Characterization of an isotactic polystyrene/poly(2,6-dimethylphenylene oxide) nanorod blend with gradient composition and crystallinity
Hui
Wu
ab,
Zhaohui
Su
c and
Atsushi
Takahara
*ab
aJapan Science and Technology Agency, ERATO, Takahara Soft Interfaces Project, Fukuoka, 819-0395, Japan. E-mail: takahara@cstf.kyushu-u.ac.jp
bInstitute for Materials Chemistry and Engineering, Kyushu University, Fukuoka, 819-0395, Japan
cState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China
First published on 16th August 2012
Abstract
Using a nanoporous alumina template, we prepared an isotactic polystyrene/poly(2,6-dimethylphenylene oxide) (iPS/PPO) nanorod blend with gradient composition and gradient crystallinity. The crystallinity, orientation and composition of the polymer in the nanorods were investigated by micro-FTIR spectroscopy. Although iPS and PPO are compatible at the molecular level, due to the viscosity difference between the two components, the polymer blend underwent phase separation during the capillary flow into the nanopores to form a gradient distributed composition in the nanorods. Along the growth direction (from the bottom to the top) of the nanorods, the iPS content increased, but the iPS crystallinity decreased due to crystallization initiated by the nuclei formed in the bulk and the constrained crystal growth. This finding provides a unique approach to the design and fabrication of novel nanomaterials with gradient properties in nanoscale engineering.
Introduction
In recent years, one-dimensional (1-D) nanomaterials have received growing attention due to their unique properties and potential applications in electronics, mechanical, optical, sensor and biomedical devices.1–4 A number of strategies, such as template infiltration,5,6 electrospinning,7,8 self-assembly of microphase-separated block copolymers,9–13 and nanoimprint lithography,2,14 have been pursued to build 1-D polymeric nanostructures. Among these approaches, one of the most intriguing fabrication methods involves the use of nanoporous anodic aluminum oxide (AAO) templates. The AAO templates consist of aligned, rigid and separated cylindrical pores, the length and diameter of which are adjustable by controlling the electrochemical anodization conditions. This offers unique opportunities for the preparation and investigation of 1-D polymer nanomaterials with various dimensions and aspect ratios. The polymer nanostructures, including nanotubes, nanowires and nanorods, are generated inside the templates by infiltrating the high surface energy nanopores with polymer melts or solutions of low surface energy, and then released from the templates by dissolving the alumina in an acidic or basic solution.For the 1-D polymer nanomaterials, supramolecular organization plays an essential role in determining their intrinsic properties, and well-controlled nanostructures with novel morphology,12,13 specific orientation,15–22 reduced crystallinity,21–23 polymorphism,24 enhanced mobility,25 and high thermal conductivity26 have been reported. A variety of unusual morphologies of phase-separated amorphous block copolymers in nanopores that are not accessible in the bulk can be observed.12,13 Nanorods of semicrystalline polymers are particularly interesting. It has been revealed that the c-axes of polymer crystals preferentially orient perpendicular to the long axis of the nanopore,15–22 and this orientation can be attributed to the kinetic crystal growth compatible with the cylindrical confinement.15 Significant depression in crystallinity and melting temperature was observed in nanorods in contrast to the bulk due to the confinement of cylindrical nanopores, and the decrease is more substantial in nanorods of smaller diameters.21–23 In smaller nanopores, stronger confinement leads to nucleation behavior different from that in the bulk, which results in polymorphic polymer nanorods.24
Generally these 1-D polymeric nanoobjects are considered uniform along the length direction. However, our recent studies have indicated otherwise for polymer nanorods fabricated using AAO templates. Composition gradient was revealed in amorphous nanorods of completely miscible poly(2,6-dimethylphenylene oxide)/polystyrene (PPO/PS) blends27 and partially miscible polycarbonate/polystyrene (PC/PS) blends.28 In semi-crystalline polymer nanorods of isotactic polystyrene (iPS), gradient distribution in both crystallinity and orientation of the crystallites along the length direction of the nanorod was observed.29 These findings inspire us to further explore a more complicated system, polymer blends with one semicrystalline component, where interplay between composition gradient and crystallization may be present. In this work, iPS/PPO blends were chosen as an example of this kind of system for investigation. Nanorods of 35 nm diameter of iPS/PPO blends were obtained by a template wetting method. Because the characteristic vibrational bands associated with various iPS and PPO microstructures and morphologies have been well established,30–34 it is convenient to investigate iPS/PPO nanorod arrays using vibrational spectroscopy techniques. The crystallinity, orientation and composition distribution in small sections of a nanorod array from top to bottom after crystallization was analyzed by micro-FTIR spectroscopy. Our results are reported here.
Experimental
Sample preparation
iPS was kindly supplied by Idemitsu Kosan Co., Ltd., and PPO was purchased from Aldrich. To minimize the influence of polymers with different molecular weight forming gradient molecular composition27,28,35 on the polymer crystallization behavior36 in nanopores, the iPS and PPO were fractionated first by successive precipitation using toluene (for iPS) and CHCl3 (for PPO) as solvents and methanol as the non-solvent,37,38 and then by a recycling preparative HPLC (Japan Analytical Industry Co., Ltd., LC-9104) equipped with an RI detector (JAI RI-7s) using CHCl3 as eluent. iPS with Mn of 121,000 (Mw/Mn = 1.16) and PPO with Mn of 24,300 (Mw/Mn = 1.16) were obtained and used in this study. The isotacticity of the iPS was about 100% as determined by 13C NMR. An AAO template with pore diameter of 35 nm (Fig. 1a, b) and length of 140 μm was prepared by a two-step anodization process using sulfuric acid as electrolyte at a voltage of 25 V.39 | ||
| Fig. 1 SEM images of an AAO template with a pore diameter of 35 nm and a slice of iPS/PPO nanorods/film prepared with the AAO template: (a) bottom view and (b) cross section of the AAO template; (c) cross section of the polymer nanorods connected with the bulk film; (d) nanorods. | ||
A film of an iPS/PPO blend (90/10 w/w) was prepared by solution casting from a CHCl3 solution into a Petri dish, which was further dried under vacuum at 353 K for 36 h to remove any residual solvent. Then, a homogeneous amorphous iPS/PPO film blend of 200 μm thickness was obtained by compression moulding the cast film between sheets of aluminum foil under vacuum at 533 K (20 K above the equilibrium melting point of iPS) for 4 min and quickly quenching the film in ice water. The AAO template was placed on top of the compression-moulded iPS/PPO film blend. The assembly was maintained under vacuum at 533 K for 8 h to yield polymer nanorods with a length of ca. 115 μm. Then the sample was crystallized at 443 K under vacuum for 40 h for the iPS crystallization to fully develop.40 Thin slices of the cross section of iPS/PPO film with protruding nanorods (Fig. 1c, d) for scanning electron microscopy (SEM) and micro-FTIR analyses were cut along the rod direction using a razor blade after removal of the AAO template by a 5 wt.% phosphoric acid solution.
Characterization
The morphologies of the AAO template and the blend nanorods were investigated using an S-4300SE (Hitachi Co., Ltd.) SEM operating at an acceleration voltage of 5 kV. Micro-FTIR measurements were performed on a PerkinElmer Spectra One spectrometer in connection with a microscope equipped with an MCT detector operating in the transmission mode. The microscope connected to the FTIR included a viewing system so that the operation of the microscope, mapping and spectra collection from the sample were controlled by the Autoimage software. A spatial resolution was maintained at 300 × 25 μm2. Polarized FTIR spectra were recorded using a wire-grid polarizer parallel or perpendicular to the rod direction (Fig. 2A). All spectra were collected at 2 cm−1 resolution with 128 scans co-added.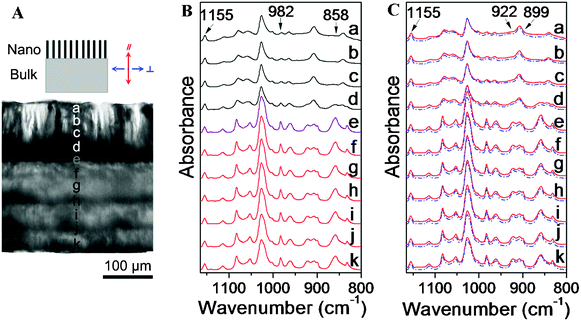 | ||
| Fig. 2 (A) Optical micrograph of a thin slice of the iPS/PPO nanorods/film. On the top is a schematic illustration of the nanorods/film with the reference direction. (B) Micro-FTIR spectra measured at different positions of the nanorods and the bulk film as labeled in (A). The spatial resolution is 300 × 25 μm2 and the spectra are normalized with respect to the 1155 cm−1 band. (C) Corresponding polarized IR spectra at these positions: parallel polarization (—); perpendicular polarization (---) against the length direction of the rods. | ||
Results and discussion
The iPS/PPO nanorod blend were prepared by wetting the porous AAO template with the polymer melt via capillary force. To minimize the influence of molecular weight distribution,35,36 both iPS and PPO were fractionated prior to use. iPS and PPO are compatible over the whole composition range,41 and the amounts of PPO in the blend have a significant effect on the crystallization of iPS.40 When examining the iPS/PPO blend films with different blending ratios, the crystallinity of the bulk film with PPO contents higher than 18 wt.% is extremely low (ca. one-tenth of that of pure iPS). Therefore, to better understand the crystallization and phase behavior of polymer blend in nanorods, an iPS/PPO blend of 90/10 (w/w) was prepared in this study. The iPS/PPO nanorod blend were fabricated by wetting the AAO template with a pore diameter of 35 nm (Fig. 1a, b) with the blend melt. After annealing at 533 K for 8 h, nanorods with a length of ca. 115 μm and a diameter of 35 nm (Fig. 1c, d) were obtained. The polymer nanorod blend in the AAO template was then annealed at 443 K for 40 h for iPS crystallization to fully develop.40 PPO is known to crystallize only in the presence of solvent or solvent vapor (acetone, 2-butanone, etc.)40 and thus remained amorphous under these conditions.To analyze the composition distribution and crystal texture of iPS along the length direction of the nanorod blend, thin slices of the iPS/PPO nanorods/film were cut along the rod direction using a razor blade after removal of the AAO template and characterized using micro-FTIR. Fig. 2A shows the optical micrograph of a thin slice of the iPS/PPO nanorod array/film. The dark area at the top is the nanorod array which is supported by the translucent bulk film at the bottom.21 During the FTIR measurements, a spatial resolution of 25 μm along the length direction of the rods was maintained to obtain the spectra from the top to the bottom of the nanorods in the thin slice. Fig. 2B shows the corresponding micro-FTIR spectra of the iPS/PPO nanorods/film in the measured positions. The top four curves are the spectra of the nanorod array at positions a–d, the bottom six curves represent the residual bulk film at positions f–k, while the curve at position e is the spectrum of the polymer blend at the bottom of the nanorod array which is connected with the bulk film.
Because many absorption bands overlap one another in the IR spectra of the iPS/PPO blends, the characteristic bands at 1155, 982, 922, 899 and 858 cm−1, which are relatively isolated and clean, were used for structural analysis.30–34 The band at 1155 cm−1 is due to the phenyl CH in-plane deformation of iPS.30 This band exhibits little intensity variation during the crystallization process31 as well as weak dichroism,32 and thus is largely independent of crystallinity and orientation. Therefore, the 1155 cm−1 band was chosen as a reference band,29,38 and all spectral data were normalized with respect to this band. The peak at 858 cm−1 is assigned to the CH out-of-plane bending vibration of the 1,2,4,6-tetrasubstituted benzene ring of PPO.33 The 858 and 1155 cm−1 bands can be utilized to determine quantitatively the composition of iPS/PPO blends, viaeqn 1 and 2,27,28
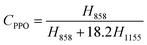 | (1) |
| CiPS = 100% − CPPO | (2) |
where CPPO and CiPS represent the contents of PPO and iPS in the iPS/PPO blends, and H858 and H1155 are the respective peak heights for the 858 and 1155 cm−1 bands. Using the above equations, PPO and iPS contents at different positions along the nanorod and in the bulk film were extracted from the IR spectra (Fig. 2B) and are plotted in Fig. 3A. It can be seen that, in various positions of the bulk film, the iPS content remains almost constant at 90%, same as that formulated for the blend as expected, and that, then into the nanorods, the iPS content rises from 88% at the bottom (position e) to 99% at the top (position a). The average iPS content in the nanorods (97%) is significantly higher than that in the bulk (90%). This indicates that a gradient composition distribution was formed in the nanopores along the long axis of the nanorod, and that the polymer blend underwent a phase-separation process, which is consistent with the findings reported previously.27,28
