First total synthesis of seimatopolide B†
U.
Nookaraju
,
Anand
Harbindu
,
Ankushkumar D.
Bhise
,
Brijesh M.
Sharma
and
Pradeep
Kumar
*
Division of Organic Chemistry, CSIR-National Chemical Laboratory, Pune 411008, India. E-mail: pk.tripathi@ncl.res.in; Fax: +91–20–25902629; Tel: +91–20–25902050
First published on 15th October 2012
Abstract
The first enantioselective total synthesis of seimatopolide B has been achieved, using ring-closing metathesis and DCC (N,N′-dicyclohexylcarbodiimide) coupling as key steps. The stereogenic centres were generated by means of iterative hydrolytic kinetic resolution (HKR) of racemic epoxides.
Introduction
Seimatopolides A and B, two polyhydroxylated 10-membered macrolides, were recently isolated by Jang and Lee et al.1 from an ethyl acetate extract of Seimatosporium discosioides culture medium, along with three known compounds: monosporascone, arthrinone and 3a,9a-deoxy-3a-hydroxy-1-dehydroxyarthrinone. Seimatopolides belong to a novel class of 10-membered lactones, as well as members of the deacarestrictine,2 microcarpalide3 and pinolidoxin4 families etc. (Fig. 1). These compounds are structurally related to each other based on their physico-chemical properties. Owing to their interesting biological properties, including inhibition of cholesterol biosynthesis,5 antimalarial and antibacterial activities,6 and microfilament formation,3 these compounds have attracted a great deal of interest among synthetic organic chemists worldwide as attractive synthetic targets towards developing new therapeutic agents. Seimatopolides exhibited significant activity in a reporter gene assay for activation of peroxisome proliferator-activated receptor γ (PPAR-γ)7a with EC50 values of 11.05 μM, which shows therapeutic potential in the treatment of type 2 diabetes, inflammatory disease and certain cancers.7b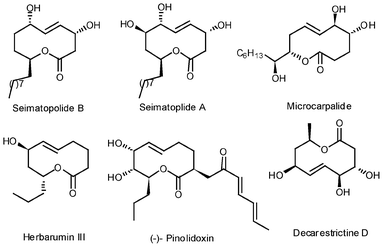 | ||
| Fig. 1 Some selected examples of 10-membered lactones. | ||
As a part of our research programme aimed at developing enantioselective syntheses of biologically active natural products8 based on hydrolytic kinetic resolution (HKR),9 we became interested in the design of a simple and concise route to seimatopolide B. Herein we report our successful endeavours towards the first total synthesis of seimatopolide B employing HKR, DCC coupling and ring-closing metathesis (RCM)10 as the key steps.
Results and discussion
In continuation of our ongoing interest in exploiting the HKR method to install the stereocentres and ring-closing metathesis for making cyclic compounds viz. 10-membered lactone rings based upon protecting group directed ring-closing metathesis protocol11 and generalizing its substrate based selectivity, we planned to synthesize seimatopolide B.Our retrosynthetic analysis for seimatopolide B is based on the convergent approach as outlined in Scheme 1. We envisioned that the natural product 1 could be obtained by ring-closing metathesis of diene precursor 2, which in turn could be prepared by intermolecular DCC coupling of acid 3 and alcohol 4. Acid 3 could be obtained from 3-butene-1-ol (5) while the alcohol fragment could be prepared from rac-epichlorohydrin (6) via iterative HKR.
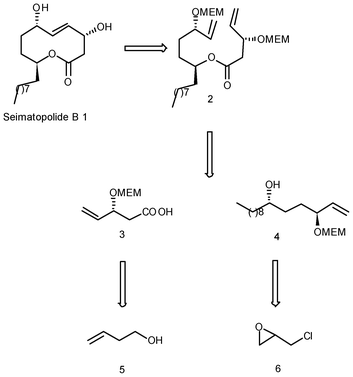 | ||
| Scheme 1 Retrosynthetic route to seimatopolide B. | ||
As depicted in Scheme 2, synthesis of acid fragment 3 commenced from commercially available 3-butene-1-ol (5). This, upon hydroxy group protection with TBSCl followed by oxidation using mCPBA, gave epoxide 7 in 90% yield. The rac-epoxide 7 was subjected to Jacobsen's HKR9 using (R,R)-salen-CoIII(OAc) catalyst to give the enantiopure epoxide12a (R)-7a in 46% yield along with diol 7b in 45% yield, which were separated by silica gel column chromatography.
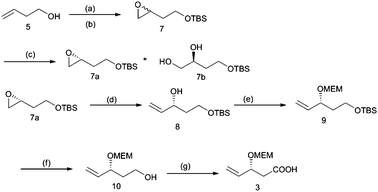 | ||
| Scheme 2 Synthesis of the acid fragment 3. Reagents and conditions: (a) TBDMSCl, imidazole, CH2Cl2, 0 °C to rt, 4h, 88%; (b) m-CPBA, CH2Cl2, 0 °C to rt, 1h, 90%; (c) R,R-salen-Co-(OAc) (0.5 mol %), dist. H2O (0.55 equiv), isopropyl alcohol, 0 °C, 24h, (46% for 7a, 45% for 7b); (d) (CH3)3SI, n-BuLi, THF, 4h, 86%; (e) MEMCl, DIPEA, CH2Cl2, 16h, 87%; (f) TBAF, THF, 1h, 88%; (g) TEMPO, NaH2PO4, NaOCl, NaClO2, CH3CN, overnight, 95%. | ||
Epoxide (R)-7a, on ring opening with dimethylsulfonium methylide,13 afforded one-carbon homologated allylic alcohol 8 in 86% yield, which was protected as the MEM ether using MEMCl and DIPEA followed by removal of the TBS group to furnish alcohol 10 in 88% yield. TEMPO-catalysed oxidation of the alcohol with NaOCl resulted in the formation of acid 3 in excellent yield.
As illustrated in Scheme 3, synthesis of alcohol fragment 4 started from commercially available (±) epichlorohydrin 6, which was converted to epoxide 12 by a known procedure.8m The racemic epoxide 12 was resolved using (S,S)-salen CoIII-OAc to give enantiopure epoxide12b12a in 45% yield. The epoxide 12a was opened with allylmagnesium bromide, followed by epoxidation and hydroxy group protection as TBS to give the epoxide 14 as mixture of syn and anti compounds (syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti, 1:1.15).14 In order to obtain the diastereomerically pure epoxide, the epoxide 14 was resolved using (S,S)-salen CoIII-OAc and water in THF to give the diastereomerically pure epoxide 14a in 48% yield. As the HKR method provided the desired epoxide 14a along with unwanted diol 14b, we thought it appropriate to convert diol 14b into the required epoxide 14avia internal nucleophilic substitution of the secondary mesylate.15 Accordingly, chemoselective pivalation of diol 14b with pivaloyl chloride was followed by mesylation of the secondary hydroxy group. Treatment of the crude mesylate with potassium carbonate in methanol led to deprotection of the pivaloyl ester and concomitant ring closure via intramolecular SN2 displacement of the mesyl group to furnish epoxide 14a in 68% overall yield.
:anti, 1:1.15).14 In order to obtain the diastereomerically pure epoxide, the epoxide 14 was resolved using (S,S)-salen CoIII-OAc and water in THF to give the diastereomerically pure epoxide 14a in 48% yield. As the HKR method provided the desired epoxide 14a along with unwanted diol 14b, we thought it appropriate to convert diol 14b into the required epoxide 14avia internal nucleophilic substitution of the secondary mesylate.15 Accordingly, chemoselective pivalation of diol 14b with pivaloyl chloride was followed by mesylation of the secondary hydroxy group. Treatment of the crude mesylate with potassium carbonate in methanol led to deprotection of the pivaloyl ester and concomitant ring closure via intramolecular SN2 displacement of the mesyl group to furnish epoxide 14a in 68% overall yield.
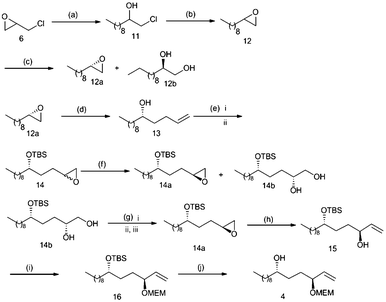 | ||
| Scheme 3 Synthesis of the alcohol fragment 4. Reagents and conditions: (a) Octylmagnesium bromide, THF, CuI, −40 °C, 12 h; (b) KOH, CH2Cl2, rt, 14h; (c) S,S-salen-Co-(OAc) (0.5 mol %), dist. H2O (0.55 equiv), isopropyl alcohol, 0 °C, 14h, (45% for 12a, 43% for 12b); (d) Allylmagnesium bromide, ether, CuI, −20 °C, 2h, 89%; (e) (i) TBDMSCl, imidazole, CH2Cl2, 0 °C to rt, 16h, 90%; (ii) m-CPBA, CH2Cl2, 0 °C to rt, 1h, 80%; (f) S,S-salen-Co-(OAc) (0.5 mol %), dist. H2O (0.55 equiv), isopropyl alcohol, 0 °C, 16h, (48% for 14a, 39% for 14b); (g) (i) PivCl, Et3N, cat. DMAP, rt, 2h; (ii) MsCl, Et3N, DMAP, 0 °C to rt, 1h; (iii) K2CO3, MeOH, rt, overnight (68% for three steps); (h) (CH3)3SI, n-BuLi, THF, 4h, 81%; (i) MEMCl, DIPEA, CH2Cl2, 14h, 80%; (j) TBAF, THF, 3h, 81%. | ||
With substantial amounts of epoxide 14a in hand we further proceeded towards the synthesis of alcohol fragment 4 by ring opening of the epoxide with dimethylsulfonium methylide to obtain the allylic alcohol 15 in excellent yield. Subsequent hydroxyl group protection as its MEM ether followed by TBS removal furnished the alcohol fragment 4 in 81% yield (Scheme 3).
With substantial amounts of both the fragments in hand the coupling of acid 3 and alcohol 4 was achieved by using intermolecular DCC coupling to afford the diene ester 2 in 91% yield, which was subjected to deprotection of MEM groups using PPTS in t-BuOH to give the naked diol ester 17. Subsequent ring-closing metathesis using Grubbs’ 2nd generation catalyst in CH2Cl2 resulted in the unnatural isomer, Z-seimatopolide B 1 exclusively in 74% yield (Scheme 4). Z-Seimatopolide B 1 was well characterized by physical and spectroscopic methods (see the ESI†). With an aim to synthesize the natural isomer E-seimatopolide B 1, we extended our studies to explore the protecting group directed ring-closing metathesis (RCM).11 We envisaged that protecting groups around the reacting centers might act as temporary constraints to adequately shape the dienes and simultaneously confer selectivity upon the stereochemistry of the newly formed double bond. With this view in mind, the suitably protected diene ester 2 was then subjected to ring-closing metathesis conditions using Grubbs’ 2nd generation catalyst in CH2Cl2 under reflux conditions. To our delight, the reaction led to the formation of cyclized product 18, albeit in only 50% yield. Subsequent deprotection of MEM ethers using TFA in CH2Cl2 afforded the natural product, seimotopolide B 1 exclusively as the trans-isomer. E-Seimatopolide B 1 was well characterized by 1H & 13C NMR, mass and IR spectral data. In 1H NMR the coupling constant between H4 and H5 clearly demonstrated the trans nature of the double bond (Scheme 4).
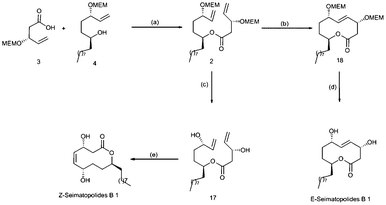 | ||
| Scheme 4 Synthesis of E-seimatopolide and Z-seimatopolide. Reagents and conditions: a) DCC, cat. DMAP, CH2Cl2, 6h, rt, 91% ; (b) Grubbs’ 2nd generation catalyst, CH2Cl2, reflux, 16h, 50% ;(c) PPTS, t-BuOH, reflux, 16h, 82%; (d) TFA, CH2Cl2, rt, 16h, 70% ; (e) Grubbs’ 2nd generation catalyst, CH2Cl2, reflux, 16h, 74%. | ||
The physical and spectroscopic data of E-seimatopolide B 1 was in full agreement with that of the natural product reported in literature1 (Fig. 2, Table 1).
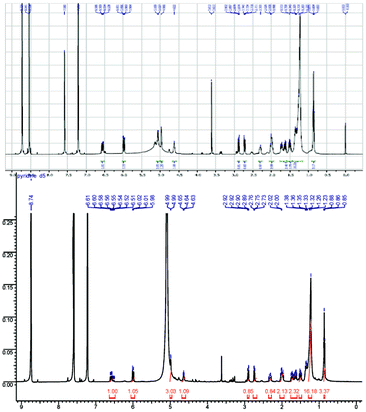 | ||
| Fig. 2 1H NMR spectra of E-seimatopolide B 1 (500 MHz, pyridine-d5): Natural (top) and synthetic (bottom). | ||
| E-Seimatopolide B (natural) spectroscopic data (500 MHz, pyridine-d5) | E-Seimatopolide B (synthetic) spectroscopic data (500 MHz, pyridine-d5) | ||
|---|---|---|---|
| 1H NMR (J in Hz) | 13C NMR | 1H NMR (J in Hz) | 13C NMR |
| 6.56, dd (8.5, 16.0) | 170.5 | 6.56, dd (8.54, 16.17) | 170.6 |
| 5.98, dd (3.0, 16.0) | 133.4 | 5.98, dd (3.05, 16.17) | 133.4 |
| 5.06, ddd (7.0, 7.0, 13.0) | 133.4 | 5.09, m | 133.4 |
| 4.96, m | 76.5 | 4.99, m | 76.5 |
| 4.62, dd (7.5, 7.5) | 74.9 | 4.64, m | 74.9 |
| 2.89, dd (3.0, 11.5) | 67.8 | 2.90, dd (3.4, 11.6) | 67.8 |
| 2.72, dd (3.0, 11.5) | 45.7 | 2.74, dd (3.9, 11.6) | 45.8 |
| 2.30, m | 38.5 | 2.32, m | 38.6 |
| 2.00, m | 36.3 | 2.00, m | 36.4 |
| 2.00, m | 32.4 | 2.00, m | 32.4 |
| 1.72, m | 31.0 | 1.72, m | 31.2 |
| 1.62, m | 30.2 | 1.63, m | 30.3 |
| 1.51, m | 30.2 | 1.51, m | 30.2 |
| 1.22, m | 30.1 | 1.23 (brs) | 30.1 |
| 1.22, m | 29.9 | 0.88, t (7.0) | 29.9 |
| 1.22, m | 26.0 | 26.1 | |
| 1.22, m | 23.2 | 23.3 | |
| 1.22, m | 14.6 | 14.6 | |
| 1.22, m | |||
| 1.22, m | |||
| 0.86, dd (7.0, 7.0) | |||
Conclusion
In conclusion, a convergent and efficient first total synthesis of seimatopolide B, both natural (E-isomer) and unnatural (Z-isomer) has been accomplished with high enantioselectivity, in which the stereocentres were generated by means of iterative Jacobsen's hydrolytic kinetic resolution, and cyclization was achieved by ring-closing metathesis. This approach could be used for the synthesis of other members of this class of macrolides for structure–activity relationship studies. Work in this direction is currently in progress.Experimental section
Z-Seimatopolide B 1
To a stirred solution of 17 (0.030 g, 0.088 mmol, 0.001 M) in freshly distilled degassed anhydrous CH2Cl2 (90 ml) was added Grubbs’ second generation catalyst (0.015 g, 0.017 mmol) and heated under reflux for 16 h under an argon atmosphere until the complete consumption of the starting material (monitored by TLC). The solvent was evaporated to a brown residue, which was purified by silica gel column chromatography using petroleum ether:EtOAc (53:47) to afford Z-seimatopolide B 1 (0.020 g, 74%) as a white amorphous solid. [α]25D: −118.53 (c 0.049, MeOH). IR (neat, cm−1): νmax 3243, 2911, 2852, 1722; 1H NMR (400 MHz, pyridine-d5): δ 7.02 (brs, 1H), 6.58 (brs, 1H), 5.94 (dd, J = 10.55, 9.78 Hz, 1H), 5.74 (dd, J = 10.29, 11.04 Hz, 1H), 5.51 (m, 1H), 5.33 (m, 1H), 5.12 (m, 1H), 3.36 (dd, J = 14.05, 6.02 Hz, 1H), 2.78 (m, 1H), 2.35–2.44 (m, 1H), 2.02–2.12 (m, 1H), 1.70–1.82 (m, 2H), 1.50–1.59 (m, 2H), 1.23 (brs, 14H), 0.88 (t, J = 7.03 Hz, 3H); 13C NMR (100 MHz, pyridine-d5): δ 170.6, 135.2, 133.9, 74.8, 66.6, 65.1, 44.6, 33.2, 32.6, 31.6, 30.3, 30.2, 30.1, 28.4, 26.8, 23.5, 14.8, 30.5; LC–MS: m/z = 335.17 [M + Na]+ HRMS (ESI) for C18H32O4Na (M + Na)+ found 335.2187, calcd 335.2193.
E-Seimatopolide B 1
To a solution of compound 18 (0.040 g, 0.082 mmol) in CH2Cl2, was added TFA (0.062 mL, 0.82 mmol) and the mixture stirred at room temperature for 16 h. The reaction mixture was quenched with a saturated aqueous solution of NaHCO3, extracted with EtOAc and the organic layer was separated, washed with brine, dried over Na2SO4, concentrated under reduced pressure and purified by column chromatography using EtOAc:petroleum ether (1:1) as eluent to afford E-seimatopolide B 1 (0.018 g, 70%) as a white amorphous solid. [α]25D: −212.60 (c 0.035, MeOH).{lit1. [α]25D −125.3 (c 0.03, MeOH)}; IR (neat, cm−1): νmax 3242, 2912, 2853, 1721; 1H NMR (500 MHz, pyridine-d5): δ 6.56 (dd, J = 8.54, 16.17 Hz 1H), 5.98 (dd, J = 3.05, 16.17 Hz 1H), 5.09 (m, 2H), 4.99 (m, 1H), 4.64 (m, 1H), 2.90 (dd, J = 3.36, 11.60 Hz, 1H), 2.74 (dd, J = 3.97, 11.60 Hz, 1H), 2.32 (m, 1H), 2.00 (m, 2H), 1.72 (m, 1H), 1.63 (m, 1H), 1.51 (m, 1H), 1.23 (m, 14H), 0.88 (t, J = 7.02 Hz, 3H); 13C NMR (125 MHz, pyridine-d5): δ 170.6, 133.4, 76.5, 74.9, 67.8, 45.8, 38.6, 36.4, 32.4, 31.2, 30.3, 30.2, 30.1, 29.9, 26.1, 23.3, 14.6; LC–MS: m/z = 335.17 [M + Na]+. HRMS (ESI) for C18H32O4 (M + Na)+ found 335.2187, calcd 335.2193.
Acknowledgements
UN and AH thank UGC New Delhi and AD and BMS thank CSIR New Delhi for research fellowships. Financial support from DST, New Delhi (grant No. SR/S1/OC-44/2009) is gratefully acknowledged.References
- N. T. Hiep, Y. Choi, N. Kim, S. S. Hong, S. Hong, B. Y. Hwang, H. Lee, S. Lee, D. S. Jang and D. Lee, J. Nat. Prod., 2012, 75, 784 CrossRef.
- M. M. Victor, V. B. Riatto and R. A. Pilli, Tetrahedron, 2008, 64, 2279 CrossRef.
- A. S. Ratnayake, W. Y. Yoshida, S. L. Mooberry and T. Hemscheidt, Org. Lett., 2001, 3, 3479 CrossRef CAS.
- A. Furstner and K. Radkowski, Chem. Commun., 2001, 671 RSC.
- (a) A. Gohrt, A. Zeeck, K. Hutter, R. Kirsch, H. Kluge and R. Thiericke, J. Antibiot., 1992, 45, 66 CrossRef CAS; (b) S. Grabley, E. Granzer, K. Hutter, D. Ludwig, M. Mayer, R. Thiericke, G. Till, J. Wink, S. Philipps and A. Zeeck, J. Antibiot., 1992, 45, 56 CrossRef CAS; (c) S. Grabley, P. Hammann, K. Hutter, R. Kirsch, H. Kluge, R. Thiericke, M. Mayer and A. Zeeck, J. Antibiot., 1992, 45, 1176 CrossRef CAS; (d) M. Mayer and R. Thiericke, J. Antibiot., 1993, 46, 1372 CrossRef CAS.
- (a) V. Rukachaisirikul, S. Pramjit, C. Pakawatchai, M. Isaka and S. Supothina, J. Nat. Prod., 2004, 67, 1953 CrossRef CAS; (b) G. Drager, A. Kirschning, R. Thiericke and M. Zerlin, Nat. Prod. Rep., 1996, 13, 365 RSC.
- (a) D. Lee, J. H. Lee, X. F. Cai, J. C. Shin, K. Lee, Y. S. Hong and J. J. Lee, J. Antibiot., 2005, 58, 615 CrossRef CAS; (b) G. J. Murphy and J. C. Holder, Trends Pharmacol. Sci., 2000, 21, 469 CrossRef CAS.
- (a) P. Kumar, P. Gupta and S. V. Naidu, Chem.–Eur. J., 2006, 12, 1397 CrossRef CAS; (b) P. Gupta, S. V. Naidu and P. Kumar, Tetrahedron Lett., 2004, 45, 849 CrossRef CAS; (c) P. Gupta, S. V. Naidu and P. Kumar, Tetrahedron Lett., 2005, 46, 6571 CrossRef CAS; (d) S. K. Pandey and P. Kumar, Tetrahedron Lett., 2005, 46, 6625 CrossRef CAS; (e) P. Kumar and S. V. Naidu, J. Org. Chem., 2006, 71, 3935 CrossRef CAS; (f) S. K. Pandey and P. Kumar, Synlett, 2007, 2894 Search PubMed; (g) S. V. Naidu and P. Kumar, Tetrahedron Lett., 2007, 48, 3793 CrossRef CAS; (h) P. Gupta and P. Kumar, Eur. J. Org. Chem., 2008, 1195 CrossRef; (i) S. K. Pandey, M. Pandey and P. Kumar, Tetrahedron Lett., 2008, 49, 3297 CrossRef CAS; (j) P. S. Chowdhury, P. Gupta and P. Kumar, Tetrahedron Lett., 2004, 50, 7188 CrossRef; (k) P. S. Chowdhury, P. Gupta and P. Kumar, Tetrahedron Lett., 2009, 50, 7018 CrossRef CAS; (l) P. Gupta and P. Kumar, Tetrahedron: Asymmetry, 2007, 18, 1688 CrossRef CAS; (m) A. Harbindu and P. Kumar, Synthesis, 2010, 9, 1479 Search PubMed.
- (a) M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936 CrossRef CAS; (b) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS.
- For reviews on ring-closing metathesis, see: (a) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413 CrossRef CAS; (b) J. Prunet, Angew. Chem., Int. Ed., 2003, 42, 2826 CrossRef CAS.
- (a) D. K. Mohapatra, D. K. Ramesh, M. A. Giardello, M. S. Chorghade, M. K. Gurjar and R. H. Grubbs, Tetrahedron Lett., 2007, 48, 2621 CrossRef CAS; (b) D. Castoldi, L. Caggiano, P. Bayón, A. M. Costa, P. Cappella, O. Sharon and C. Gennari, Tetrahedron, 2005, 61, 2123 CrossRef CAS; (c) L. Caggiano, D. Castoldi, R. Beumer, P. Bayón, J. Telser and C. Gennari, Tetrahedron Lett., 2003, 44, 7913 CrossRef CAS; (d) N. A. Sheddan, V. B. Arion and J. Mulzer, Tetrahedron Lett., 2006, 47, 6689 CrossRef CAS; (e) N. A. Sheddan and J. Mulzer, Org. Lett., 2006, 8, 3101 CrossRef CAS.
- The enantiomeric purity of epoxides 7a and 12a was determined by comparing optical rotation with known literature values: Compound 7a: [α]25D: +10.85 (c 2, CHCl3) lit.12a [α]24D: +11.0 (c 2, CHCl3); Compound 12a: [α]25D: -6.96 (c 1, CHCl3); Compound ent-12a: lit.12b [α]24D: +6.34 (c 1, CH); (a) N. P. H. Tan and C. D. Donner, Tetrahedron Lett., 2009, 65, 4007 CAS; (b) M. Amatore, T. D. Beeson, S. P. Brown and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 5121 CrossRef CAS.
- L. Alcaraz, J. J. Harnett, C. Mioskowski, J. P. Martel, T. Le Gall, D. Shin and J. R. Falck, Tetrahedron Lett., 1994, 30, 5449 CrossRef.
- Diastereomeric ratio (syn/anti ratio) was determined by 1H and 13C NMR analysis.
- (a) K. C. Nicolaou and S. E. Webber, Synthesis, 1986, 453 CrossRef CAS; (b) K. Takao, H. Ochiai, K. Yoshida, T. Hashizuka, H. Koshimura, K. Tadano and S. Ogawa, J. Org. Chem., 1995, 60, 8179 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H NMR, 13C NMR spectra of compounds 7a, 8, 9, 10, 3, 12a, 13, 14, 14a, 15, 4, 16, 2, 17, 18, Exp. See DOI: 10.1039/c2ra21838k |
| This journal is © The Royal Society of Chemistry 2012 |