A simple diamine as ligand in iron-catalyzed regioselective allylic alkylation†
Chenguang
Yu
,
Aihua
Zhou
and
Jing
He
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: jinghe@263.net.cn; Fax: +86-10-64425385; Tel: +86-10-64425280
First published on 2nd August 2012
Abstract
A simple and efficient diamine was found to be an efficient ligand for the iron-catalyzed regioselective allylic alkylation between various allyl carbonates and nucleophiles, affording excellent yields and good regioselectivity (up to 94% and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Allylic alkylation is among the most powerful methods for carbon–carbon bond formation in organic chemistry.1 Regioselective nucleophilic addition on the 1- or 3-carbon of an allylic entity is often required in total synthesis, yet is the most challenging.2 Significant endeavour has been devoted to addressing this issue through exploring effective metals and developing valid ligands. Various transition metal catalysts have been found to possess different regioselectivity propensities in allylic alkylation. Pd(0) catalysis3 preferentially forms linear products while iridium(I),4 ruthenium(II),5 or molybdenum(0)6 catalysis favor the formation of branched products. Unfortunately, all the metal complexes mentioned above show deficiencies in the regioselective nucleophilic addition to unsymmetric allylic carbonyl derivatives containing different alkyl or aryl substituents on the 1- and 3-carbon. In this case, rhodium(I)7 complexes exhibit high regioselectivity and the nucleophilic addition predominantly takes place at the carbon bearing the leaving group. Although rhodium complexes have shown excellent regioselectivity, activity and broad substrate tolerance, searching for inexpensive and abundant metals as alternatives for rhodium(I) is still desirable. Fe(−II) possesses ten valence electrons and is isoelectronic with Pd(0). Not surprisingly, Fe(−II) complexes8 catalyze allylic alkylation smoothly, but the reaction proceeds in a way more similar to that with rhodium than with palladium. Fe(−II) complexes form a σ-enyl–iron intermediate in the reaction process and hence show the same regioselective propensity as rhodium(I) complexes.8,9 From both economical and ecological points of view, iron catalysis is obviously more significant.10
In 1979, Roustan and coworkers8a discovered that Na[Fe(CO)3(NO)] regioselectively catalyzed allylic alkylation. The nucleophilic addition preferentially took place at the carbon bearing the leaving group. Then Xu and Zhou8b improved Roustan's procedure through employing shelf-stable tetrabutylammonium salt [Bu4N][Fe(CO)3(NO)]. In this procedure, a higher regioselectivity was obtained, but a CO atmosphere was indispensable to successfully operate the catalytic process. The large catalyst loading and utilization of a toxic CO atmosphere hindered further study on iron-catalyzed allylic alkylation. No progress was made until 2006 when Plietker8c found that triphenylphosphine, a σ-donor ligand, was effective in iron-catalyzed regioselective allylic alkylation without involving a CO atmosphere. However, DMF had to be employed as solvent to increase the catalyst nucleophilicity. Inclusion of DMF as solvent resulted in a more tedious work-up procedure and exclusion of some active nucleophiles. Later, Plietker8d replaced DMF with methyl tert-butyl ether and improved the reaction efficiency by employing N-heterocyclic carbenes as more effective ligands. Despite all the successes,11 simple and efficient ligands are still in high demand.
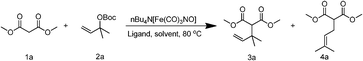
Compared with prevalent phosphorus ligands, amine ligands present many advantages in their availability, ease of recovery, and stability against oxidation, and thus have been widely used as ligands for metal catalytic centers.12 Although no empty orbital exists for electron retrodonation from transition metals, amine ligands still show efficient coordination with transition metals.13 Absence of electron retrodonation orbitals can be expected to result in an accumulation of electron density in the core metal. Electron-rich iron centers could facilitate the formation of σ-enyl-iron complexes, which are the key intermediates in iron-catalyzed regioselective allylic alkylations. In this work, commercially available simple amines are proposed to act as ligands of the Fe(−II) center for regioselective allylic alkylation. It has been found that, with N,N,N′,N′-tetramethylethylenediamine (TMEDA), a simple and inexpensive diamine as ligand, the regioselective allylic alkylation between various allyl carbonates and nucleophiles can be effectively catalyzed, affording excellent yields and good regioselectivity (up to 94% and >20:1). More significantly, a sequential allylic alkylation has been successfully catalyzed by this Fe(−II)–diamine system, producing a synthetically useful cyclopentene unit.
Initially, the allylic alkylation was performed with dimethyl malonate (1a) and tert-butyl (2-methylbut-3-en-2-yl) carbonate (2a) in DMF, the optimal solvent for the Fe(−II)–PPh3 system according to a previous report,8c using TMEDA, triethylamine (Et3N), or N,N-diisopropylethylamine (i-Pr2NEt) as the ligand of [Bu4N][Fe(CO)3NO]. Inspiringly, the desired product was afforded in 68% yield and 6.3:1 regioselectivity with TMEDA (Table 1, entry 2), in 58% yield and 6.3:1 regioselectivity with Et3N (Table 1, entry 3), and in 65% yield and 6.7:1 regioselectivity with i-Pr2NEt (Table 1, entry 4). In contrast, only a trace amount of product was observed in the absence of any ligand (Table 1, entry 1), indicating that the amines employed here are all effective ligands of Fe(−II) for allylic alkylation. More encouragingly, the regioselectivity with amine ligands is similar to that with PPh3 ligand (Table 1, entry 5), even though DMF is a medium fit for the Fe(−II)–PPh3 system. However, the yields need to be improved with amine ligands, so the optimization of the reaction solvent was further performed (Table 1, entries 6–11). When the solvent was altered from strongly coordinating DMF to weakly coordinating or even non-coordinating solvents, the yield was visibly improved with either i-Pr2NEt, a monoamine ligand (Table 1, entries 4, 6–9), or TMEDA, a diamine ligand (Table 1, entries 2, 10–11). An increase in regioselectivity was also observed with decreasing coordinating capacity of the solvents. The solvent optimization was found to impart the most visible positive effects on the diamine ligand. A yield of 92% and regioselectivity of 8.0:1 were achieved with TMEDA in toluene, a non-coordinating solvent (Table 1, entry 11). When the catalyst loading was decreased to 5 mol%, the yield and regioselectivity were well preserved (Table 1, entry 12). Even when the catalyst loading was further decreased to 2.5 mol%, 88% yield and 7.7:1 regioselectivity were obtained in 24 h (Table 1, entry 13). 5 mol% was considered as the optimal catalyst loading. Due to a more convenient work-up procedure and better tolerance of active nucleophile, toluene is preferred to DMF. However, when triphenylphosphine was tested in toluene, a lower yield (79%) and similar regioselectivity (8.9:1) were obtained (Table 1, entry 14), which indicates the advantages of TMEDA as ligand.
| Entry | Ligand | Solvent | t/h |
3a:4ab |
Yield [%]c |
|---|---|---|---|---|---|
| a Conditions: 1a (0.6 mmol), 2a (0.3 mmol) and 10 mol% [Bu4N][Fe(CO)3NO] modified with 11 mol% ligand; 0.3 mL solvent. b Determined by 1H NMR. c Isolated yield. d 5 mol% [Bu4N][Fe(CO)3NO] and 6 mol% PPh3 used. e 5 mol% [Bu4N][Fe(CO)3NO] and 5.5 mol% TMEDA used. f 2.5 mol% [Bu4N][Fe(CO)3NO] and 2.75 mol% TMEDA used. | |||||
| 1 | — | DMF | 66 | — | Trace |
| 2 | TMEDA | DMF | 10 | 6.3:1 |
68 |
| 3 | Et3N | DMF | 10 | 6.3:1 |
58 |
| 4 | i-Pr2NEt | DMF | 10 | 6.7:1 |
65 |
| 5d | PPh3 | DMF | 10 | 6.5:1 |
88 |
| 6 | i-Pr2NEt | CH3CN | 10 | 7.4:1 |
80 |
| 7 | i-Pr2NEt | Dioxane | 10 | 7.4:1 |
76 |
| 8 | i-Pr2NEt | THF | 10 | 7.2:1 |
82 |
| 9 | i-Pr2NEt | Toluene | 10 | 8.3:1 |
81 |
| 10 | TMEDA | THF | 10 | 6.0:1 |
91 |
| 11 | TMEDA | Toluene | 10 | 8.0:1 |
92 |
| 12e | TMEDA | Toluene | 10 | 8.3:1 |
93 |
| 13f | TMEDA | Toluene | 24 | 7.7:1 |
88 |
| 14d | PPh3 | Toluene | 10 | 8.9:1 |
79 |
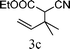
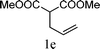
The substrate tolerance for Fe(−II)-TMEDA catalyzed regioselective allylic alkylation in toluene was then investigated. In the screening of nucleophiles (Table 2), the steric hindrance around the reactive carbon atom was altered, which is supposed to affect reaction regioselectivity as well as yield. The results show that, with malonitrile (1b) as substrate, a nucleophile with less steric hindrance than dimethyl malonate (1a), 87% yield and more than 20:1 regioselectivity were afforded (entry 1). On increasing the steric hindrance of the nucleophile by replacing one cyano group with an ester group (1c), a similar yield and regioselectivity to those observed for dimethyl malonate (1a) were produced (90% and 8.3:1, entry 2). When the steric hindrance of the nucleophile was further increased through changing the methyl groups of dimethyl malonate (1a) to isopropyl (1d) groups, 88% yield and 5.5:1 regioselectivity (Table 2, entry 3) were afforded. Even for dimethyl allylmalonate (1e), with a tertiary reactive carbon atom, Fe(−II)-TMEDA catalysis afforded the desired product in 67% yield and 5.9:1 regioselectivity in 72 h (Table 2, entry 4). Two contiguous quaternary carbon centers were formed herein, which is always a challenge. The reaction between 1e and but-3-en-2-yl tert-butyl carbonate on Fe(−II)-TMEDA was further performed, and 82% yield and 4.8:1 regioselectivity were afforded (see ESI,† eqn (1)). Although regioselectivity decreased slightly with the increase of nucleophile steric hindrance, nucleophilic additions all predominantly took place at the carbon bearing the leaving group.
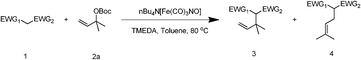
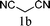
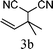
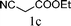
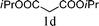
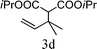
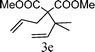
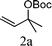
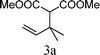
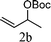
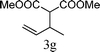
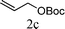
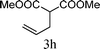
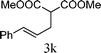
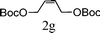
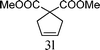
From the point of view of the mechanism, substitution on C1 or C3 of allylic carbonates might affect the yield, so allylic carbonates bearing various C1 and C3 substituents were screened (Table 3) with the Fe(−II)-TMEDA catalyst in toluene. First, the allylic carbonates bearing no substituent on C3 and dimethyl (2a), monomethyl (2b), and no substitution (2c) on C1 were used as substrates. The desired products were afforded in 93% yield and 8.3:1 regioselectivity for 2a (entry 1), 94% yield and 8.3:1 regioselectivity for 2b (entry 2) and 88% yield for 2c (entry 3). When there are one alkyl (2d), two alkyl (2e) or even one aryl substituent (2f) on the C3 of allylic carbonate, Fe(−II)-TMEDA successfully catalyzes all reactions, giving 91% yield and 7.1:1 regioselectivity for 2d (entry 4), 85% yield and 9.0:1 regioselectivity for 2e (entry 5) and 84% yield and 11.0:1 regioselectivity for 2f (entry 6). Therefore, it can be concluded that Fe(−II)-TMEDA has good tolerance for substituted allylic carbonates. More significantly, Fe(−II)-TMEDA was found to successfully catalyze a sequential allylic alkylation between dimethyl malonate (1a) and (Z)-but-2-ene-1,4-diyl di-tert-butyl dicarbonate (2g), yielding synthetically useful substituted cyclopentene (3l) in 41% yield (entry 7). The lower yield was due to the non-cyclization product (35%) remaining in the reaction mixture.

| Entry | Substrate 2 | Product 3 | t/h |
3:4b |
Yield [%]c |
|---|---|---|---|---|---|
| a Conditions: unless specified, see footnote e in Table 1 and ESI. b Determined by 1H NMR. c Isolated yield. d 1a (0.2 mmol) and 2g (0.3 mmol) in 1.0 mL toluene. | |||||
| 1 |
|
|
10 | 8.3:1 |
93 |
| 2 |
|
|
10 | 8.3:1 |
94 |
| 3 |
|
|
48 | — | 88 |
| 4 |
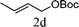
|

|
10 | 7.1:1 |
91 |
| 5 |
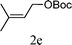
|
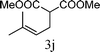
|
48 | 9.0:1 |
85 |
| 6 |
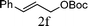
|
|
24 | 11.0:1 |
84 |
| 7d |
|
|
72 | — | 41 |
To investigate the role of the diamine as a ligand in Fe(−II) catalysis, the FT-IR spectra of the iron catalyst before and after addition of TMEDA as ligand and allylic carbonate as substrate were recorded (Fig. 1). For [Bu4N][Fe(CO)3(NO)], the bands assigned to the stretching vibrations of coordinated carbonyl groups appeared at 1981 and 1877 cm−1 (Fig. 1a), which is in agreement with the previous report.8b After adding TMEDA and stirring for half an hour, the bands at 1981 and 1877 cm−1 are preserved, while a new set of bands at 1779 and 1722 cm−1 (Fig. 1b) is observed, owing to the interference of TMEDA in the stretching vibrations of the coordinated carbonyl groups. The band shifting to lower wavenumbers indicates that the Fe(−II) center becomes more electron-rich due to the coordination with TMEDA. This phenomenon was also observed for the coordination between Mo(CO)6 and TMEDA; in that case, more electron density was accumulated at the molybdenum centre compared with coordination with dppe (1,2-bis(diphenylphosphino)ethane).14 When allylic carbonate 2a was added into the THF solution containing [Bu4N][Fe(CO)3NO] and TMEDA, the bands at 1779 and 1722 cm−1 disappeared (Fig. 1c), indicating that the Fe(−II)–TMEDA complex is the catalytically active species. According to previous reports,12b the stability of the Fe–amine complex is strengthened with an increase in the number of nitrogen atoms available for bonding in the ligand, which explains the better performance of diamine over monoamine as the ligand of Fe(−II).
![FT-IR spectra of (a) [Bu4N][Fe(CO)3NO], (b) [Bu4N][Fe(CO)3NO]/TMEDA, and (c) [Bu4N][Fe(CO)3NO]/TMEDA/allylic carbonate 2a.](/image/article/2012/RA/c2ra21646a/c2ra21646a-f1.gif) | ||
| Fig. 1 FT-IR spectra of (a) [Bu4N][Fe(CO)3NO], (b) [Bu4N][Fe(CO)3NO]/TMEDA, and (c) [Bu4N][Fe(CO)3NO]/TMEDA/allylic carbonate 2a. | ||
In conclusion, a simple, inexpensive and efficient diamine ligand has been used for iron-catalyzed regioselective allylic alkylation. The Fe(−II)-diamine complex can effectively catalyze the regioselective allylic alkylation between various allyl carbonates and nucleophiles in excellent yields and with good regioselectivity (up to 94% and >20:1). Extension of the catalytic applications of Fe(−II)–diamine is underway in our laboratory.
Acknowledgements
The authors wish to thank the NSFC and 973 Project (2011CBA00504) for financial support. J. H. particularly appreciates the financial aid of the China National Funds for Distinguished Young Scientists from the NSFC.Notes and references
- For selected reviews regarding application of allylic alkylation in total synthesis, see: (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS; (b) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476 CrossRef CAS.
- For selected examples about application of regioselective allylic alkylation in total synthesis, see: (a) P. A. Evans, D. K. Leahy, W. J. Andrews and D. Uraguchi, Angew. Chem., Int. Ed., 2004, 43, 4788 CrossRef CAS; (b) P. A. Evans, J. Qin, J. E. Robinson and B. Bazin, Angew. Chem., Int. Ed., 2007, 46, 7417 CrossRef CAS; (c) P. A. Evans, K. W. Lai, H.-R. Zhang and J. C. Huffman, Chem. Commun., 2006, 844 RSC.
- For selected reviews regarding palladium catalyzed allylic alkylation, see: (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS; (b) B. M. Trost, Acc. Chem. Res., 1996, 29, 355 CrossRef CAS; (c) B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747 CrossRef CAS.
- For selected reviews regarding iridium catalyzed allylic alkylation, see: (a) G. n. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675 RSC; (b) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS.
- For selected examples regarding ruthenium catalyzed allylic alkylation, see: (a) R. Hermatschweiler, I. Fernández, F. Breher, P. S. Pregosin, L. F. Veiros and M. J. Calhorda, Angew. Chem., Int. Ed., 2005, 44, 4397 CrossRef CAS; (b) N. Kanbayashi and K. Onitsuka, J. Am. Chem. Soc., 2010, 132, 1206 CrossRef CAS.
- For selected examples regarding molybdenum catalyzed allylic alkylation, see: (a) B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1987, 109, 1469 CrossRef CAS; (b) B. M. Trost and I. Hachiya, J. Am. Chem. Soc., 1998, 120, 1104 CrossRef CAS.
- For selected examples regarding rhodium catalyzed allylic alkylation, see: (a) P. A. Evans, J. E. Robinson and J. D. Nelson, J. Am. Chem. Soc., 1999, 121, 6761 CrossRef CAS; (b) P. A. Evans and M. J. Lawler, J. Am. Chem. Soc., 2004, 126, 8642 CrossRef CAS; (c) P. A. Evans and E. A. Clizbe, J. Am. Chem. Soc., 2009, 131, 8722 CrossRef CAS.
- For selected examples regarding iron catalyzed allylic alkylation, see: (a) J. L. Roustan, J. Y. Mérour and F. Houlihan, Tetrahedron Lett., 1979, 20, 3721 CrossRef; (b) Y. Xu and B. Zhou, J. Org. Chem., 1987, 52, 974 CrossRef CAS; (c) B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 1469 CrossRef CAS; (d) B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 6053 CrossRef CAS; (e) B. Plietker, A. Dieskau, K. Möws and A. Jatsch, Angew. Chem., Int. Ed., 2008, 47, 198 CrossRef CAS; (f) M. Holzwarth, A. Dieskau, M. Tabassam and B. Plietker, Angew. Chem., Int. Ed., 2009, 48, 7251 CrossRef CAS; (g) M. Jegelka and B. Plietker, Org. Lett., 2009, 11, 3462 CrossRef CAS; (h) R. Trivedi and J. A. Tunge, Org. Lett., 2009, 11, 5650 CrossRef CAS; (i) D.-C. M. Jegelka and B. Plietker, Chem.–Eur. J., 2011, 17, 10417 CrossRef; (j) M. S. Holzwarth, W. Frey and B. Plietker, Chem. Commun., 2011, 47, 11113 RSC; (k) M. Jegelka and B. Plietker, ChemCatChem, 2012, 4, 329 CrossRef CAS; (l) A. P. Dieskau, M. S. Holzwarth and B. Plietker, Chem.–Eur. J., 2012, 18, 2423 CrossRef CAS.
- P. A. Evans and J. D. Nelson, J. Am. Chem. Soc., 1998, 120, 5581 CrossRef CAS.
- For recent reviews regarding iron catalyzed reactions, see: (a) A. Correa, O. García Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (b) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS; (c) W. M. Czaplik, M. Mayer, J. Cvengroš and A. J. von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS; (d) O. García Mancheño, Angew. Chem., Int. Ed., 2011, 50, 2216 Search PubMed; (e) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (f) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS; (g) M. J. Ingleson and R. A. Layfield, Chem. Commun., 2012, 48, 3579 RSC.
- (a) B. Plietker and A. Dieskau, Eur. J. Org. Chem., 2009, 775 CrossRef CAS; (b) D.-. S. Magens and B. Plietker, Chem.–Eur. J., 2011, 17, 8807 CrossRef; (c) A. P. Dieskau, M. S. Holzwarth and B. Plietker, J. Am. Chem. Soc., 2012, 134, 5048 CrossRef CAS.
- (a) B. J. Bulkina and J. A. Lynch, Inorg. Chem., 1968, 7, 2654 CrossRef; (b) F. Birencwaig, H. Shamai and Y. Shvo, Tetrahedron Lett., 1979, 20, 2947 CrossRef; (c) J. R. Miller, B. D. Podd and M. O. Sanchez, J. Chem. Soc., Dalton Trans., 1980, 1461 RSC.
- For selected reviews regarding amines as metal ligand in various transformations, see: (a) F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS; (b) J.-C. Kizirian, Chem. Rev., 2008, 108, 140 CrossRef CAS.
- B. M. Trost and C. A. Merlic, J. Am. Chem. Soc., 1990, 112, 9590 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical and spectroscopic data for synthetic compounds, copies of NMR spectra. See DOI: 10.1039/c2ra21646a |
| This journal is © The Royal Society of Chemistry 2012 |