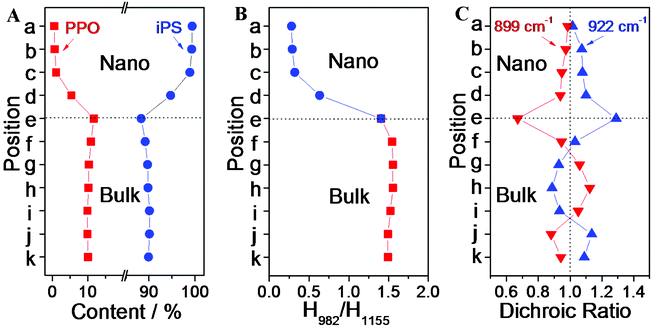 | ||
| Fig. 3 (A) iPS and PPO contents at different measured positions. (B) Relative intensities of the crystalline 982 cm−1 band with respect to the 1155 cm−1 band as indirect indices of iPS crystallinity at the different measured positions. (C) Dichroic ratios of the 922 cm−1 band (▲) and 899 cm−1 band (▼) in the nanorods at different positions. | ||
The degree of crystallinity of iPS in the blends was examined by the band at 982 cm−1. This band is assigned to the CH out-of-plane bending vibration of the benzene ring of iPS crystal,30,32 and is most sensitive to crystallization, with an absorbance increment in proportion to the degree of crystallinity. Therefore, the ratio of the absorptions at 982 and 1155 cm−1, H982/H1155, can be used as an indirect index of crystallinity of iPS.29,38Fig. 3B displays the values of H982/H1155 at different positions in the nanorods and the bulk film, which were extracted from the data in Fig. 2B. The H982/H1155 ratio is ∼1.5 and barely varies at different positions in the bulk, but declines sharply in the nanorods from the bottom (position e) to the top (position a) to a value of ∼0.3. This result indicates a gradient crystallite formation in the nanorods, and the crystallinity of the iPS in the nanorods is obviously lower than that in the bulk. The crystallinity at position e (bottom of the nanorods) is much higher than that at positions a–d (upper sections of the nanorods), showing that the polymer in the nanopores mainly crystallized at the bottom of the nanorods. The iPS content at the top of the nanorods is the highest and declines from top to bottom, yet the iPS crystallinity at the top is the lowest and increases downward, indicating an unusual crystallization process in the nanorods.
To gain further understanding of the crystallization process, the nanorods were analyzed by polarized FTIR, and the spectra collected at different positions along the nanorods and in the bulk are shown in Fig. 2C. The growth direction of the nanorods was defined as the reference direction (Fig. 2A). The solid traces are the spectra measured in the parallel polarization, whereas the dashed lines are those in the perpendicular polarization. To examine the orientation order in the crystalline domains, dichroic ratios of the bands of 922 and 899 cm−1 at different positions were analyzed. These bands are absent in the spectra of amorphous iPS, and are assigned to CH out-of-plane bending vibrations of crystalline iPS with appropriate perpendicular and parallel dichroism, respectively.30,34 The dichroism of 922 and 899 cm−1 bands was calculated as 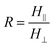 , with
, with 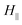 and
and 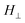 the measured absorbances (peak heights) in the parallel and perpendicular spectra, respectively. The dichroic ratios of these two bands at different positions are plotted in Fig. 3C. It can be observed that the dichroic ratios of the 922 cm−1 band in the nanorods (positions a–e) are greater than 1, indicating that the iPS crystallites tend to orient perpendicularly, with the c-axis perpendicular to the long axis of the nanorods. In addition, the dichroic ratios of the 922 cm−1 band decrease from position e to a, which implies that the degree of orientation of the crystallites in the nanorods declines along the growth direction. Meanwhile, the trend observed for the dichroic ratios of the parallel band at 899 cm−1 are just the opposite, further confirming the above derived results based on the perpendicular 922 cm−1 band.
the measured absorbances (peak heights) in the parallel and perpendicular spectra, respectively. The dichroic ratios of these two bands at different positions are plotted in Fig. 3C. It can be observed that the dichroic ratios of the 922 cm−1 band in the nanorods (positions a–e) are greater than 1, indicating that the iPS crystallites tend to orient perpendicularly, with the c-axis perpendicular to the long axis of the nanorods. In addition, the dichroic ratios of the 922 cm−1 band decrease from position e to a, which implies that the degree of orientation of the crystallites in the nanorods declines along the growth direction. Meanwhile, the trend observed for the dichroic ratios of the parallel band at 899 cm−1 are just the opposite, further confirming the above derived results based on the perpendicular 922 cm−1 band.
From the results discussed above, it is clear that a gradient composition and gradient crystallinity are present simultaneously in the blend nanorods. A schematic illustration of the formation process of the iPS/PPO nanorod blend in the nanopores is shown in Fig. 4. Two steps contribute to the formation of this complicated and novel nanostructure. The first step is the formation of gradient composition in the nanorods. Before the nanopores are infiltrated by the polymer melt, both iPS and PPO in the blend are in the molten state (the experimental temperature is well above the Tm of iPS). Because the melt viscosity of PPO is much higher than PS, the addition of PS can improve the processability of PPO.42 However, during the capillary flow of the polymer blend melt into the nanopores, the iPS component with lower viscosity has higher mobility to enter the nanopores than PPO, leading to higher iPS content in the nanorods than that in the bulk film. Within the nanopores, the iPS component continues to move upwards faster than PPO, resulting in the greatest iPS content at the top of the nanorods and a composition gradient along the growth direction of the nanorods. Although iPS and PPO are compatible at molecular level over the entire composition range,41 the difference in viscosity between the two components drives the phase separation of iPS/PPO during the capillary flow in the nanopores, which leads to the formation of nanorods with a composition gradient.27
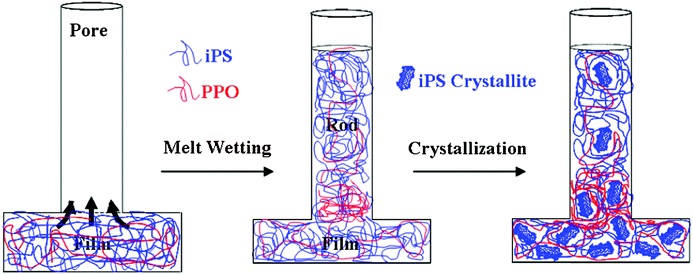 | ||
| Fig. 4 Schematic illustration of structure formation in the iPS/PPO blend in the nanopores. Two steps contribute to the development of gradient composition and gradient crystallinity in the nanorods. The first step involves the formation of gradient composition due to viscosity differences between the two components during the capillary flow into the nanopores. The second step is the formation of gradient distributed crystallites owing to the crystallization initiated by the nuclei formed in the bulk and crystal growth constrained by the nanopores. | ||
The second step is the formation of gradient crystallinity in the nanorods. For semicrystalline polymers crystallized in nanopores, the nucleation process is significantly suppressed.23 In a separate experiment, we observed by polarized light microscopy (PLM) an iPS/PPO film blend isothermally crystallized at 443 K, and found that the nucleation density was ca. 1.8 × 10−6 nuclei/μm3 (data not shown). For a nanorod with a diameter of 35 nm and a length of 115 μm, it contains only 2.0 × 10−7 nuclei. Thus due to the lack of heterogeneous nuclei, it is difficult for the polymer to crystallize in isolated nanopores, and in many nanorods the polymer remains amorphous and the degree of crystallinity in the nanorods is lower than that in the bulk.21,22 However, in this study the nanorods are connected with a bulk film that experiences an identical thermal history. Thus, the crystals (lamellae in the spherulites) formed in the bulk can hit the nanorods and serve as nuclei to initiate crystallization at the bottom of the nanorods (e.g. position e). Due to the ‘gate effect’ imposed by the nanopores,15 only a fraction of the lamellae can grow into the nanopores to form crystals with a perpendicular or near perpendicular orientation. Therefore, the polymer crystallized at the bottom of the nanorods (position e) exhibits lower crystallinity than that in the bulk (positions f–k) and higher preferred perpendicular orientation with the c-axis perpendicular to the long axis of the nanopores. Further into the upper sections of the nanorods (along the rod direction from position d to a), only crystallites oriented along the pore direction can still grow and growth of other crystals is suppressed by the pore wall. Therefore, the iPS crystallinity decreases rapidly from the bottom to the top of the nanorods, causing a gradient distribution of crystallites in the nanopores, although the iPS content increases from the bottom to the top at the same time.
Conclusions
In summary, we have studied a blend of nanorods of semicrystalline iPS and amorphous PPO fabricated using an AAO template, and revealed the co-existence of gradient composition and gradient crystallinity in these blended nanorods. Although iPS and PPO are compatible at the molecular level over the entire composition range, the polymer blend in the nanopores phase separates to form a composition gradient along the growth direction due to viscosity differences between the two components when the nanorods develop driven by capillary forces. Then in the subsequent crystallization process, because of the geometric constraint imposed by the nanopores, crystallization initiated by the nuclei in the bulk into the nanorods results in decreases in both degrees of crystallinity and perpendicular orientation of the iPS crystallites from the bottom to the top of the nanorods, although the iPS content rises at the same time. This outcome could pave a way to the design of novel 1-D nanomaterials with a gradient of properties such as refractive index and modulus for applications in functional optical and mechanical nanodevices.References
- M. Steinhart, Adv. Polym. Sci., 2008, 220, 123 CAS.
- Z. J. Hu, M. W. Tian, B. Nysten and A. M. Jonas, Nat. Mater., 2009, 8, 62 CrossRef CAS.
- J. S. Kim, Y. Park, D. Y. Lee, J. H. Lee, J. H. Park, J. K. Kim and K. Cho, Adv. Funct. Mater., 2010, 20, 540 CrossRef CAS.
- V. Callegari and S. Demoustier-Champagne, Macromol. Rapid Commun., 2011, 32, 25 CrossRef CAS.
- V. M. Cepak and C. R. Martin, Chem. Mater., 1999, 11, 1363 CrossRef CAS.
- M. Steinhart, R. B. Wehrspohn, U. Gösele and J. H. Wendorff, Angew. Chem., Int. Ed., 2004, 43, 1334 CrossRef CAS.
- D. Li and Y. N. Xia, Adv. Mater., 2004, 16, 1151 CrossRef CAS.
- Y. Liu, L. Cui, F. X. Guan, Y. Gao, N. E. Hedin, L. Zhu and H. Fong, Macromolecules, 2007, 40, 6283 CrossRef CAS.
- P. Huang, Y. Guo, R. P. Quirk, J. J. Ruan, B. Lotz, E. L. Thomas, B. S. Hsiao, C. A. Avila-Orta, I. Sics and S. Z. D. Cheng, Polymer, 2006, 47, 5457 CrossRef CAS.
- S. Nojima, Y. Ohguma, K. Kadena, T. Ishizone, Y. Iwasaki and K. Yamaguchi, Macromolecules, 2010, 43, 3916 CrossRef CAS.
- T. M. Chung, T. C. Wang, R. M. Ho, Y. S. Sun and B. T. Ko, Macromolecules, 2010, 43, 6237 CrossRef CAS.
- K. Shin, H. Q. Xiang, S. I. Moon, T. Kim, T. J. McCarthy and T. P. Russell, Science, 2004, 306, 76 CrossRef CAS.
- P. Dobriyal, H. Xiang, M. Kazuyuki, J.-T. Chen, H. Jinnai and T. P. Russell, Macromolecules, 2009, 42, 9082 CrossRef CAS.
- K. Honda, M. Morita, H. Masunaga, S. Sasaki, M. Takata and A. Takahara, Soft Matter, 2010, 6, 870 RSC.
- M. Steinhart, P. Göring, H. Dernaika, M. Prabhukaran, U. Gösele, E. Hempel and T. Thurn-Albrecht, Phys. Rev. Lett., 2006, 97, 027801 CrossRef.
- J. Martin, C. Mijangos, A. Sanz, T. A. Ezquerra and A. Nogales, Macromolecules, 2009, 42, 5395 CrossRef CAS.
- M. C. Garcia-Gutierrez, A. Linares, J. J. Hernandez, D. R. Rueda, T. A. Ezquerra, P. Poza and R. J. Davies, Nano Lett., 2010, 10, 1472 CrossRef CAS.
- J. L. Lutkenhaus, K. McEnnis, A. Serghei and T. P. Russell, Macromolecules, 2010, 43, 3844 CrossRef CAS.
- K. Park, K. Choi, J. H. Lee, S. H. Park, S. C. Lee and H. S. Lee, ACS Macro Lett., 2012, 1, 110 CrossRef CAS.
- R. M. Michell, A. T. Lorenzo, A. J. Muller, M.-C. Lin, H.-L. Chen, I. Blaszczyk-Lezak, J. Martin and C. Mijangos, Macromolecules, 2012, 45, 1517 CrossRef CAS.
- H. Wu, W. Wang, H. Yang and Z. Su, Macromolecules, 2007, 40, 4244 CrossRef CAS.
- K. Shin, E. Woo, Y. G. Jeong, C. Kim, J. Huh and K. W. Kim, Macromolecules, 2007, 40, 6617 CrossRef CAS.
- H. Duran, M. Steinhart, H. J. Butt and G. Floudas, Nano Lett., 2011, 11, 1671 CrossRef CAS.
- H. Wu, W. Wang, Y. Huang, C. Wang and Z. Su, Macromolecules, 2008, 41, 7755 CrossRef CAS.
- K. Shin, S. Obukhov, J. T. Chen, J. Huh, Y. Hwang, S. Mok, P. Dobriyal, P. Thiyagarajan and T. P. Russell, Nat. Mater., 2007, 6, 961 CrossRef CAS.
- B.-Y. Cao, Y.-W. Li, J. Kong, H. Chen, Y. Xu, K.-L. Yung and A. Cai, Polymer, 2011, 52, 1711 CrossRef CAS.
- H. Wu, Z. Su and A. Takahara, Soft Matter, 2011, 7, 1868 RSC.
- H. Wu, Z. Su and A. Takahara, Polym. J., 2011, 43, 600 CrossRef CAS.
- H. Wu, Z. Su and A. Takahara, Soft Matter, 2012, 8, 3180 RSC.
- H. Tadokoro, Y. Nishiyama, S. Nozakura and S. Murahashi, Bull. Chem. Soc. Jpn., 1961, 34, 381 CrossRef CAS.
- T. Kimura, H. Ezure, S. Tanaka and E. Ito, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1227 CrossRef CAS.
- P. C. Painter and J. L. Koenig, J. Polym. Sci., Part B: Polym. Phys., 1977, 15, 1885 CAS.
- S. T. Wellinghoff, J. L. Koenig and E. Baer, J. Polym. Sci., Part B: Polym. Phys., 1977, 15, 1913 CAS.
- B. Jasse and J. L. Koenig, Polymer, 1981, 22, 1040 CrossRef CAS.
- M. F. Zhang, P. Dobriyal, J. T. Chen, T. P. Russell, J. Olmo and A. Merry, Nano Lett., 2006, 6, 1075 CrossRef CAS.
- E. Ergoz, J. G. Fatou and L. Mandelkern, Macromolecules, 1972, 5, 147 CrossRef CAS.
- F. Danusso and G. Moraglio, J. Polym. Sci., 1957, 24, 161 CrossRef CAS.
- K. Kawahara and R. Okada, J. Polym. Sci., 1962, 56, S7 CrossRef CAS.
- H. Masuda, F. Hasegwa and S. Ono, J. Electrochem. Soc., 1997, 144, L127 CrossRef CAS.
- R. Hammel, W. J. MacKnight and F. E. Karasz, J. Appl. Phys., 1975, 46, 4199 CrossRef CAS.
- W. Wenig, F. E. Karasz and W. J. Macknight, J. Appl. Phys., 1975, 46, 4194 CrossRef CAS.
- L. A. Utracki, Polymer Alloys and Blends, Hanser Publishers, New York, 1989 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |