Bioinspired and sustainable chitosan-based monoliths for antibody capture and release
Telma
Barroso
,
Ana C. A.
Roque
* and
Ana
Aguiar-Ricardo
*
REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal. E-mail: cecilia.roque@fct.unl.pt; air@fct.unl.pt
First published on 18th September 2012
Abstract
Chitosan-based monoliths activated by plasma technology induced the coupling of a robust biomimetic ligand, previously reported as an artificial Protein A, with high yields while minimizing the environmental impact of the procedure. Due to the high porosity, good mechanical and tunable physicochemical properties of the affinity chitosan-based monoliths, it is possible to achieve high binding capacities (150 ± 10 mg antibody per gram support), and to recover 90 ± 5% of the bound protein with 98% purity directly from cell-culture extracts. Therefore, the chitosan-based monoliths prepared by clean processes exhibit a remarkable performance for the one-step capture and recovery of pure antibodies or other biological molecules with biopharmaceutical relevance.
1. Introduction
Antibody-based biopharmaceuticals will be a major source of new therapies for at least the next 10 years.1 The large number of antibody products in development certainly supports the case for optimized antibody manufacturing approaches. However, the large quantities in which some antibody products are required put considerable pressure on current manufacturing facilities.2,3 In particular, current purification processes are struggling to equal the high productivities already obtained in upstream antibody production. Affinity chromatography using Protein A from Staphylococcus aureus is the method of choice for antibody capture during the purification process.1 However, protein A resins utilized in chromatography are expensive adsorbents with low stability, often causing the leaching of the affinity ligand together with the antibody product.1,2,4 Thus, synthetic affinity ligands mimicking biological receptors have been developed in an attempt to overcome Protein A drawbacks.5 Ligands based on a triazine scaffold, in particular ligand 22/8 or artificial protein A, are known to be low cost and highly resistant to the harsh conditions employed during cleaning-in-place (CIP) and sterilization-in-place (SIP).5,6 Ligand 22/8 has been immobilized on agarose,5 cellulose membranes,7 and magnetic particles,8 and shown to selectively bind to human Immunoglobulin G (hIgG) when employed in chromatographic processes. With the need to treat large volumes of concentrated antibody solutions, fast processes with high selectivity are desired.9,10 Monoliths are porous materials cast in a single block and inserted into a chromatography housing. These porous supports have been successfully employed in the purification of biological species.11–14 As convective transport is prevalent when using monoliths, faster volumetric throughput rates are achieved thus increasing the process speed and productivity.9The aim of this work was to combine the robustness of synthetic affinity ligands, namely ligand 22/8, with monolith blocks prepared from biopolymeric materials, in particular chitosan (CHT). In addition, sustainable chemistry options have been adopted for the preparation and functionalization of the materials based on freeze-drying method15,16 and plasma technology,17,18 respectively. In particular, freeze drying processes work by freezing the casting solution and then reducing the surrounding pressure to allow the frozen water in the material to sublimate directly from the solid phase to the gas phase, resulting in attractive porous structures.19 On the other hand, plasma technology is a fast and solvent free technique which allows surface modification through the introduction of chemical species.17–20 Recently, plasma surface activation was combined for the first time with supercritical fluid technology to surface-graft stimuli-responsive hydrogels on differently shaped devices.21 Herein, plasma surface activation was extended to introduce radicals on monolith surfaces for further coupling with the affinity ligand. The materials produced have been characterized in terms of morphological, mechanical and physico-chemical properties, and have shown to recover antibodies directly from crude cell samples. Chitosan (CHT) has been selected due to its nontoxic, antimicrobial, biocompatible, and biodegradable properties, as well as its pH sensitive behavior.22 Owing to a high molecular weight, chitosan forms viscous solutions in dilute aqueous acetic acid that can be used to produce porous structures. The tensile strength of these structures can be improved through crosslinking with epichlorohydrin23 or glutaraldehyde,24 copolymerization with other monomers, or via blending. In this work chitosan was copolymerized (cryo-polymerization) with glycidyl methacrylate (GMA) at low temperatures in order to produce well defined porous monoliths with good tensile strength. Poly(vinyl alcohol) (PVA) was also blended with chitosan as an alternative strategy to improve the mechanical performance of chitosan monoliths. The processing of these renewable materials into 3D structures with tunable and controlled morphological and mechanical properties, large surface area and their subsequent functionalization with a low cost affinity ligand through green methodologies will offer a great contribution in materials design for bioseparation processes. This integrated process aims to replace traditional practices of monolith preparation and functionalization by new ones where the integrity of the support is maintained from its conception to its application even at industrial scale.
2. Results and discussion
2.1 Preparation and characterization of native chitosan-based monoliths
Supports for protein separation must ideally possess high hydrophilicity, chemical and mechanical resistance, a narrow pore size distribution, as well as enough reactive functional groups for ligand attachment.1,9 In this work, chitosan-based monoliths were prepared by a combination of freezing and lyophilization methods and further evaluated according to their morphological and mechanical properties. It is known that the macromolecular morphology of monoliths is influenced by the conditions employed during their preparation—in general, the freezing and lyophilization processes generate open pore microstructures with a high degree of interconnecting pores in the composite materials. Large pores and high interconnectivities contribute to high fluxes across the materials and faster bioseparation processes.15Native monoliths prepared only with chitosan (CHT_N) present regular and spherical pores (data not shown); the addition of PVA facilitates the formation of monoliths (CP_N) with large and semi-spherical pores (Fig. 1A), while the copolymerization with GMA (CG_N) generates even larger and more elongated pores (Fig. 1B). In order to look deeper into the porous network prepared, analysis by mercury intrusion porosimetry was performed. The average pore size diameter for CHT_N monoliths is 17 μm, while those of CP_N and CG_N monoliths are 53 ± 5 and 123 ± 5 μm, respectively (Table 1), which corroborate the conclusions taken from SEM image analysis. The porosity values are similar for all chitosan-based monoliths, varying between (70–75) ± 5%, which was expected due to the constant temperature conditions employed during monolith preparation. Typically, the pore size decreases with the decrease of freezing temperature.15,16 During the freezing process, water ice crystals are formed and separate from the solutes which will be confined to the interstitial regions between ice crystals. Large ice crystal formation is expected in less viscous casting solutions and higher freezing temperatures, and the opposite is also true.16 In this work, chitosan-based casting solutions were frozen at −80 °C, a temperature at which a low average pore size diameter—between 20 and 100 μm—is obtained.9,16 This average pore size diameter is within the range of interest for the purpose of the work. Concerning bioseparation applications, monoliths with large pores and high porosities are required to enable the fast permeation of cellular components and proteins present in crude samples.25 Therefore a delicate balance between pore size diameter, porosity and surface area is needed during monolith design. If monoliths have larger pores (≥150 μm) the surface area available for further functionalization decreases, leading to a low density of the affinity ligand.9,16,26 Also, a decrease of specific surface area with increasing average pore size diameter is expected. The monoliths inner surface area (total pore area) is within the range of values of similar 3D porous structures reported in the literature.25 The permeability and the compressive modulus of native chitosan-based monoliths are included in Table 1. The highest permeability value is registered to CG_N monolith, 390 L m−2 h−1 atm−1, followed by CP_N and CHT_N monoliths with permeability values of 294 and 142 L m−2 h−1 atm−1, respectively. These results indicate a clear concordance between the permeability of the materials and their morphological properties and also a good interconnectivity between the pores. The density of the dried and wet chitosan-based monoliths (Table 1), was calculated by measuring the weight and dimensions of the prepared materials. Dried native chitosan-based monoliths present similar values of density. Wet native monoliths also showed similar densities although 50 times greater than the dried ones due to their huge water uptake ability. The compressive mechanical properties were studied by uniaxial compression measurements also under dry and hydrated conditions. The compression modulus (kPa), given by the slope of stress–strain curves, translates the material stiffness (Table 1). In general, a high compression modulus indicates a stiffer material.27 Native CHT-based monoliths present lower compression modulus values when compared to the functionalized materials. In more detail, CP_N monoliths at different stages, wet or dry, always present higher values of compression modulus than CG ones. The hydrated CG_N monolith exhibits a huge swelling and consequently becomes fragile, losing the initial integrity. These results are consistent with the morphological and mechanical properties previously discussed—supports with lower average pore size diameter and porosity are more rigid and present higher compression modulus. It is also known that the compression modulus of porous structures prepared at higher freezing temperatures (e.g. −30 and −20 °C) are lower than those prepared at −80 °C, due to the smaller pore size attained at this temperature.25 Another factor that can influence the support stiffness is the water absorption ability, and the proof of it is the dramatic decrease of mechanical properties observed for monoliths in the hydrated sate.
 | ||
| Fig. 1 SEM images of chitosan (CHT) based-monoliths before and after functionalization with the ligand 22/8: (A) CP_N, native monolith prepared from chitosan and polyvinyl alcohol (CP); (B) CG_N, native monolith prepared from chitosan and glycidyl methacrylate (CG); (C) CP_22/8, CP monolith functionalized with ligand 22/8 and (D) CG_22/8, CG monolith functionalized with ligand 22/8. All the images have a magnification of 300 and the scale bar in white indicates 10 μm. | ||
| Monolith | Average pore size diameter a (μm) | Porosity a (%) | Surface Areaa (m2 g−1 monolith) | Permeability (L m−2 h−1 atm−1) | Density ×10−3 (g cm−3) | Compressive Modulus (kPa) | ||
|---|---|---|---|---|---|---|---|---|
| Dry | Wet | Dry | Wet | |||||
| a Determined for dried monoliths by mercury porosimetry analysis. b n.a. The mechanical assay was impossible to perform due to the high swelling of CG_N. | ||||||||
| CP_N | 53 ± 5 | 70 ± 5 | 1.3 ± 0.1 | 294 ± 15 | 4 ± 2 | 165 ± 10 | 0.41 ± 0.5 | 0.2 ± 5 |
| CG_N | 123 ± 5 | 75 ± 5 | 0.9 ± 0.1 | 390 ± 5 | 3 ± 2 | 177 ± 5 | 0.26 ± 0.5 | n.a.b |
| CP_22/8 | 45 ± 5 | 68 ± 5 | 2.3 ± 0.2 | 123 ± 15 | 3 ± 2 | 146 ± 5 | 14 ± 5 | 9 ± 4 |
| CG_22/8 | 85 ± 10 | 60 ± 10 | 1.5 ± 0.2 | 323 ± 10 | 5 ± 2 | 131 ± 10 | 8 ± 2 | 4 ± 2 |
2.2 Preparation and characterization of affinity chitosan-based monoliths
Several methodologies for affinity monoliths activation and ligand coupling have been described by diverse authors.9,24 Herein the functionalization of chitosan monoliths with a triazine-based ligand (ligand 22/8) followed two different strategies. The first one was based on the optimized procedure for agarose5 and was also applied to cellulose membranes7 due to the similarity in the chemical composition of these supports, namely the high content of free hydroxyl groups.Native chitosan-based monoliths were activated with epichlorohydrin in order to add epoxide functionality for subsequent amination. As this epoxyactivation procedure occurs through the reaction of epichlorohydrin with free OH groups, the density and availability of OH groups determine the extension of the epoxide functionality achieved. The epoxy activation yielded densities for CHT, CP and CG were 187 ± 51, 226 ± 72 and 364 ± 100 μmol g−1 of moist monolith, respectively. These epoxy densities are significantly higher than those reported in the literature for agarose (50 μmol of epoxy per gram of moist weight gel)5 and for cellulose membranes (130 ± 10 μmol of epoxy per gram of moist membrane). The second strategy consisted of the activation of native chitosan-based monoliths by argon (Ar) plasma treatment. Plasma treatment is a solvent free technique which allows the surface modification according to the gas and power applied.17 In this work, Ar plasma treatment introduced radicals onto the chitosan materials for subsequent reaction with 1,6-diaminohexane. The latter acts as a spacer arm3,5 between the surface of the epoxy or plasma activated chitosan-based monoliths and the small biomimetic ligand 22/8, contributing to an optimal interaction between the ligand and the target protein (Scheme 1).
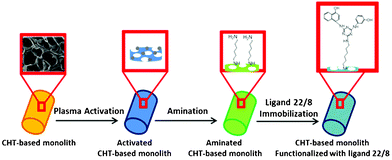 | ||
| Scheme 1 Schematic showing the functionalization strategy of chitosan-based monoliths with ligand 22/8 using non-thermal plasma surface activation. | ||
The amination values achieved were significantly higher for all chitosan-based scaffolds activated by plasma treatment (Table 2). The maximum amount of amines introduced using epoxy activated chitosan-based monoliths was 364 ± 50 μmol g−1 support. In marked contrast, using the new approach based on plasma surface activation this value increased almost 5-fold (1747 ± 47 μmol g−1 support). Due to these remarkable yields of amination, chitosan-based monoliths activated by plasma treatment were subsequently coupled with ligand 22/8. This ligand was prepared following the procedure described by Lowe and colleagues5 and was immobilized onto the aminated chitosan-based monoliths still containing a labile chloride on the triazine ring for nucleophilic substitution. Different yields of ligand immobilization were obtained (Table 2), according to the monolith materials. Although the highest value of ligand immobilization was obtained for the CP monolith, 910 ± 17 μmol g−1 support, CG monolith also presented a very high functionalization ratio, 677 ± 24 μmol g−1 support. The different ligand densities on CP and CG monoliths can be related with the surface area available in each support. CP monoliths have a smaller pore size diameter and porosity than CG monoliths suggesting that CP supports have higher surface area available for further functionalization. These facts contributed to a decrease of possible active sites in CG monoliths and consequently to lower ligand density compared to the CP ones. In this work, the density of ligand 22/8 immobilized in chitosan-based monoliths was 10-fold the value reported for ligand 22/8 immobilized in agarose beads,5,6 70-fold on cellulose membranes,7 and less than 2-fold on magnetic nanoparticles.8 It can also be noted that, in general, the density of immobilized ligand 22/8 is quite high when compared to the values obtained for immobilized natural ligands.23,28 To the best of our knowledge, this is the first report on the preparation of affinity monoliths with such a high ligand density. After ligand 22/8 immobilization, the morphological and mechanical properties of chitosan-based monoliths were evaluated. SEM images represented in Fig. 1C and D show that the porous network and architecture did not change significantly after the functionalization procedure proving the potential of the new strategy developed in this work. The average pore size diameter, porosity and surface area values obtained for affinity CP_22/8 and CG_22/8 monoliths registered in Table 1 were kept approximately within the combined experimental errors. Permeability values obtained for CP and CG monoliths decreased from 294 to 123 L m−2 h−1 atm−1 for CP_22/8, and from 390 to 323 L m−2 h−1 atm−1 for CG_22/8 monoliths. The flow reduction can be explained by the presence of the affinity ligand which confers a hydrophobic character to the supports. However, it should be noted that the permeability still shows the target values for separation processes whilst allowing enough residence time for the contact between the protein and the support.3 The apparent density of dried affinity monolithic supports did not change compared to the native ones at the same conditions, while wet monoliths showed a slight increase.
| Monolith | Epoxy activation | Plasma activation | Plasma activation |
|---|---|---|---|
| [NH2] (μmol g−1 support) | [NH2] (μmol g−1 support) | Ligand 22/8 immobilization (μmol g−1 support) | |
| CP | 226 ± 72 | 1747 ± 47 | 910 ± 17 |
| CG | 364 ± 100 | 906 ± 246 | 667 ± 24 |
After functionalization, CP_22/8 and CG_22/8 registered a decrease in density values which can be also explained by the hydrophobic nature of ligand 22/8 leading to a reduction in water uptake capability. The compressive modulus increased (∼10-fold) for all monoliths after ligand coupling (refer to Table 1). The improvement of the monolith mechanical properties whether in dry or wet conditions was expected as the incorporation of a synthetic ligand confers rigidity and robustness to the monoliths. Therefore, the sustainable functionalization strategy of the monoliths based on plasma activation and further ligand 22/8 coupling is able to keep most morphological properties of the native supports while improving their mechanical properties.
In order to evaluate the stability of the affinity monoliths at typical operating conditions used during purification processes, monoliths were incubated at several pH solutions (from pH 1 to 12), corresponding to those employed in equilibration, elution, regeneration and cleaning-in-place of chromatographic matrices. The amount of ligand released from the support was monitored (Fig. 2). The maximum amount of ligand 22/8 released from CP_22/8 and CG_22/8 was 12% and 7%, after 4 and 2 h of incubation, respectively (Fig. 2A and C). In particular, at the pH conditions used in the binding and elution of antibodies from ligand 22/8 supports, pH 3 and 8 respectively, the greatest amount of ligand leaching was observed after 2 and 4 h, for CP_22/8 and CG_22/8. In general, the maximum time of monolith exposure to these conditions during a typical purification run is 30 min, far below the threshold time for ligand leaching, which was only observed after 2 h of exposure. On the other hand, the highest loss of ligand from CP_22/8 and CG_22/8 occurs with sodium hydroxide solutions after 2 h (Fig. 2B and D). Nevertheless the maximum contact time of each functionalized monolith at these harsh conditions (CIP conditions) is about 10–20 min, the time at which no ligand release was registered.
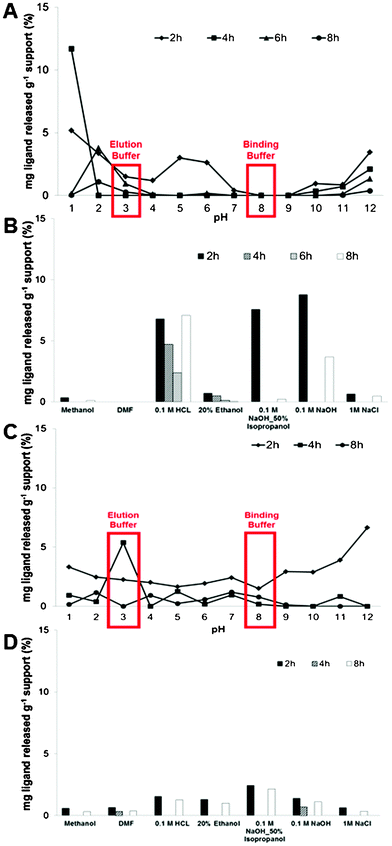 | ||
| Fig. 2 Stability evaluation of CP_22/8 (A and B) and CG_22/8 (C and D) monoliths immersed over 12 h in solutions typically used during cleaning-in-place (CIP) procedures, including solutions with pH values between 1 and 12. All data was obtained from duplicated measurements with errors of ±5. | ||
2.3 Evaluation of affinity monoliths for antibody purification
To evaluate the affinity constants for hIgG and BSA on native and functionalized chitosan-based monoliths, static partition equilibrium measurements were performed in duplicate. To attain the equilibrium between proteins and affinity supports,10 partition equilibrium studies were performed in a batch system over 12 h, varying the protein concentration.The experimental data was fitted using a Langmuir–Freundlich isotherm that is the indicated model for porous structures with heterogeneous morphology. Different affinity constants (Ka), theoretical maximum capacity (Qmax) and Langmuir–Freundlich coefficients (n) were obtained (Fig. 3) with an error of ±0.5, ±40.0 and ±0.1, respectively. The affinity monoliths always presented higher values of Ka and Qmax (14 and 3-fold, respectively) when compared to the native ones. The CG_22/8 monolith registered the highest value of Ka and Qmax, 4.0 × 104 M−1 and 590 mg hIgG g−1 support, respectively. Calculated Ka values exhibit the same order of magnitude usually obtained for similar affinity devices fitted with Langmuir–Freundlich isotherm.27,28,30,31 In addition, Ka values obtained for CP_22/8 and CG_22/8 are an order of magnitude lower than the Ka values reported for agarose,5 magnetic nanoparticles8 and cellulose membranes7 functionalized with ligand 22/8 and fitted with a Langmuir isotherm model. It should be remarked that values of Ka in a range between 104 and 106 M−1 indicate a medium affinity interaction, considered optimal for bioseparation processes while facilitating protein recovery. The values of Qmax for CP_22/8 and CG_22/8, 475 and 590 mg hIgG g−1 support respectively, are 2-fold those for agarose5 and magnetic particles8 functionalized with ligand 22/8, and very close to the one obtained for cellulose membrane.7 Values of Langmuir–Freundlich coefficients higher than 1 (n > 1) were registered for all functionalized monoliths suggesting a positive cooperativity in binding (attractive forces due to lateral interactions) and a heterogeneous nature of protein adsorption.
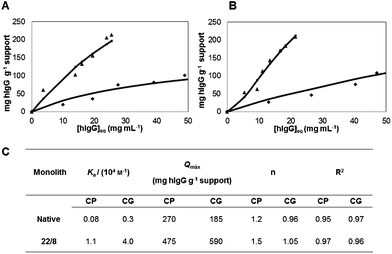 | ||
| Fig. 3 Langmuir–Freundlich adsorption isotherms for (A) CP and (B) CG monoliths: (◆) native and (▲) functionalized. (C) summarizes the estimated parameters of the Langmuir–Freundlich isotherms and standard errors for CP and CG monoliths before and after functionalization with ligand 22/8. | ||
The cooperativity depends on the macromolecular nature and multiple functional groups, which usually results in multiple interactions.27,28 It should be noted that the same adsorption assays were performed with bovine serum albumin (BSA), a model contaminant protein of antibodies in serum and cell culture supernatants and none of the tested isotherms could fit the experimental data. This result indicates a nonspecific adsorption of BSA.
The dynamic binding capacity of a stationary phase is one of the most critical factors to evaluate its chromatographic performance. To assess the mass transfer and dynamic binding properties of the affinity monoliths, breakthrough curves of purified hIgG were measured in duplicates for two flow rates (Fig. 4), as the binding capacity and elution efficiency of supports are affected by the residence time on the monolithic chromatographic column.
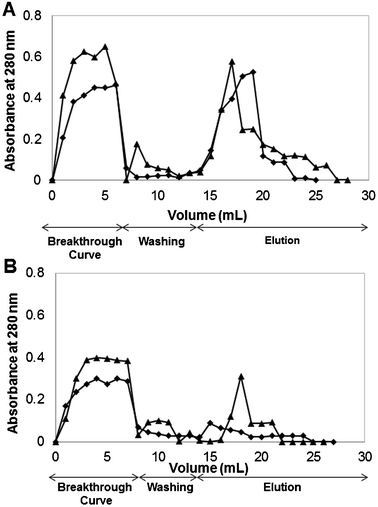 | ||
| Fig. 4 Breakthrough profiles for human IgG upon (A) CP_22/8 and (B) CG_22/8 monoliths at different flow rates: (◆) 1 mL min−1 and (▲) 2 mL min−1. | ||
Both supports, CP_22/8 and CG_22/8, duplicated the binding and elution capacities when the flow rate decreased from 2 to 1 mL min−1 (Fig. 4). The estimated binding capacities for CP_22/8 and CG_22/8 were 57 ± 10 and 15 ± 5 mg hIgG g−1 support respectively, when using a flow rate of 2 mL min−1 and 110 ± 15 and 26 ± 8 mg hIgG g−1 support for a flow rate of 1 mL min−1, respectively. The elution capacity for CP_22/8 and CG_22/8 increased from 40 to 90 ± 5% and from 30 to 60 ± 5% when the flow rate decreased from 2 to 1 mL min−1.
2.4 Optimization of an affinity monolith for antibody recovery
Due to the promising morphological, mechanical and physico-chemical properties, high stability at different pH conditions and high static and dynamic binding capacities towards hIgG, CP monoliths were selected for the optimization of IgG purification process. In order to evaluate and enhance the efficiency of affinity CP monoliths for antibody capture and recovery, three consecutive chromatographic cycles and a fourth one after monolith autoclaving (After_AC) were performed in duplicates at two different flow rates, 2 and 1 mL min−1. BSA, the model contaminant, was used as a control in the same experiments in order to evaluate the selectivity and affinity of CP_N and CP_22/8 towards hIgG. CP monoliths exhibited different performances on capturing and eluting hIgG and BSA (Fig. 5 and Fig. 6).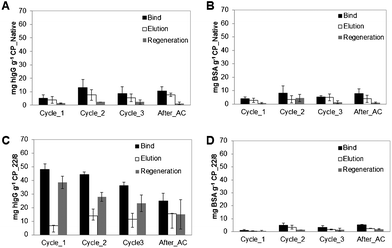 | ||
| Fig. 5 Evaluation of chromatographic performance for (A, B) native and (C, D) functionalized CP monoliths using pure IgG and BSA solutions, respectively. The chromatographic procedures (bind, elution and regeneration steps) were performed consecutively along four cycles at a flow rate of 2 mL min−1. The last cycle was performed after autoclaving (After_AC). | ||
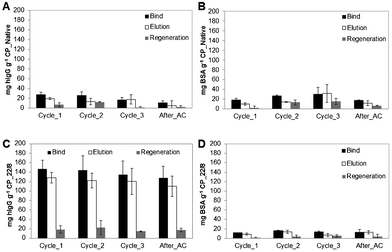 | ||
| Fig. 6 Evaluation of chromatographic performance for (A, B) native and (C, D) functionalized CP monoliths using pure IgG and BSA solutions, respectively. The chromatographic procedures (bind, elution and regeneration steps) were performed consecutively along four cycles at a flow rate of 1 mL min−1. The last cycle was performed after autoclaving (After_AC). | ||
In detail, Fig. 5A and B show that CP_N has approximately the same low capacity to capture and elute hIgG and BSA over the four cycles, suggesting residual non-specific interactions between the support and the proteins. In marked contrast, the functionalized CP_22/8 monoliths showed a substantial increase in binding and elution capacity towards hIgG (Fig. 5C) while only a negligible amount of BSA was retained (Fig. 5 D). Although presenting affinity and selectivity to hIgG molecule, the affinity monoliths lost their capacity for re-utilization, especially after autoclaving which is translated by a slight decrease in the bind and elution values. At these operational conditions, CP_22/8 loses around 20% of capacity after 3 chromatographic cycles, and 50% after autoclaving (Fig. 5C).
On the other hand, only 25% of the captured hIgG was recovered in the elution step, whereas 75% was washed during the regeneration step under extremely harsh conditions deleterious for the protein. Subsequent assays, performed at a lower flow rate (1 mL min−1), allowed an increase of the residence time and the achievement of affinity equilibrium between the attached ligand and the receptor in the solution (Fig. 6). Comparing the chromatographic runs showed in Fig. 5C and 6C, it is clear that CP_22/8 exhibited a great improvement in the binding and elution of hIgG as only reducing the flow rate to a half led to an over 3-fold increase of captured and eluted hIgG, 150 ± 10 and 135 ± 5 mg g−1 support, respectively. Consequently, the amount of hIgG recovered during the regeneration step was significantly reduced, 10 ± 5 mg g−1 support. The bind and elution capacity of the monoliths was maintained with a slight decrease after autoclaving (a decrease of 15%). This remarkable performance of monoliths for recovering IgG using the flow rate of 1 mL min−1 led to results that depart considerably from the reported outcomes obtained with affinity membranes, particles and other monoliths used in antibody purification.5,7,8,26,29
The most critical part of the study was to evaluate the direct capture of monoclonal antibodies (Mabs) from a non-clarified homogenate. Fig. 7 presents the chromatogram of non-clarified crude extract of Mabs using CP_N and CP_22/8 monoliths (A) as well as the SDS-PAGE gel,32 revealed using a silver staining kit from BIO-RAD, obtained from the recovered samples during the chromatographic experiment (B). The chromatogram obtained from crude extract sample shows that CP_N did not capture any protein (Fig. 7A) while the affinity monolith CP_22/8 captured 61 ± 10% of Mabs from a mammalian crude extract with initially 1.3 mg of total protein per millilitre and eluted 80 ± 8% of bounded protein. When considering the purity of the samples, the flow-through (3) and the wash (4) contained contaminant proteins and antibody, while the elution fractions (5,6) showed a high purity of the antibody (estimated as 98% by ImageJ analysis). These results confirm the efficacy of CP_22/8 in capturing and eluting Mabs with great purity from a real crude extract.
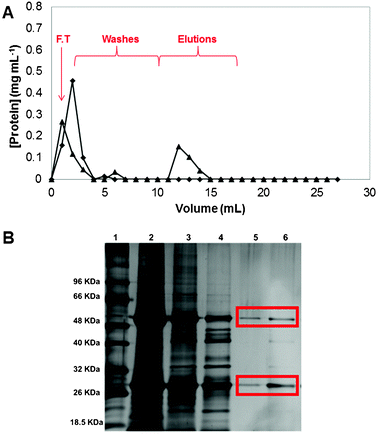 | ||
| Fig. 7 Chromatogram of Mabs purification from crude extract using (A) CP monoliths: (◆) native and (▲) functionalized, at a flow rate of 1 mL min−1. The fractions collected included the flowthrough (F.T.) followed by the washing and elution steps. The acrylamide gel from SDS-PAGE performed with the fractions collected during the Mabs purification (B): lane 1 corresponds to the molecular weight marker, lane 2 represents the loading, lane 3 is the flowthrough, lane 4 corresponds to the first wash (phosphate buffer (50 mm, pH 8.0)), and lane 5 and lane 6 are the first and second elution fractions (sodium citrate buffer (50 mm, pH 3.0)). | ||
3. Conclusions
Herein we report for the first time the preparation of chitosan-based monoliths by combining a freeze drying process with surface activation by plasma treatment to modify them with a robust biomimetic affinity ligand, previously reported as an artificial Protein A for antibody purification. It was demonstrated that the morphological and mechanical properties of monoliths can be tuned according to the polymer blend composition, while the new strategy to couple the affinity ligand was able to improve the chemical and physical stability towards the final application. The functionalization strategy based on plasma activation proved to be safer and more efficient than traditional activation procedures which involve multiple steps and innumerous organic solvents. The main practical advantages of the strategy described are that by fast and solventless plasma activation of different substrates, a direct procedure of functionalization could be applied leading to robust affinity monoliths able to selectively capture and elute antibodies even from homogenate crude extracts. Preliminary results of monoclonal antibody (Mabs) capture from a non-clarified homogenate suggested that CP_22/8 is able to recover Mabs with 98% purity.A straightforward extension of the materials and functionalization approach based on plasma technology herein described can be envisaged for the production of polymeric devices with different geometries and properties, and for the conjugation of a wide range of biological and artificial receptors. The properties of affinity polymeric devices can be tuned towards different applications in addition to the example here presented. For instance, they can find applications on the pre-concentration and solid-phase extraction of compounds, on organo- and bio-catalysis processes, on analytical and biosensing systems, on tissue engineering and as supports for cellular growth and expansion.
4. Experimental
4.1 Materials
Ammonium peroxodisulphate (APS, purity ≥98%), citric acid (purity ≥99%), disodium hydrogen phosphate monodibasic (pro analysis), disodium hydrogen phosphate dibasic (pro analysis), disodium tetraborate, ethanol absolute and sodium citrate dihydrate were purchased from Merck. Isopropanol and sodium bicarbonate were purchased from Riedel-de-Haën. Acetone (purity ≥99%), and ethyl acetate were supplied by Roth. Acetic acid (purity ≥99%), Aminocaproic acid, 3-aminophenol, 4-amino-1-naphthol hydrochloride, cyanuric chloride (purity ≥98%), 1,6-hexanodiamine (purity ≥98%), N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), glycine, ninhydrin, potassium cyanide, pyridine, sodium hydroxide (purity ≥99%) were purchased from Sigma Aldrich. Chitosan (75∼85% deacetylated, medium molecular weight), poly(vinyl alcohol) (purity ≈ 99%), N,N-methylenebisacrylamide (MBAm, purity ≥85%), N,N,N′,N′-tetramethylethylenediamine (TEMED, purity ≈ 99%), bicinchoninic acid (BCA) kit, bovine serum albumin (BSA) (purity ≥98%) were supplied by Sigma Aldrich. Human IgG was purchased by Gammanorm (purity ≥99%).4.2 Monoliths preparation
Chitosan-based monoliths were prepared blending different ratios of chitosan (0–100% (wt per wt)) with PVA (0–50%) and GMA (10%) in acetic acid aqueous solution (1% v per v). The crosslinker agent, MBA, was also added (2% (wt per wt)) and different casting solutions were placed in glass tubes and stirred until homogeneous casting solutions were obtained. After the initiator and catalyst, TEMED (23 μL) and APS (40 μL), were added to promote the crosslinking process. The crosslinking process occurred at 0 °C during 30 min under stirring. The casting solutions were frozen at −80 °C during 12 h and were lyophilized (Telstar cryodos-50) until dry.25,264.3 Monoliths characterization
Chitosan-based monoliths morphology was investigated using scanning electron microscopy (SEM) in Hitachi S 2400 equipment with an accelerating voltage set to 15 kV. The samples were frozen and fractured in liquid nitrogen for cross-sectional analysis and gold coated before analysis. Chitosan-based monoliths porosity, average pore size diameter and surface area were determined by mercury porosimetry (Micromeritics, autopore IV). Water fluxes were determined at 25 °C and by varying the applied hydrostatic pressure (within 0–0.4 MPa) using a stainless steel high-pressure cell (with an effective volume of 1.2 cm3). At least three measurements of distilled water flux were performed for each monolith. The permeability (Lp) of chitosan-based monoliths was obtained by the slope of linear relation between flux (F) and pressure (p), and is given by Darcy Law,7 represented by:| F = Lp × Δp | (1) |
The wet and dry densities were determined through the ratio of wet weight as well as dry weight of the chitosan-based monoliths with respect to their volume. The apparent density in g cm−3 was calculated by the equation below as described by Tripathi et al.14
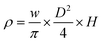
| (2) |
Uniaxial compression was used to determine the mechanical properties of the monoliths using tensile testing equipment (MINIMAT firmware v.3.1) at room temperature. Samples were prepared in a cylindrical shape (10 mm in diameter thickness). The length between clamps was set at 5 mm, the speed set to 0 1 mm min−1, a full scale load of 20 N and maximum extension of 90 mm was used. The compression modulus was calculated from the slope of the linear portion of the stress-strain curve:7
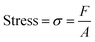 | (3) |
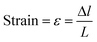 | (4) |
4.4 Monolith functionalization
The chitosan-based monoliths followed two different activation strategies for ligand 22/8 immobilization. The first one involved the introduction of reactive epoxy groups on monoliths by epoxy activation. CP and CG monoliths were covered with distilled water (10 mL), NaOH (72 μL, 0.1 M) and epichlorohydrin (0.83 mL) and then incubated for 3 h at 35 °C on a rotary shaker (140 rpm). After incubation, the epoxy-activated chitosan-based monoliths were washed with distilled water (200 mL). The epoxy activation content was determined by adding sodium thiosulfate (3 mL, 1.3 M) to 1 gram of epoxy-activated chitosan-based monoliths and incubating them at room temperature for 20 min. This mixture was neutralized with HCl (0.1 M) and the amount of HCl was registered. The volume of HCl added corresponded to the amount of hydroxyl ions released. The second strategy (Scheme 1) involved the use of plasma treatment which was carried out in a radio frequency plasma reactor (Plasma system FEMTO, version 5). Native chitosan-based monoliths were introduced in a plasma chamber which was thoroughly purged with a continuous flow of argon to reduce trace amounts of air and moisture. During the treatment, the argon flow was adjusted in order to keep a constant pressure of 0.3 Torr inside the chamber. A power of 60 W was applied over 5 min. At the end of the experiment the plasma chamber was ventilated and the activated samples were immediately introduced in an aqueous solution of hexane-1,6-diamine to be aminated. Activated chitosan-based monoliths reacted with an excess of hexane-1,6-diamine in water for 24 h at 45 °C, and then thoroughly washed with water in order to remove residues of hexane-1,6-diamine which did not react. The extent of amination was determined using the Kaiser test. This test is a colorimetric assay to quantify free amine groups and is based on the reaction of ninhydrin with primary amines, which gives a characteristic dark blue color. To perform the Kaiser test, each following reagents i) 80% crystalline phenol in ethanol (w/v), ii) 2% aqueous solution of potassium cyanide (0.001 M) in pyridine (v/v) and iii) 5% ninhydrin in ethanol (w/v) were added (50 μl of each one) to the aminated chitosan-based monolith samples (1 mL). The samples were then placed in a water-bath at 100 °C during 5 min. The calibration curve was represented by standard solutions of glycine (0–5 μmol mL−1) and the absorbance measurements of the samples (diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18) were performed at 560 nm. For ligand 22/8 immobilization, an excess of ligand 22/8 (3 equiv. to the amination content, 530 μmol, 200 mg), was added to the aminated chitosan-based monolith samples (approximately 60 mg) in water–DMF (1:2). The volume of solvent used was sufficient to cover the chitosan-based monoliths, and the solution was incubated on a rotary shaker (140 rpm) for 72 h at 85 °C. The synthesis of ligand 22/8 followed the procedure described by Teng et al.5 and Barroso et.al.7 The synthesis of ligand 22/8 was confirmed by 1H and C-NMR and FT-IR. Functionalized chitosan-based monolith samples were then washed with water–DMF (1:2) until the absorbance at 270 nm was zero.
:18) were performed at 560 nm. For ligand 22/8 immobilization, an excess of ligand 22/8 (3 equiv. to the amination content, 530 μmol, 200 mg), was added to the aminated chitosan-based monolith samples (approximately 60 mg) in water–DMF (1:2). The volume of solvent used was sufficient to cover the chitosan-based monoliths, and the solution was incubated on a rotary shaker (140 rpm) for 72 h at 85 °C. The synthesis of ligand 22/8 followed the procedure described by Teng et al.5 and Barroso et.al.7 The synthesis of ligand 22/8 was confirmed by 1H and C-NMR and FT-IR. Functionalized chitosan-based monolith samples were then washed with water–DMF (1:2) until the absorbance at 270 nm was zero.
In order to assure a complete exclusion of non-absorbed ligands into and onto chitosan-based monoliths, functionalized samples were introduced in a Varian column (a reservoir with 3 mL of capacity) and were submitted to several washes with water–DMF (1:2) and to clean-in-place-procedure. The clean-in-place-procedure consisted of washing functionalized chitosan-based monoliths with NaOH (1 M) until the absorbance at 270 nm be zero, with water (10 mL), then with regeneration buffer (NaOH 0.1 M in 30% of isopropanol) until the absorbance at 270 nm be zero, and finally, again with distilled water (10 mL). The functionalized chitosan-based monoliths were equilibrated with sodium phosphate buffer (50 mM, pH 8.0) and sodium citrate buffer (50 mM, pH 3.0) until the absorbance at 270 nm be zero. The extinction coefficient of ligand 22/8 was determined at its maximum absorbance wavelength (ε270 = 10.72 L g−1 cm−1), respectively. The ligand 22/8 density was determined by subtracting the amount of ligand contained in all the washing liquors from the initial ligand used in the immobilization step. The chitosan-based monoliths containing ligand 22/8 were characterized in terms of morphological and mechanical properties as referred previously.
4.5 Static partition equilibrium experiments
Partition equilibrium experiments were performed in a batch system as described by Barroso et al.7 The adsorption of hIgG and BSA on the chitosan-based monoliths was investigated using a sample of native and functionalized chitosan-based (10 mg) monoliths varying the concentration of hIgG and BSA (0.0–45 mg mL−1, 400 μL) in phosphate buffer (50 mM, pH 8.0) solutions. All experiments were conducted in duplicates at 25 °C, at a stirring rate of 200 rpm during 12 h. At the end of this period, the native and functionalized chitosan-based monoliths were removed from the medium. The amount of adsorbed hIgG and BSA was determined by measuring the initial and final concentrations of hIgG within the adsorption medium. A calibration curve was prepared using hIgG and BSA in phosphate buffer solutions (50 mM, pH 8.0) (0.0–45 mg mL−1). The concentration of protein was measured at 280 nm on a microplate reader (Tecan Infinite F200, filter, λ = 280 nm). The adsorption phenomenon followed the Langmuir–Freundlich model27,28 and it was represented by: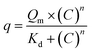 | (5) |
4.6 Frontal Analysis—breakthrough curves and binding capacity
The dynamic loading capacity of the packed columns with native and functionalized chitosan-based monoliths was determined using frontal analysis according with the equation33 below.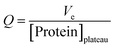 | (6) |
Where Q corresponds to the estimated adsorbent capacity and Ve corresponds to the elution volume.
This technique consisted in loading hIgG and BSA solutions (8 mL, 0.5 mg mL−1) in sodium phosphate buffer (50 mM, pH 8.0) through the equilibrated monolithic packed columns at different flow rates, 1 and 2 mL min−1 until the protein concentration of the output and input streams were identical. Then, packed monolithic columns were washed with phosphate buffer (50 mM, pH 8.0) and the bound protein was eluted with sodium citrate buffer (50 mM, pH 3.0). Each collected milliliter during loading, washing and elution steps were analyzed by absorbance at 280 nm, using a microplate reader, in order to estimate the amount of protein bounded and eluted.
4.7 Chromatographic experiments
In order to estimate the capacity of functionalized chitosan-based monoliths, Varian columns (with a capacity of 3 mL and an effective volume of 1.2 cm3) were packed with these polymeric affinity structures. The columns were connected to a peristaltic pump using rubber tubing in order to allow flow control. The affinity capturing experiments were performed in a step-wise adsorption–desorption process by switching eluents at room temperature and at atmospheric pressure. Packed columns with functionalized chitosan-based monoliths were loaded with 1 mL of hIgG and BSA solution (5 mg mL−1) in order to estimate the capacity of the columns for each protein. Then, columns were washed with sodium phosphate buffer (50 mM, pH 8.0) until the absorbance measured at 280 nm reached ≤0.005, in order to remove all the protein physically adsorbed. The bound protein was eluted and recovered with sodium citrate buffer (50 mM, pH 3.0). Packed columns with functionalized chitosan-based monoliths were then regenerated as described above in order to be reused. These chromatographic experiments were repeated during 3 cycles and one more after auto-claving in order to evaluate the column capacity over time. The total amount of IgG bound, eluted and regenerated from the chitosan-based monoliths was initially determined by absorbance measured at 280 nm and by the BCA (microplate reader assay). As the results were consistent (data not shown) only absorbance measured at 280 nm was further used for protein quantification.4.8 Purification of monoclonal antibodies from mammalian crude extracts
In order to evaluate the possibility to capture directly antibodies from non-clarified crude extracts, packed columns with native and functionalized CP monoliths were loaded with a mammalian crude extract solution (1 mL, 1.3 mg of total protein per milliliter). After crude extract loading, packed columns were washed with the sodium phosphate buffer (15 mL, 50 mM, pH 8.0) until the absorbance measured at 280 nm reached ≤0.005 and the bound protein was after eluted and recovered with sodium citrate buffer (50 mM, pH 3.0). All collected samples were analyzed by the BCA assay in order to quantify the amount of total protein bound and eluted from the solid support. The BCA assay consists in adding the BCA working reagent (200 μL) to the samples (25 μL). The microplates were incubated in the dark for 30 min at 37 °C. A standard curve was determined for each assay (200–1000 μg mL−1). SDS-PAGE was performed on acrylamide gel (12.5%) in Tris-Glycine buffer system pH 8.3. Electrophoresis apparatus (from BIO-RAD) was connected with power supply at 120 V, 190 mA for 1 h. The gel was revealed using a silver staining kit from BIO-RAD.Acknowledgements
The authors would like to thank the financial support from Fundação para a Ciência e Tecnologia, Portugal, through contracts MIT-Pt/BS-CTRM/0051/2008, PTDC/EBB-BIO/102163/2008, PTDC/EBB-BIO/098961/2008 and PTDC/EBB-BIO/118317/2010 and doctoral grant SFRH/BD/62475/2009 (T. B.), Bioengineering Systems Focus Area, Fundação Calouste Gulbenkian, FEDER and FSE. We wish to thank the Analytical Services Laboratory of REQUIMTE for the characterization of materials. Authors are also thankful to Lonza Biologics, UK (Dr Richard Alldread) and the Animal Cell Technology Unit of ITQB-UNL/IBET (Dr Paula M Alves and Dr Ana Teixeira) for providing the cell culture bulks of antibodies.References
- (a) D. Low, R. O'Leary and N. S. Pujar, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 848, 48 CrossRef CAS; (b) M. Fuentes, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2006, 7, 540 CrossRef CAS.
- (a) H. Alkan, N. Bereli, Z. Baysal and A. Denizli, Biochem. Eng. J., 2009, 45, 201 CrossRef CAS; (b) Y. Goto, R. Matsuno, T. Konno, M. Takai and K. Ishihara, Biomacromolecules, 2008, 9, 828 CrossRef CAS.
- K. Mondal and M. N. Gupta, Biomol. Eng., 2006, 23, 59 CrossRef CAS.
- A. Jungbauer and R. Hahn, J. Chromatogr., A, 2008, 1184, 62 CrossRef CAS.
- S. F. Teng, K. Sproule, A. Husain and C. R. Lowe, J. Chromatogr., Biomed. Appl., 2000, 740, 1 CrossRef CAS.
- S. F. Teng, K. Sproule, A. Husain and C. R. Lowe, J. Mol. Recognit., 1999, 12, 67 CrossRef CAS.
- T. Barroso, M. Temtem, A. Husain, A. Aguiar-Ricardo and A. C. A. Roque, J. Membr. Sci., 2010, 348, 224 CrossRef CAS.
- I. L. Batalha, A. Hussain and A. C. A. Roque, J. Mol. Recognit., 2010, 23, 462 CrossRef CAS.
- (a) R. Mallik and D. S. Hage, J. Sep. Sci., 2006, 29, 1686 CrossRef CAS; (b) M. J. Hortigüela, I. Aranaz, M. C. M. Gutieürrez and L. Del Monte, Biomacromolecules, 2011, 12, 179 CrossRef.
- G. Bayramoglu, A. U. Senel and M. Y. Arica, Sep. Purif. Technol., 2006, 50, 229 CrossRef CAS.
- A. Jungbauer and R. Hahn, J. Chromatogr., A, 2008, 1184, 62 CrossRef CAS.
- J. L. Williams, Catal. Today, 2001, 69, 3 CrossRef CAS.
- C. Peskoller, R. Niessner and M. Seidel, J. Chromatogr., A, 2009, 1216, 3794 CrossRef CAS.
- (a) F. M. Plieva, H. Kirsebom and B. Mattiasson, J. Sep. Sci., 2011, 34, 2164 CAS; (b) A. Tripathi and A. Kumar, Macromol. Biosci., 2011, 11, 22 CrossRef CAS.
- T. Nge, M. Nogi, H. Yano and J. Sugiyama, Cellulose, 2010, 17, 349 CrossRef CAS.
- S. Deville, Materials, 2010, 3, 1913 CrossRef CAS.
- (a) T. Desmet, R. Morent, N. Geyter, C. Leys, E. Schacht and P. Dubruel, Biomacromolecules, 2009, 10, 2351 CrossRef CAS; (b) J. Cho, F. S. Denes and R. B. Timmons, Chem. Mater., 2006, 18, 2989 CrossRef CAS.
- M. Schulze, A. Yanguas-Gil, A. Von Keudell and P. Awakowicz, J. Phys. D: Appl. Phys., 2008, 41, 065206 CrossRef.
- F. J. ÓBrien, B. A. Harley, Y. V. Yannas and L. Gibson, Biomaterials, 2006, 25, 1077 CrossRef.
- J. J. A. Barry, M. M. C. G. Silva, K. M. Shakesheff, S. M. Howdle and M. R. Alexander, Adv. Funct. Mater., 2005, 15, 1134 CrossRef CAS.
- T. Barroso, R. Viveiros, M. Temtem, T. Casimiro, A. M. Botelho do Rego and A. Aguiar-Ricardo, ACS Macro Lett., 2012, 1, 356 CrossRef CAS.
- (a) S.-W. Choi, J. Xie and Y. Xia, Adv. Mater., 2009, 21, 2997 CrossRef CAS; (b) P. Sabrina, K. Tommeras, K. M. Varum and K. Ostgaard, Biomacromolecules, 2001, 2, 1313 Search PubMed; (c) Y. Cheng, X. Luo, G. F. Payne and G. W. Rubloff, J. Mater. Chem., 2012, 22, 7659 RSC.
- Y. C. Wei, S. M. Hudson, J. M. Mayer and D. L. Kaplan, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2187 CrossRef CAS.
- (a) M. Temtem, T. Barroso, T. Casimiro, J. F. Mano and A. Aguiar-Ricardo, J. Supercrit. Fluids, 2012, 66, 398 CrossRef CAS; (b) M. Temtem, L. M. C. Silva, P. Z. Andrade, F. dos Santos, C. L. da Silva, J. M. S. Cabral, M. M. Abecasis and A. Aguiar-Ricardo, J. Supercrit. Fluids, 2009, 48, 269 CrossRef CAS.
- (a) P. Arvidsson, F. M. Plieve, V. I. Lozinsky, I. Y. Galaev and B. Mattiasson, J. Chromatogr., A, 2003, 986, 275 CrossRef CAS; (b) N. Kathuria, A. Tripathi, K. K. Kar and A. Kumar, Acta Biomater., 2009, 5, 406 CrossRef CAS; (c) A. Wang, Q. Ao, W. Cao, M. Yu, Q. He, L. Kong, L. Zhang, Y. Gong and X. Zhang, J. Biomed. Mater. Res., Part A, 2006, 1, 36 CrossRef.
- (a) D. Josic and A. Buchacher, J. Biochem. Biophys. Methods, 2011, 49, 153 CrossRef; (b) A. Bronshtein, N. Aharonson, A. Turnianski and M. Altstein, Chem. Mater., 2000, 12, 2050 CrossRef CAS.
- C-F. Dai, C.-J. Weng, P.-R. Li and J.-M. Yeh, Polym. Degrad. Stab., 2010, 95, 600 CrossRef CAS.
- C. Boi, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 848, 19 CrossRef CAS.
- L. C. L.Aquino, E. A. Miranda, I. S. Duarte, P. T. V. Rosa and S. M. A. Bueno, Braz. J. Chem. Eng., 2003, 20, 3 Search PubMed.
- R. Wongchuphan, B. T. Tey, W. S. Tan, F. S. Taip, S. M. M. Kamal and T. C. Ling, Biochem. Eng. J., 2009, 45, 232 CrossRef CAS.
- L. G. Berruex, R. Freitag and T. B. Tennikova, J. Pharm. Biomed. Anal., 2000, 24, 95 CrossRef CAS.
- (a) R. J. Carreira, F. M. Cordeiro, A. J. Moro, M. G. Rivas, R. Rial-Otero, E. M. Gaspar, I. Moura and J. L. Capelo, J. Chromatogr., A, 2007, 1153, 291 CrossRef CAS; (b) V. Neuhoff, N. Arold, D. Taube and W. Ehrhardt, Electrophoresis, 1988, 9, 255–262 CrossRef CAS.
- D. J. Winzor, J. Biochem. Biophys. Methods, 2001, 49, 99 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |