hair dryer to shape thin films of samples. The film samples were mounted onto a sample bar, and then put into the beamline chamber. The chamber was evacuated to a pressure of less than 10−7 mbar. The spectra were recorded through adjusting the incident photon energy across the oxygen K-edge (520–560 eV) with a step of 0.5 eV. The spectra were acquired in the total electron yield mode (TEY). The TEY signals were normalized by the concurrent signal from the reference grid coated with clean gold.
Results and discussion
Absorption efficiency and reabsorption efficiency of SO2
To verify the desulfurization performance of FA solution and understand the mechanisms of SO2 absorption, we performed absorption experiments in FA solution, regenerated FA solution, purified FA solution and deionized water. Fig. 2 shows the SO2 absorption efficiency with time. It can be seen that a high SO2 absorption efficiency (95–97.5%) is maintained in the first 20.4 min for the FA solution (Fig. 2a). Then the SO2 absorption efficiency starts to decline, when the pH of the FA solution is 3.1 (Fig. S2, see ESI†). It is finally reduced to 75% at 29.4 min, when the pH is 2.7 (Fig. S2, see ESI†) and the concentrations of SO42− and SO32− are 772.8 and 69.3 mg L−1 (Table 1), respectively. Besides the concentrations of SO42− and SO32−, Table 1 also presents the total SO2 absorption and the molar ratio of S-containing ions to total SO2 absorption. These results indicate that the FA solution exhibits excellent absorption performance and confirms the feasibility of FA as a SO2 absorbent. It should be pointed out that our FA sample contains small amounts of SO42− (98 mg L−1). Furthermore, Table 2 shows the ratios of both sulfur and oxygen increase from 4.482 to 6.706% and from 49.386 to 51.957%, respectively, which indicate that FA has interacted with SO2 in the absorption process.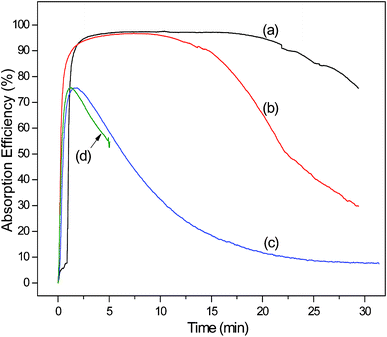 | ||
| Fig. 2 The SO2 absorption efficiency with time for the FA solution of Cycle 1 (a), regenerated FA solution of Cycle 8 (b), (c) and purified FA solution (d). Experimental conditions: 100 mL solution, gas flow of 0.14 m3 h−1, inlet SO2 of 2200 ppm, inlet O2 of 5 vol%, at 298 K and ambient pressure. | ||
| Sample | Maximum absorption efficiency of SO2 (%) | DTHE (min) | Total SO2 absorptionb (mmol) | S-containing ion concentration after absorption (mg L−1) | Molar ratio of S-containing ion to total SO2 absorption (%) | |
|---|---|---|---|---|---|---|
| SO32− ion | SO42− ion | |||||
| a FA solution (100 mL, 0.04 g mL−1, the initial pH 5.5); gas flow of 0.14 m3 h−1; inlet SO2 of 2200 ppm; 5 vol% O2; 298 K; ambient pressure; the same SO2 absorption time of 29.4 min for Cycles 1–8. b Total SO2 absorption is calculated by multiplying the integral of the absorption efficiency curve by inlet SO2 concentration and by gas flow. | ||||||
| Cycle 1 | 97.5 | 20.4 | 6.06 | 69.3 | 772.8 | 14.7 |
| Cycle 2 | 97.3 | 16.2 | 6.05 | 75.2 | 876.3 | 16.6 |
| Cycle 3 | 96.8 | 15.2 | 5.91 | 87.9 | 926.0 | 18.2 |
| Cycle 4 | 97.5 | 16.4 | 5.90 | 104.2 | 948.5 | 19.0 |
| Cycle 5 | 96.6 | 14.3 | 5.77 | 109.8 | 952.3 | 19.6 |
| Cycle 6 | 96.6 | 12.7 | 5.51 | 124.6 | 974.8 | 21.3 |
| Cycle 7 | 96.2 | 12.8 | 5.38 | 167.9 | 977.5 | 22.8 |
| Cycle 8 | 96.7 | 11.6 | 5.02 | 223.1 | 1059.0 | 27.5 |
| Element | FA | FA desulfurization productsa | Purified FA |
|---|---|---|---|
| a Gas flow, 0.14 m3 h−1; inlet SO2, 2200 ppm; O2, 5 vol%; 298 K; ambient pressure; FA absorption solution of Cycle 1, 100 mL, 0.04 g mL−1. b nd: not determined. | |||
| Ultimate analysis | |||
| Carbon | 41.935 | 38.020 | ndbb |
| Nitrogen | 0.1215 | 0.102 | ndbb |
| Sulfur | 4.482 | 6.706 | ndbb |
| Oxygen | 49.386 | 51.957 | ndbb |
| ICP–AES analysis | |||
| Calcium | 3.657 | ndbb | 0.110 |
| Sodium | 3.486 | ndbb | 0.098 |
| Magnesium | 0.202 | ndbb | 0.015 |
By heating, 97% of the absorbed SO2 was driven off from the loaded solution, and a regenerated FA solution was obtained. Fig. 2 shows the reabsorption performance of SO2 in the regenerated FA solution of Cycle 8. For absorption curves of Cycles 2–7 see Fig. S3, ESI.† Although the FA solution has absorbed SO2 gas seven times, the regenerated FA solution still exhibits good absorption performance with a Duration Time of High Efficiency (DTHE, the time of the SO2 absorption efficiency above 95%) of 11.6 min and a maximum absorption efficiency of 96.7% (Table 1). The total SO2 absorption of this regenerated solution is 83% of the FA solution of Cycle 1. The results of eight successive absorption/desorption cycles indicate that the interaction between FA and SO2 is neither too strong nor too weak, and absorption and desorption can occur. Therefore, SO2 absorption in FA solution is a viable method for flue gas desulfurization.
As can be seen from Table 1, the highest SO2 absorption by 4 g FA is 6.06 mmol among Cycles 1–8, which translates to 0.10 g SO2 g−1 FA. This value is lower than results obtained by Wu et al. (0.31 g SO2 g−1 [TMG]L)1 and Heldebrant et al. (0.54 g SO2 g−1 DBUA),7 but higher than Huang et al. (0.02 g SO2 g−1 [TMG][BF4]]).4a It should be noted that the absorption of 6.06 mmol is not the maximum SO2 absorption capacity of FA. Because too low SO2 absorption efficiency has no engineering value, we terminate the SO2 absorption of Cycle 1 when the efficiency decreases to 75%. However, the FA absorption liquid still has SO2 absorption capacity. So FA is an absorbent with high desulfurization capacity. Interestingly, the pH of all regenerated FA solutions is almost 2.7—the same as that of the FA solution at the end of SO2 absorption. When the eighth absorption finishes, the concentrations of SO42− and SO32− are 1059.0 and 223.1 mg L−1 (Table 1), respectively. As shown in Fig. 2, this regenerated FA solution of Cycle 8 has a shorter DTHE than the FA solution of Cycle 1. In other words, the SO2 absorption capacity of the regenerated FA solution is weaker than the FA. This is because a certain amount of sulfate radical generated during SO2 absorption causes an important effect on SO2 reabsorption.
When SO2 is dissolved in FA buffer solution (eqn (1)), a SO2–FA–H2O system forms and the following chemical equilibrium should be considered:
| SO2(g) ↔ SO2(aq) | (1) |
| SO2(aq) + H2O(aq) ↔ H2SO3(aq) | (2) |
| H2SO3(aq) ↔ HSO3−(aq) + H+(aq) | (3) |
| HSO3−(aq) ↔ SO32−(aq) + H+(aq) | (4) |
| FA−(aq) + H+(aq) ↔ FA(aq) | (5) |
| nFA–M(aq) ↔ nFA−(aq) + Mn+(aq) | (6) |
| 2SO32−(aq) + O2(g) → 2SO42−(aq) | (7) |
By comparing the (c) and (d) curves in Fig. 2, we find that deionized water and purified FA solution (0.04 g mL−1) have a similar poor SO2 absorption performance with no DTHE, in which the absorption can be considered as a physical absorption process controlled by molecular diffusion. The findings suggest that trace metal ions binding to FA play a decisive role in the SO2 absorption. In other words, SO2 absorption depends on carboxylate cation-binding groups of FA.
Chemical analysis of FA absorbent before and after absorbing SO2
To obtain some information about the chemical interaction between SO2 and FA, we characterized the FA and FA desulfurization products by FTIR, XPS and NEXAFS. Fig. 3 shows the FTIR spectra of the FA, FA desulfurization products and purified FA. The interpretations of the spectra are based on numerous studies, which are summarized in Table S1 (see ESI†). In general, the spectra of the three samples are very similar and show the same pattern of bands. In spite of the similarities, the differences among the spectra are still obvious. The first important difference between the spectra of FA (Fig. 3a) and FA desulfurization products (Fig. 3b) is that the absorption band at 3405 cm−1 broadens slightly in the latter. This finding suggests an increase in the content of –OH groups for FA after absorbing SO2. A second difference is that the carboxylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (υCO(COOH)) band at 1705 cm−1 shifts to a slightly higher frequency (1717 cm−1), and the absorption intensity of the υCO(COOH) increases markedly in the latter (Fig. 3b). The modifications signify an increase in the content of carboxyl group. A third pronounced difference is that the asymmetric stretch υas(COO)− band of the carboxylate functional groups overlapped by aromatic CC (Table S1, ESI†) is shifted towards the υCO(COOH) from 1600 to 1617 cm−1 owing to SO2 absorption; additionally, as compared to the FA spectra, the υas(COO)− stretch of FA desulfurization products at 1617 cm−1 becomes sharper with a slight intensity decrease, as well as a decrease in the intensity of the symmetric stretch υs(COO)− at 1426 cm−1, which suggests a decrease of the content of COO− groups. This result is ascribed to the partial protonation of COO− groups of the FA.
O stretching (υCO(COOH)) band at 1705 cm−1 shifts to a slightly higher frequency (1717 cm−1), and the absorption intensity of the υCO(COOH) increases markedly in the latter (Fig. 3b). The modifications signify an increase in the content of carboxyl group. A third pronounced difference is that the asymmetric stretch υas(COO)− band of the carboxylate functional groups overlapped by aromatic CC (Table S1, ESI†) is shifted towards the υCO(COOH) from 1600 to 1617 cm−1 owing to SO2 absorption; additionally, as compared to the FA spectra, the υas(COO)− stretch of FA desulfurization products at 1617 cm−1 becomes sharper with a slight intensity decrease, as well as a decrease in the intensity of the symmetric stretch υs(COO)− at 1426 cm−1, which suggests a decrease of the content of COO− groups. This result is ascribed to the partial protonation of COO− groups of the FA.
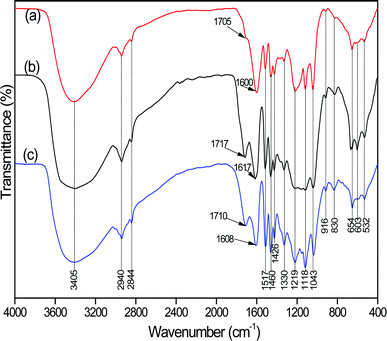 | ||
| Fig. 3 FTIR spectra of FA (a), FA desulfurization products (b) and purified FA (c). | ||
On the basis of the above findings, we conclude that the carboxylate groups of FA are protonated to carboxylic groups after SO2 absorption. Because of one important acidic binding site of carboxylic hydroxyl groups, FA is capable of interacting with trace metal ions to form metal fulvates. However, at low pH, carboxylate groups (COO−) can be protonated to carboxylic groups (COOH). Importantly, with SO2 absorption in FA solution, a large number of hydrogen ions are supplied by the dissociation of sulfurous acid (eqn (3) and (4)), which facilitates the protonation of COO−. To further corroborate the conclusion, we also recorded the spectra of the purified FA (Fig. 3c). Interestingly, a similar frequency shift of υCO(COOH) for the purified FA is observed at 1710 cm−1, where the absorption intensity also increases compared to the FA. Moreover, we also notice a simultaneous decrease and shift of υas(COO)− at 1608 cm−1 (Fig. 3c). This is because COO− was transformed into COOH by proton-saturated resin in the purification of FA, which also triggered a strong loss of cations (see ICP-AES results in Table 2). Hence, the explanation for spectral changes of carboxylate groups and protonated carboxylic groups is well supported by the spectra of purified FA.
Finally, a fourth noticeable difference is that the 603 cm−1 band of FA desulfurization products is more prominent than FA, and simultaneously the 1118 cm−1 band broadens. A reasonable explanation is that more sulfates are generated, since bands at 603 and 1118 cm−1 can be assigned to sulfate (SO42−).19 Note that the generation of the sulfate radical prevented a few carboxylate groups from being regenerated during desorption, which would explain why the regenerated FA solution has shorter DTHE than the FA through successive absorption/desorption cycles, as shown in Fig. 2. Unfortunately, we can not find evidence for the HSO3− and SO32− ions, whose intense absorptions are centered at 1025 and 950 cm−1,20 respectively. To detect the HSO3−, we adopt the new method of rhodamine-based fluorescent probe selective for the bisulfite anion in aqueous ethanol media, which is proposed by Yang et al.21 Fig. S4 (ESI†) shows changes in the absorption spectra of a rhodamine-based fluorescent probe upon mixing with FA solution and SO2-loaded FA solution. We observe that the solution of the rhodamine-based probe with FA exhibits almost no absorption peak in the visible region (400–680 nm), as shown in Fig. S4 (see ESI†). However, an obvious peak at around 565 nm appears when mixing with the SO2-loaded FA solution, especially with a SO2-saturated FA solution. This confirms that HSO3− ions are generated during SO2 absorption. For detailed results, see ESI.† With respect to the SO32− ions, ion chromatography has provided evidence for their existence (Table 1).
As an ideal tool for identifying differences in surface chemistry, XPS was used to characterize the surface composition and chemical environment of FA before and after absorbing SO2. Fig. S5 (ESI†) shows the survey spectra of FA and FA desulfurization products. The most prominent photoelectron peaks at 286 eV and 533 eV are assigned to C1s and O1s. Other peaks include the Na1s peak at 1072 eV, as well as Ca2s, Ca2p, S2s, S2p, Na2s and O2s. In addition, auger lines of O and Na are present. For the two samples, we find no noticeable difference in the relative intensity of peaks (Fig. S5, ESI†). However, Table S2 (ESI†) presents several changes in relative atomic concentrations, which are calculated using the intensity of an appropriate line and XPS cross sections. After SO2 absorption, the surface atomic concentrations of both S and O increase from 2.0 to 2.6% and from 33.0 to 36.3%, respectively, which is consistent with the results of the elemental analysis (Table 2). This suggests that an interaction of FA and SO2 occurs. In spite of detailed FTIR analysis, we do not completely understand the speciation transformation of sulfur from SO2 and the changes of functional groups on FA, which would determine the nature of the interaction between SO2 and FA. In addition to the compositional information, XPS also provides information on the chemical state of each element. Thus, we can investigate surface chemical changes that occur on the FA surface exposed to SO2 by qualitative and quantitative analysis of the C1s, O1s and S2p XPS spectra.
Fig. 4 shows the deconvolution of high-resolution XPS spectra of C1s, O1s and S2p along with fit peak parameters and assignments, of both the FA and FA desulfurization products. In general, the two samples are very heterogeneous. The C1s spectra can be deconvoluted into five different component peaks: C–C/C–H (284.7 eV), C–O (286.3 eV), COO− (288.2 eV), COOH (288.6 eV) and π–π* shake-up transitions in aromatic rings (291.7 or 292.0 eV), as shown in Fig. 4a and b. We assign COO− (deprotonated carboxyl group) and COOH (protonated carboxyl group) on the basis of the results of Yoshida et al.22 In fact, the FTIR results discussed above also confirm both COO− and COOH in the two samples. In Fig. 4a and b, the peak profiles representing carboxyl groups at 288–290 eV are obviously different, which means the component changes in carboxyl groups. Inspection of Fig. 4a and b also shows that the amount of deprotonated carboxyl carbon decreases (from 3.2 to 1.2%), while protonated carboxyl carbon increases (from 2.9 to 5.1%) after SO2 absorption, indicating that carboxyl groups bonded to metal ions were cleaved and free carboxylic acid groups were formed. As expected, with the decrease of pH, there is a transition from COO− to COOH on FA. However, the ratio of peak areas of C–C/C–H and C–O for the C1s peaks remains essentially constant. This reveals that these bonds are not directly involved in the interaction of FA with SO2, which is in good agreement with the FTIR result that no changes in the C–H band (2940 and 2844 cm−1), the C–O band (1330 cm−1) and the C–C band (532 cm−1) are observed in Fig. 3.
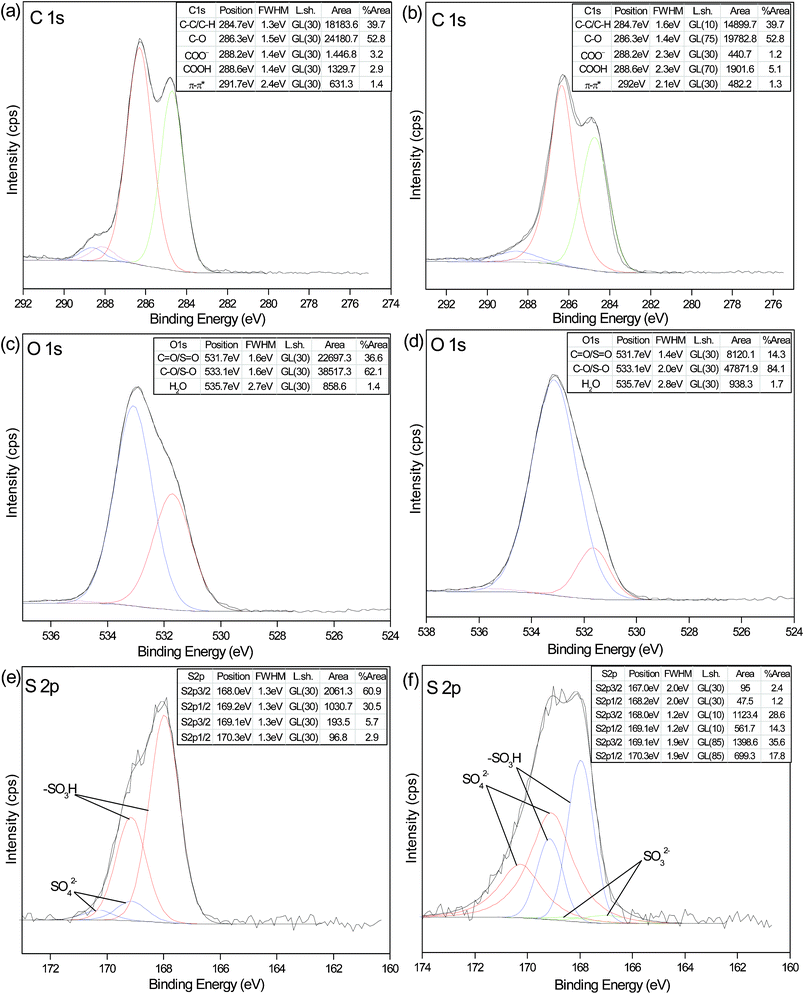 | ||
Fig. 4 High-resolution XPS spectra of the C1s, O1s and S2p regions for FA (a, c and e) and FA desulfurization products (b, d and f) with the corresponding fits. The fit parameters, assignments and relative areas of the peaks are given in the insert Tables. The S2p doublets were constrained to a separation energy of 1.18 eV with an area ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and equivalent full-width-at-half-maximum (FWHM) for both components (S2p3/2 and S2p1/2). :1 and equivalent full-width-at-half-maximum (FWHM) for both components (S2p3/2 and S2p1/2). | ||
The O1s spectra show the presence of three oxygen components: oxygen atoms from CO in carboxyl or carbonyl groups at 531.7 eV, the one related to C–O in carboxyl or phenolic groups at 533.1 eV, oxygen atoms in water or chemisorbed oxygen species at 535.7 eV. Note that binding energies for OS and O–S (sulfonic groups, sulfate and sulfite), appear in the same range as those for OC and O–C, respectively.23 From the insert tables, it can be observed the ratio of peak areas at 531.7 eV decreases significantly after SO2 absorption, while the ratio at 533.1 eV increases obviously. Since oxygen atoms in the carboxyl group have both single and double bonds, we can hardly determine the contribution of COOH according to the variation of area ratio. However, we could tentatively attribute it to the introduction of sulfate and sulfite, which would be verified in the following O K-edge NEXAFS and S2p analysis.
Oxygen K-edge NEXAFS spectra are highly sensitive to the chemical nature around the oxygen atom and are capable of detecting some significant differences in the spectra. For comparison, the O K-edge spectra of FA and FA desulfurization products are shown in Fig. 5. The spectrum of FA exhibits a prominent pre-edge peak at 532 eV, which is assigned to the 1s → π∗(CO) transition from a carbonyl oxygen atom of the carboxylic group,24 a strong main-edge peak at 535.6 eV, which corresponds to the 1s → σ*(C–O) transition from the carboxylate (COO−) group,24a and a weak post-edge peak at 537.4 eV, which is attributed to the transition from the sulfate oxygen O(1s) → S(3p,4s)–O(2p) anti-bonding orbitals.25 After SO2 absorption, a new weak shoulder arises at 530.7 eV; this small resonance can be assigned to the 1s → π∗ transitions of SO bonds from the absorbed SO2 molecules.24b Importantly, we observe that the absorption feature of 1s → σ*(C–O) at 535.6 eV disappears and a new absorption feature of σ*(CO, OH) at 538.2 eV appears,24b which result from the fact that the carboxylate group was protonated to the carboxyl group. Meanwhile, the intensity of 1s → S(3p, 4s)–O(2p) at 537.4 eV increases, indicating more sulfates in the FA desulfurization products. Note that sulfite species might also contribute to the increase. Interestingly, the spectra of FA desulfurization products show both a red shift of 0.24 eV and a slight broadening. The results agree with the findings of Messer et al.24a As previously discussed, the carboxylate group was protonated at low pH, resulting in the breaking of the degeneracy of the π*(COO) orbital, lifting the degeneracy of the two CO bonds and creating a C–O bond.
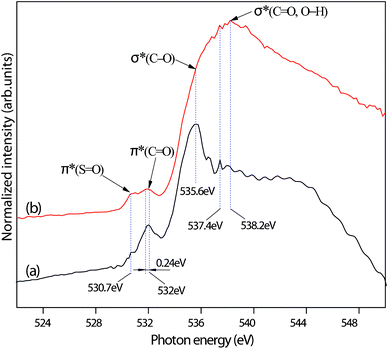 | ||
| Fig. 5 O K-edge NEXAFS of FA (a) and FA desulfurization products (b). | ||
To determine the chemical state of the sulfur atom and to gain an insight into the nature of sulfur-containing reaction products following the interaction of SO2 and FA, we carefully deconvoluted the S2p spectra and referred to the many sulfur standards published.23a,26 The S2p3/2 binding energies are used to elucidate the chemical state of sulfur. Fig. 4e shows two doublets of S2p, and there are two types of sulfur species in FA: one at low binding energy (BE) (168.0 eV), attributed to the sulfonic acid group (–SO3H),23a and another at a higher BE (169.1 eV), corresponding to sulfate.26 Sulfate salts exist as inorganic impurities in the FA sample, which has been confirmed by our previous analysis by ion chromatography and FTIR. Likewise, the sulfonic acid group is detected at two bands (1043 and 654 cm−1) in Fig. 3. A new doublet of S2p3/2 (167.0 eV) and S2p1/2 (168.2 eV) appears in Fig. 4f, which implies sulfite formation.26 Apart from the aforementioned three types of sulfur species, we cannot detect any other sulfur species in the process of fitting the S2p spectra, as shown in Fig. 4f. A comparison of quantitative results in Fig. 4e and f clearly shows that the contribution of sulfite is much smaller than sulfate for FA desulfurization products, and the ratio of sulfate increases dramatically from 8.6 to 53.4%, which likely results from the partial oxidation of sulfite prior to analysis.27 The results demonstrate that the absorbed SO2 is transformed into sulfite and sulfate. Although the ratio of sulfonic acid groups dramatically reduces from 91.4 to 42.9%, this does not imply that some sulfonic acid groups are transformed into other forms of sulfur-containing groups.
Conclusions
Based on the above analysis, we draw a conclusion that the buffer of fulvate plays a decisive role in the process of SO2 absorption. Because of the lower acidity of FA (pKa1 = 3.8, carboxyl groups;28 pKa2 = 8.6, phenolic groups29) compared to that of SO2 (pKa1 = 1.8, H2SO330), deprotonated carboxylate in FA can indirectly interact with SO2. The absorption of SO2 results in an increase in the H+ concentration, which triggers the protonation of COO− to COOH. Namely, the Na+, Ca2+ and Mg2+ ions binding to the COO− groups are replaced by H+. No other chemical changes are found for FA after SO2 absorption in spite of detailed chemical characterization, which demonstrates that FA is stable as a regenerable desulfurization absorbent. The pH of the FA solution is below 5.5, so the bisulfite ions are the principal reactive sulfite species in solution,31 and the absorption mechanism of SO2 can be described as follows:| R–COONa + SO2 + H2O → R–COOH + NaHSO3 | (8) |
| R–(COO)2Ca + 2SO2 + 2H2O → R–(COOH)2 + Ca(HSO3)2 | (9) |
In this paper, we focused on the SO2 absorption mechanisms by FA solution without considering the influences of other components in the flue gas, such as water vapour, CO2, NO and the ash on the absorption, desorption and reabsorption processes. In further research work, we will investigate their influence in detail. The experimental conditions in this work are appropriate for a batch reactor, but the results can also be applied to a continuous reactor where the FA absorption solution can be circulated between the absorption tower and a desorber with the pumps.
Overall, the FA solution can efficiently absorb SO2, the absorbed SO2 can be reversibly desorbed by heating, and the FA solution can be reused. This novel desulfurization method with low costs and benign environmental effects is feasible and potentially an alternative to the WFGD processes based on limestone scrubbing.
Acknowledgements
We gratefully acknowledge financial support by the National Science Foundation of China (No. 50876062, No. 51176113) and the Ministry of Science and Technology of China (No. 2007AA05Z313). We thank the Instrumental Analysis Center of SJTU for FT-IR, ICP-AES and ion chromatography measurements. We also thank Shanghai Synchrotron Radiation Facility (SSRF) for providing beam time at the BL08U beamline.References
- W. Wu, B. Han, H. Gao, Z. Liu, T. Jiang and J. Huang, Angew. Chem., Int. Ed., 2004, 43, 2415–2417 CrossRef CAS.
- (a) Y. Zhong, X. Gao, W. Huo, Z.-y. Luo, M.-j. Ni and K.-f. Cen, Fuel Process. Technol., 2008, 89, 1025–1032 CrossRef CAS; (b) M. Gustin and K. Ladwig, Environ. Sci. Technol., 2010, 44, 4012–4018 CrossRef CAS.
- (a) X. Xu, C. Chen, H. Qi, R. He, C. You and G. Xiang, Fuel Process. Technol., 2000, 62, 153–160 CrossRef CAS; (b) Y. Li, C. Song and C. You, Energy Fuels, 2010, 24, 1682–1686 CrossRef CAS; (c) I. Dahlan, K. T. Lee, A. H. Kamaruddin and A. R. Mohamed, Environ. Sci. Technol., 2006, 40, 6032–6037 CrossRef CAS; (d) A. Srinivasan and M. W. Grutzeck, Environ. Sci. Technol., 1999, 33, 1464–1469 CrossRef CAS.
- (a) J. Huang, A. Riisager, P. Wasserscheid and R. Fehrmann, Chem. Commun., 2006, 4027–4029 RSC; (b) J. Huang, A. Riisager, R. W. Berg and R. Fehrmann, J. Mol. Catal. A: Chem., 2008, 279, 170–176 CrossRef CAS; (c) Y. Shang, H. Li, S. Zhang, H. Xu, Z. Wang, L. Zhang and J. Zhang, Chem. Eng. J., 2011, 175, 324–329 CAS.
- X. L. Yuan, S. J. Zhang and X. M. Lu, J. Chem. Eng. Data, 2007, 52, 596–599 CrossRef CAS.
- S. Y. Hong, J. Im, J. Palgunadi, S. D. Lee, J. S. Lee, H. S. Kim, M. Cheong and K.-D. Jung, Energy Environ. Sci., 2011, 4, 1802–1806 CAS.
- D. J. Heldebrant, P. K. Koech and C. R. Yonker, Energy Environ. Sci., 2010, 3, 111–113 CAS.
- E. M. Peña-Méndez, J. Havel and J. Patočka, J. Appl. Biomed., 2005, 3, 13–24 Search PubMed.
- Pat. China., CN. Patent 200710045443.2, 2008 Search PubMed.
- (a) Z. Sun, Y. Zhao, H. Gao and G. Hu, Energy Fuels, 2010, 24, 1013–1019 CrossRef CAS; (b) G. Hu, Z. Sun and H. Gao, Environ. Sci. Technol., 2010, 44, 6712–6717 CrossRef CAS.
- (a) F. Coccia, L. Tonucci, D. Bosco, M. Bressan and N. d'Alessandro, Green Chem., 2012, 14, 1073–1078 RSC; (b) F. J. Stevenson, Humus chemistry: genesis, composition, reactions, John Wiley and Sons, New York, 2nd edn, 1994 Search PubMed; (c) J. C. García-Gil, S. B. Ceppi, M. I. Velasco, A. Polo and N. Senesi, Geoderma, 2004, 121, 135–142 CrossRef; (d) M. Schnitzer and S. U. Khan, Humic substances in the environment, Marcel Dekker, New York, 1972 Search PubMed.
- (a) R. Khanna, M. Witt, M. Khalid Anwer, S. P. Agarwal and B. P. Koch, Org. Geochem., 2008, 39, 1719–1724 CrossRef CAS; (b) D. Gondar, R. Lopez, S. Fiol, J. M. Antelo and F. Arce, Geoderma, 2005, 126, 367–374 CrossRef CAS; (c) F. J. Stevenson and K. M. Goh, Geochim. Cosmochim. Acta, 1971, 35, 471–483 CrossRef CAS; (d) G. Ait Baddi, M. Hafidi, J. Cegarra, J. A. Alburquerque, J. Gonzálvez, V. Gilard and J.-C. Revel, Bioresour. Technol., 2004, 93, 285–290 CrossRef CAS; (e) A. E. Eneji, T. Honna, S. Yamamoto, T. Masuda, T. Endo and M. Irshad, Commun. Soil Sci. Plant Anal., 2003, 34, 2303–2314 CrossRef; (f) C. P. Singh and A. Amberger, Agrochimica, 1997, 41, 221–228 CAS.
- E. M. Thurman and R. L. Malcolm, Environ. Sci. Technol., 1981, 15, 463–466 CrossRef CAS.
- Y. Zhao, G. Hu, Z. Sun and J. Yang, Energy Fuels, 2011, 25, 2927–2931 CrossRef CAS.
- J.-Y. Lee, S.-J. Khang, C.-H. Tseng and T. C. Keener, Ind. Eng. Chem. Res., 2001, 40, 5822–5830 CrossRef CAS.
- E. Tipping, Cation binding by humic substances, Cambridge University Press, Cambridge, U.K., 2002 Search PubMed.
- (a) M. M. de Souza Sierra, K. Arend, A. N. Fernandes, M. Giovanela and B. Szpoganicz, Anal. Chim. Acta, 2001, 445, 89–98 CrossRef CAS; (b) G.-P. Sheng, M.-L. Zhang and H.-Q. Yu, Anal. Chim. Acta, 2007, 592, 162–167 CrossRef CAS.
- S. Beilke and D. Lamb, Tellus, 1974, 26, 268–271 CrossRef CAS.
- S. M. Doncea, R. M. Ion, R. C. Fierascui, E. Bacalum, A. A. Bunaciu and H. Y. Aboul-Enein, Instrum. Sci. Technol., 2009, 38, 96–106 CrossRef.
- Z. Zhang and G. E. Ewing, Spectrochim. Acta, Part A, 2002, 58, 2105–2113 CrossRef.
- X.-F. Yang, M. Zhao and G. Wang, Sens. Actuators, B, 2011, 152, 8–13 CrossRef.
- Y. Yoshida, B. Van Meerbeek, Y. Nakayama, J. Snauwaert, L. Hellemans, P. Lambrechts, G. Vanherle and K. Wakasa, J. Dent. Res., 2000, 79, 709 CrossRef CAS.
- (a) C. Petit, K. Kante and T. J. Bandosz, Carbon, 2010, 48, 654–667 CrossRef CAS; (b) J. G. C. Shen, T. H. Kalantar, R. G. Herman, J. E. Roberts and K. Klier, Chem. Mater., 2001, 13, 4479–4485 CrossRef CAS.
- (a) B. M. Messer, C. D. Cappa, J. D. Smith, K. R. Wilson, M. K. Gilles, R. C. Cohen and R. J. Saykally, J. Phys. Chem. B, 2005, 109, 5375–5382 CrossRef CAS; (b) S. C. B. Myneni, Rev. Mineral. Geochem., 2002, 49, 485–579 CrossRef CAS.
- E. C. Todd, D. M. Sherman and J. A. Purton, Geochim. Cosmochim. Acta, 2003, 67, 881–893 CrossRef CAS.
- J. Baltrusaitis, C. R. Usher and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 3011–3024 RSC.
- J. J. Burdeniuc and R. H. Crabtree, Green Chem., 1999, 1, 61–64 RSC.
- J. D. Ritchie and E. M. Perdue, Geochim. Cosmochim. Acta, 2003, 67, 85–96 CrossRef CAS.
- C. J. Milne, D. G. Kinniburgh and E. Tipping, Environ. Sci. Technol., 2001, 35, 2049–2059 CrossRef CAS.
- G. V. Buxton, S. McGowan, G. A. Salmon, J. E. Williams and N. D. Wood, Atmos. Environ., 1996, 30, 2483–2493 CrossRef CAS.
- (a) M. R. Hoffmann and J. O. Edwards, J. Phys. Chem., 1975, 79, 2096–2098 CrossRef CAS; (b) H. Li, H. Tu, Q. Cai, Y. Xian and L. Jin, Analyst, 2001, 126, 669–672 RSC; (c) A. Lancia and D. Musmarra, Environ. Sci. Technol., 1999, 33, 1931–1935 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental apparatus, pH curve of the FA solution and regenerated FA solution, SO2 absorption efficiency curve of Cycles 1–8, and characterization data. See DOI: 10.1039/c2ra21536e |
| This journal is © The Royal Society of Chemistry 2012 |