Density functional theory study on a 1.4 nm silicon nanocrystal coated with carbon
Zhenyi
Ni
,
Xiaodong
Pi
* and
Deren
Yang
State Key Laboratory of Silicon Materials and Department of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: xdpi@zju.edu.cn
First published on 25th September 2012
Abstract
Charge carrier transport associated with silicon nanocrystals (Si NCs) can be improved by removing hydrocarbon chains that are routinely attached to the NC surface by means of hydrosilylation. Thermal annealing for the hydrocarbon-chain removal may lead to carbon-coated Si NCs. But the optical behavior of carbon-coated Si NCs has not been clearly understood. By comparing a carbon-coated Si NC with those fully passivated by hydrogen (H) or coated with silicon oxide (SiO2) in the framework of density functional theory, we find that carbon coating causes both the excitation energy and emission energy of the Si NC to significantly decrease. The carbon-coated Si NC exhibits a smaller Stokes shift than the fully H-passivated and SiO2-coated Si NCs. The radiative recombination rate of the carbon-coated Si NC is two orders of magnitude lower than those of the fully H-passivated and SiO2-coated Si NCs. The thermal removal of hydrocarbon chains at the NC surface is not recommended for Si-NC-based light-emitting devices because carbon-coated Si NCs with rather low light emission efficiency may be produced. In contrast, the carbon coating of Si NCs may be beneficial for Si-NC-based solar cells.
It is well known that surface chemistry is critical to the optical performance of silicon nanocrystals (Si NCs) that are a few nanometers large.1–6 Up to now one of the most remarkable achievements in the tuning of the optical properties of Si NCs by means of surface chemistry is hydrosilylation.7–13 Impressive high-efficiency light emission from hydrosilylated Si NCs has been demonstrated.7 Hydrosilylated Si NCs also exhibit excellent dispersibility in desired media and enhanced resistance to oxidation in air,10,11 signifying facilitated processing for Si-NC-based optoelectronic devices.
Hydrosilylation usually results in hydrocarbon chains attached to the surface of Si NCs via Si-C bonds.13–15 Since hydrocarbon chains hinder charge carrier transport between NCs,16,17 the removal of hydrocarbon chains from the surface of Si NCs is highly desired in the final device structures. However, the strong covalent Si–C bonds may prevent this removal during ligand exchange and chemical treatment, which are normally employed in the control of the surface of compound semiconductor nanocrystals.18,19 For NCs of another group IV element—Ge, researchers have turned to more energetic approaches such as thermal annealing to remove hydrocarbon chains at the NC surface.16,20 It has been shown that hydrocarbon chains can only be decomposed rather than cleaved from the NC surface in the temperature range where the quantum confinement effect of NCs is maintained.16 This implies that the thermal removal of hydrocarbon chains at the NC surface may finally lead to carbon-coated NCs, given the strong bonding of C to the NC surface and the volatility of CHx. Therefore, it is imperative to know the optical behavior of the resultant carbon-coated NCs since the technological importance of these NCs centers on their remarkable optical properties.21,22
In this work we build a model to investigate the electronic and optical properties of a carbon-coated Si NC in the framework of density functional theory (DFT). By comparing the carbon-coated Si NC with conventional ones that are fully passivated by hydrogen (H) or coated with silicon oxide (SiO2),23–25 we find that carbon coating causes both the excitation energy and emission energy of a Si NC to significantly decrease. Both the Stokes shift and radiative recombination rate of the carbon-coated Si NC are different from those of the fully H-passivated Si NC and SiO2-coated Si NC.
The size of Si NCs studied here is 1.4 nm. When the coating of SiO2 or carbon at the NC surface is considered, a larger NC size that is closer to routinely experimentally obtained ones is simply beyond the capacity of supercomputers commonly used for first-principle calculations. In fact, insightful results on the electronic structures and optical behavior of Si NCs have already been obtained by modeling 1.4 nm Si NCs.2,13,26 The models of conventional Si NCs that are fully passivated by H (Si@H) and coated with SiO2 (Si@SiO2) are shown in Fig. 1 (a) and (b), respectively. The construction of the model for Si@H (Si71H84) has been detailed in our previous work.27 In the construction of the model for Si@SiO2, every H atom in SiH and SiH3 at the surface of Si@H is first replaced with an O atom, which is then outwardly bonded to an added Si atom. Two Si atoms in the neighboring SiH2 molecules are linked via an O atom because the distance between them is too small to insert additional Si atoms. H is finally used to passivate unsaturated Si at the surface of the resultant structure. This gives rise to Si123H100O96 for Si@SiO2. The coating layer of SiO2 is ∼0.25 nm thick.
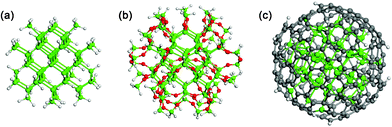 | ||
| Fig. 1 Model of a 1.4 nm Si NC, which is (a) fully passivated by H (Si@H), (b) coated with SiO2 (Si@SiO2) or (c) coated with carbon (Si@C). Si, H, O and C atoms are denoted by green, grey, red and dark grey balls, respectively. The formulas of Si@H, Si@SiO2 and Si@C are Si71H84, Si123H100O96 and Si71C216H36, respectively. | ||
Hydrosilylation only leads to the partial surface coverage of Si NCs with hydrocarbon chains.11 A Si atom that is only passivated by one H atom at the NC surface (i.e. Si in SiH) is very likely not involved in the reaction of hydrosilylation from the point of view of thermodynamics.11,13 Therefore, SiH is left intact when we construct the model for Si@C based on Si@H. H atoms in SiH2 and SiH3 at the NC surface are first all replaced with C atoms. Neighboring C atoms are then linked to each other to form six-carbon-atom rings wherever appropriate. After this treatment H is used to passivate unsaturated C. The resulting coating layer contains ∼16 at.% H and ∼90% sp2 bonds. The obtained Si@C is in the form of Si71C216H36 with ∼0.2 nm thick carbon coating (Fig. 1 (c)). Please note that the current Si@C is distinctly different from both SiC NCs and SiC-coated Si NCs because the unit cell of a SiC crystal does not exist at all. Although the configuration of the carbon coating may vary, it is not of current importance that all the possible configurations of carbon coating are strenuously examined. We believe that the aforementioned Si@C obtained by considering both the experimental and theoretical facts should be representative of carbon-coated Si NCs. We would like to stress that for Si@H, Si@SiO2 and Si@C, the crystalline Si is all 1.4 nm in diameter. Therefore, the electronic and optical changes of Si NCs can not be due to size difference.
The optimization of structures and the calculation of total energies are performed at 0 K with the modeling program of DMol3 in the framework of all-electron DFT. Once the optimization is finished, the resulting structures are thermodynamically stable at 0 K. The Becke-Lee–Yang–Parr (BLYP) correlation exchange functional at the generalized gradient approximation (GGA) level is adopted. Double numerical basis sets augmented with p-polarization functions (DNP basis sets) are used as the atomic orbital basis functions. A high self-consistent field (SCF) convergence threshold of 10−6 is employed to ensure accurate calculation. The maximum forces on all of the atoms in the optimized structures are less than 0.002 Ha/Å. When a Si NC is excited, an electron in the highest occupied molecular orbital (HOMO) transits to the lowest unoccupied molecular orbital (LUMO), leaving a hole in the HOMO. After the HOMO–LOMO transition, the relaxed geometry of Si NC at the excited state is obtained by structure and electronic optimization. For both the ground state and excited state, the HOMO–LUMO gaps are readily calculated once the Si NC optimization is finished. For a Si NC larger than 1 nm, the excitation energy of the NC is similar to the HOMO–LUMO gap of the NC at the ground state. The emission energy of the NC is similar to the HOMO–LUMO gap of the NC at the excited state.23,25,28 Such similarities are adopted to determine the excitation energy and emission energy of Si NCs in this work. Radiative recombination rates for Si NCs are numerically calculated in momentum space using Fermi's golden rule.27,29,30 We would like to mention that we consider the overall effect of coating on the electronic and optical properties of Si NCs in this work. Coating may introduce interface strain, which is implicitly dealt with in the optimization of Si NCs. Further work is needed to elucidate the specific role interface strain plays.
Fig. 2 shows the energy-level diagrams for Si@C at both the (a) ground state and (b) excited state. Those for Si@H and Si@SiO2 are also included for comparison. At the ground state the HOMO–LUMO gap of Si@C is 0.73 eV, which is significantly smaller than that of Si@H (3.13 eV) (Fig. 2 (a)). The reduction of the HOMO–LUMO gap is due to the carbon-coating-induced increase of the HOMO and decrease of the LUMO. Fig. 3 shows the distribution of electron wave functions of the HUMO and LUMO of Si NCs at both the ground state and excited state. We can see that the carbon coating causes the HOMO and LUMO of Si NCs to be mainly located in the coating layer at the ground state. It is this localization that leads to the energy shifts of the HOMO and LUMO. The SiO2 coating also results in the decrease of the HOMO–LUMO gap by increasing the HOMO and decreasing the LUMO (Fig. 2 (a)). But the HOMO–LUMO gap of Si@SiO2 is still much larger than that of Si@C (1.59 vs. 0.73 eV). The SiO2 coating causes the HOMO to be less localized inside the NC core and the LUMO to be more localized inside the NC core, in contrast to what happens due to carbon coating. Clearly, the effect of coating on the localization/delocalization of the HOMO and LUMO is responsible for the energy shifts of the HOMO and LUMO. However, the exact contribution of different elements to the HOMO/LUMO can not be obtained with the current modeling program of DMol3. High-resolution density of states (DOS) should be investigated by use of improved calculation methodology in the future.
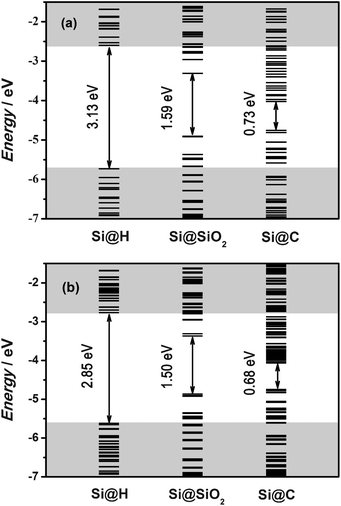 | ||
| Fig. 2 Energy-level diagrams of Si@H, Si@SiO2 and Si@C at (a) the ground state (b) the excited state. | ||
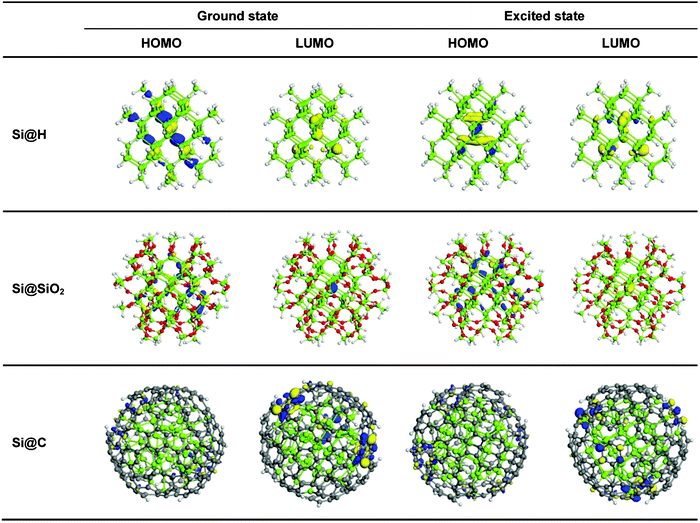 | ||
| Fig. 3 Distribution of the electron wave functions of the HOMO and LUMO of Si@H, Si@SiO2 and Si@C at the ground state and the excited state. | ||
At the excited state the HOMO–LUMO gap of Si@C is 0.68 eV (Fig. 2 (b)), indicating a small Stokes shift (0.05 eV). The Stokes shifts for Si@H and Si@SiO2 are 0.28 and 0.09 eV, respectively. It has been shown that the Stokes shift of a Si NC depends on the NC size.25 The current work demonstrates that coating also impacts the Stokes shift of a Si NC. The smallest Stokes shift for Si@C may be due to the fact that excitation introduces minimum structural distortion to Si@C. On average the excitation-induced change of bond length from center to edge is 0.05% for Si@C, while those for Si@SiO2 and Si@H are −0.08% and 0.26%, respectively.
We should point out that the above-mentioned HOMO–LUMO gaps are underestimated by ∼1–2 eV because of the BLYP-based DFT employed in this work.31 However, the relative order between the HOMO–LUMO gaps (and the approximate excitation/emission energies) is not affected by the underestimation. Now we compare the radiative recombination rate (R) of Si@C to those of Si@H and Si@SiO2 (1.4 × 104 s−1 for Si@C, 1.0 × 106 s−1 for Si@H and 1.2 × 106 s−1 for Si@SiO2). In contrast to a small change of R (20%) induced by SiO2 coating, a decrease of R as large as two orders of magnitude is induced by carbon coating. This result calls for the avoidance of the formation of carbon-coated Si NCs in Si-NC-based light-emitting structures. Once hydrosilylated Si NCs are incorporated in Si-NC-based light-emitting structures, the thermal removal of the hydrocarbon chains at the NC surface to improve the charge carrier transport is not an effective method because carbon-coated Si NCs may be produced. A solution to the contradiction between hydrosilylation and charge carrier transport is to precisely control the coverage of the hydrocarbon chains at the NC surface during hydrosilylation. Optimum coverage can not only make Si NCs soluble in solvents and stable in air for a reasonably long time, but also allows efficient charge carrier transport through uncovered sites (i.e., Si–H).
We would like to comment that the carbon coating of Si NCs may be beneficial for the photovoltaic application of Si NCs, instead. Firstly, the carbon-coating-induced decrease in the HOMO–LUMO gap extends the spectral response of Si-NC-based solar cells to the low-energy range of sunlight. Secondly, the carbon-coating-induced decrease in the recombination rate means that photo-induced carriers in Si NCs may be extracted with increased probability. Finally, the carbon coating may actually promote carrier transport in Si-NC-based solar cells because thin carbon films are usually electrically conductive.32 It will be interesting to investigate the carrier transport associated with Si@C in the future.
In conclusion, the effect of carbon coating for a 1.4 nm Si NC has been theoretically studied. By comparing a carbon-coated Si NC with those fully passivated by hydrogen (H) or coated with silicon oxide (SiO2), we find that carbon coating causes both the excitation energy and emission energy of the Si NC to significantly decrease. The carbon-coated Si NC exhibits a Stokes shift that is different from the fully H-passivated Si NC and SiO2-coated Si NC. The radiative recombination rate of the carbon-coated Si NC is two orders of magnitude smaller than those of the fully H-passivated Si NC and SiO2-coated Si NC. We think that the thermal removal of hydrocarbon chains at the NC surface to improve the charge carrier transport in Si-NC-based light-emitting structures should not be recommended because carbon-coated Si NCs with rather low light emission efficiency may be produced. However, the carbon coating of Si NCs may be beneficial for Si-NC-based solar cells. The current results should encourage experimentalists to start preparing carbon-coated Si NCs and evaluating their potential in device structures.
Acknowledgements
Shanghai Supercomputer Center is thanked for providing computation resources. This study was mainly supported by National Natural Science Foundation of China (Grant 50902122). Partial support from R&D Program of Ministry of Education of China (Grant 62501040202), Innovation Team Project of Zhejiang Province (Grant 2009R50005), Basic Funding for Research at Zhejiang University (2011FZA4005), and Major Scientific program of Zhejiang Province (Grant 2009C01024-2) is acknowledged.References
- A. Puzder, A. J. Williamson, J. C. Grossman and G. Galli, Phys. Rev. Lett., 2002, 88, 097401 CrossRef.
- Y. S. Ma, X. D. Pi and D. Yang, J. Phys. Chem. C, 2012, 116, 5401–5406 CAS.
- J. G. C. Veinot, Chem. Commun., 2006, 40, 4160–4168 RSC.
- M. V. Wolkin, J. Jorne, P. M. Fauchet, G. Allan and C. Delerue, Phys. Rev. Lett., 1999, 82, 197 CrossRef CAS.
- D. Konig, J. Rudd, M. A. Green and G. Conibeer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 035339 CrossRef.
- F. A. Reboredo and G. Galli, J. Phys. Chem. B, 2005, 109, 1072–1078 CrossRef CAS.
- D. Jurbergs, E. Rogojina, L. Mangolini and U. Kortshagen, Appl. Phys. Lett., 2006, 88, 233116 CrossRef.
- J. A. Kelly and J. G. C. Veinot, ACS Nano, 2010, 4, 4645–4656 CrossRef CAS.
- Y.-C. Liao and J. T. Roberts, J. Am. Chem. Soc., 2006, 128, 9061–9065 CrossRef CAS.
- A. Gupta, M. T. Swihart and H. Wiggers, Adv. Funct. Mater., 2009, 19, 696–703 CrossRef CAS.
- B. N. Jariwala, O. S. Dewey, P. Stradins, C. V. Ciobanu and S. Agarwal, ACS Appl. Mater. Interfaces, 2011, 3, 3033–3041 CAS.
- M. Rosso-Vasic, E. Spruijt, B. van Lagen, L. De Cola and H. Zuilhof, Small, 2008, 4, 1835–1841 CrossRef CAS.
- R. Wang, X. D. Pi and D. Yang, J. Phys. Chem. C, 2012, 116, 19434–19443 CAS.
- F. J. Hua, M. T. Swihart and E. Ruckenstein, Langmuir, 2005, 21, 6054–6062 CrossRef CAS.
- S. Calder, A. Boies, P. Y. Lei, S. Girshick and J. Roberts, Chem. Mater., 2011, 23, 2917–2921 CrossRef CAS.
- Z. C. Holman and U. R. Kortshagen, Langmuir, 2009, 25, 11883–11889 CrossRef CAS.
- J. Nelles, D. Sendor, F.-M. Petrat and U. Simon, J. Nanopart. Res., 2010, 12, 1367–1375 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Science, 1995, 270, 1335–1338 CAS.
- J. M. Luther, M. Law, Q. Song, C. L. Perkins, M. C. Beard and A. J. Nozik, ACS Nano, 2008, 2, 271–280 CrossRef CAS.
- Z. C. Holman, C.-Y. Liu and U. R. Kortshagen, Nano Lett., 2010, 10, 2661–2666 CrossRef CAS.
- X. D. Pi, R. W. liptak, J. D. Nowak, N. P. Wells, C.B. Carter, S. A. Campbell and U. Kortshagen, Nanotechnology, 2008, 19, 245603 CrossRef CAS.
- D. C. Lee, J. M. Pietryga, I. Robel, D. J. Werder, R. D. Schaller and V. I. Klimov, J. Am. Chem. Soc., 2009, 131, 3436–3437 CrossRef CAS.
- E. Degoli, G. Cantele, E. Luppi, R. Magri, D. Ninno, O. Bisi and S. Ossicini, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155411 CrossRef.
- K. Seino, F. Bechstedt and P. Kroll, Nanotechnology, 2009, 20, 135702 CrossRef CAS.
- M. L. del Puerto, M. Jain and J. R. Chelikowsky, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035309 CrossRef.
- Z. Y. Zhou, L. Brus and R. Friesner, Nano Lett., 2003, 3, 163 CrossRef CAS.
- X. D. Pi, X. B. Chen and D. Yang, J. Phys. Chem. C, 2011, 115, 9838–9843 CAS.
- C. Delerue, M. Lannoo and G. Allan, Phys. Rev. Lett., 2000, 84, 2457 CrossRef CAS.
- C. Delerue, G. Allan and M. Lannoo, Phys. Rev. B: Condens. Matter, 1993, 48, 11024–11036 CrossRef CAS.
- D. L. Dexter, Solid State Phys. Adv. Res. Appl., 1958, 6, 361 Search PubMed.
- A. J. Williamson, J. C. Grossman, R. Q. Hood, A. Puzder and G. Galli, Phys. Rev. Lett., 2002, 89, 196803 CrossRef.
- S. H. Ng, J. Z. Wang, D. Wexler, K. Konstantinov, Z. P. Guo and H. K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |