Improved synthesis of 14-hydroxy opioid pharmaceuticals and intermediates†
Gaik B.
Kok
and
Peter J.
Scammells
*
Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade Parkville, 3052, Victoria, Australia. E-mail: peter.scammells@monash.edu; Fax: (+61) 3 9903 9582; Tel: (+61) 3 9903 9542
First published on 25th September 2012
Abstract
Significantly improved reaction conditions for the synthesis of oxycodone (1), via the conventional two-step process involving oxidation of thebaine (3) into 14-hydroxycodeinone (5) and the subsequent reduction of 5via catalytic hydrogenation, are reported. Employing the hydrochloride salt of thebaine (3) in the oxidation step, in place of the more traditionally used free base form of this opiate, now provides 5 in high yield and purity. For the reduction step, aqueous acetic acid has typically been employed as the solvent. However, this was found to generate varying amounts of 14-hydroxydihydrocodeine (16) as the dominant by-product. Instead, using 5% Pd/BaSO4 as the catalyst and MeOH as the solvent completely eliminated the formation of 16, giving oxycodone (1) in high yield and purity, without the need to purify the intermediates. These improved conditions have also proved effective in the synthesis of other 14-hydroxyopiates, such as oxymorphone (2) and N-noroxymorphone (9). In the latter case, a high overall yield was achieved by starting from N-nororipavine (10), without the need to employ protecting groups.
Introduction
Oxycodone (14β-hydroxydihydrocodeinone, 1) and oxymorphone (14β-hydroxydihydromorphinone, 2) (Fig. 1) are well-known analgesics, with potencies many times higher than that of morphine itself.1,2 The most direct route for the synthesis of these 14-hydroxyopiates comprises two steps starting from thebaine (3) or oripavine (4), respectively. Firstly, the Δ6, Δ8-morphinane is oxidized, followed by the reduction of the resultant α,β-unsaturated ketone 5 or 6 (Scheme 1).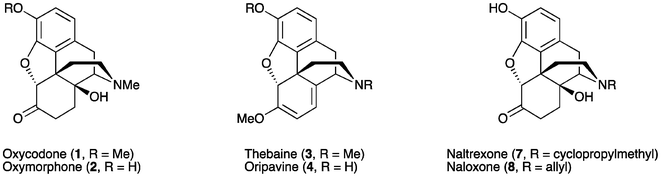 | ||
| Fig. 1 Examples of 14-hydroxymorphinane-based pharmaceuticals and precursors. | ||
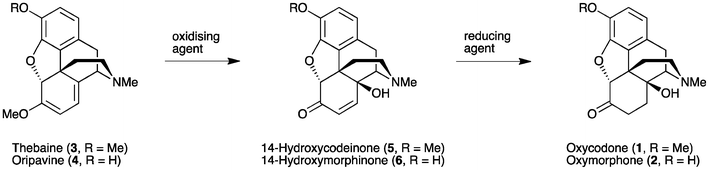 | ||
| Scheme 1 General approach for the synthesis of 14-hydroxy opioids. | ||
This method has been used routinely on an industrial scale for the production of oxycodone (1).3 Whilst the chemistry, as summarised in Scheme 1, is well established,4,5 this process continues to present significant challenges due to the range of side-products formed, particularly on the large scale. Consequently, strategies to improve the yield and reduce impurities are still desirable. In addition, when oripavine (4) is used as the starting material rather than thebaine (3), the reduction step (Scheme 1) has been reported to result in significant by-products that cannot be easily isolated or removed.6 As such, the low yield in transforming oripavine (4) to oxymorphone (2) has rendered this synthetic route commercially unviable.
Oxycodone (1) and oxymorphone (2) are also key intermediates for the synthesis of narcotic antagonists such as naltrexone (7) and naloxone (8).7 For these ‘nal’ compounds, additional steps involving N-demethylation and realkylation are required in the synthesis. Hence, N-noroxymorphone (9), the N-demethylated derivative of 2, is a centrally important active pharmaceutical intermediate (API) to N-alkylated 14-hydroxyopiates such as 7 and 8.8
The synthetic route depicted in Scheme 1 has also been used for the synthesis of oxymorphone (2) and N-noroxymorphone (9) from oxycodone (1) and N-northebaine, respectively.9 This necessitates O-demethylation of oxycodone/N-noroxycodone using reagents such as BBr3, MeSO3H/methionine or HBr, and only proceeds in modest yields.9,10 However, a practical and efficient synthesis of 2 and 9, especially from oripavine (4) and N-nororipavine (10), would be particularly advantageous since it would eliminate the harsh reaction conditions associated with O-demethylation.
The synthesis of N-noroxymorphone (9) from N-nororipavine (10) was first reported by Sipos and coworkers (Scheme 2).11 Thebaine (3) was N-demethylated using diethylazodicarboxylate (DEAD) and O-demethylated using L-selectride to afford 10 in a 43% yield. The subsequent conversion of 10 to 9 followed the method outlined in Scheme 2. However, prior benzyl protection of the 3-phenol and secondary amine of N-nororipavine (10) was necessary, giving 3,17-dibenzyloripavine (11) in a 71% yield. Oxidation of 11 was performed using HCOOH/H2O2 to afford 3,17-dibenzylnoroxymorphinone (12) in a 60% yield. Finally, catalytic hydrogenation of 12 over Pd/C gave 9 in an 84% yield, resulting in an overall yield of around 15% when starting from 3.
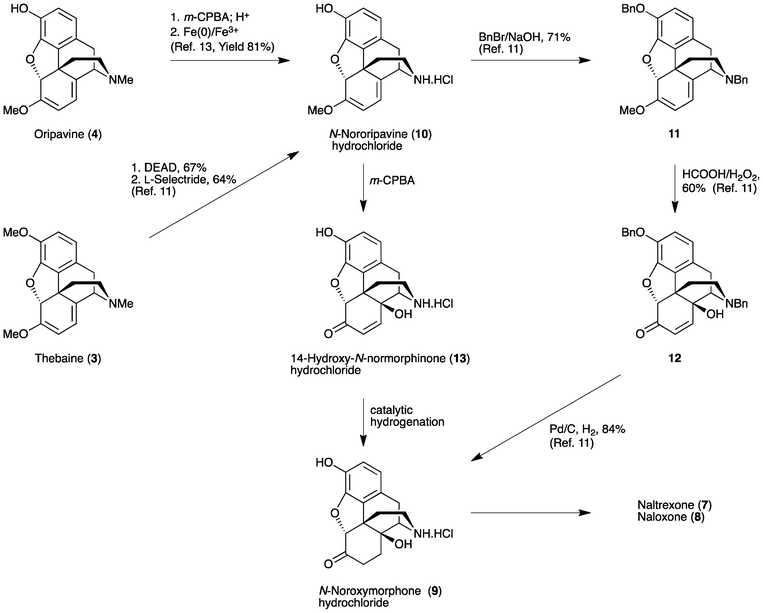 | ||
| Scheme 2 Synthetic approaches to naltrexone (7) and naloxone (8). | ||
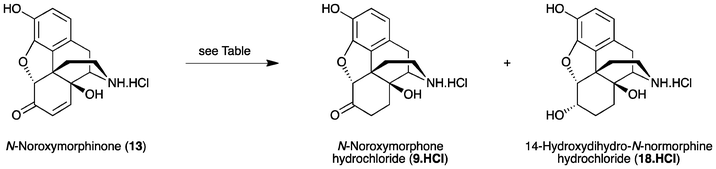
Recently, we described the first direct synthesis of N-nororipavine hydrochloride (10·HCl) from oripavine (4).12,13 Hence, by using Fe(0),12 or more effectively a combination of Fe(0) and Fe(III),13 oripavine (via the hydrochloride salt of the corresponding N-oxide) was N-demethylated under Polonovski-type conditions affording 10 in an 81% yield. The need for a facile and efficient synthesis of N-noroxymorphone (9), without the requirement for protecting group chemistry, prompted us to re-investigate the direct synthesis of this API from 10 using the sequence shown in Scheme 1. Since there is also a need to improve the synthesis of oxycodone (1) and oxymorphone (2), we have investigated in some detail the reaction conditions for the synthesis of these compounds, using the methodology outlined in Scheme 1. Improved reaction conditions with reduced side-product formation would be particularly beneficial for the synthesis of the intermediate 14-hydroxy-N-normorphinone (13) en route to 9, as it is anticipated that the increased polarity of 13 and its derivatives would make purification particularly difficult.
Results and discussion
Our study was initiated by investigating the conditions routinely used for the oxidation of thebaine (3) to 14-hydroxycodeinone (5) (Scheme 3). Conventionally, common oxidants, such as hydrogen peroxide, peracetic acid or m-CPBA are employed, with HOAc or a combination of HOAc and H2O as the solvent. Previous studies have found that the yield for this step can be improved to 74% using a combination of TFA and HOAc,14 rather than just HOAc4 alone. Nevertheless, depending upon the exact reaction conditions, the formation of various by-products has been reported. During the optimization of the oxycodone synthesis, the N-oxide of 5 was found to be a major impurity of thebaine oxidation.15 Purification by recrystallization has also allowed (8S)-hydroxyoxycodone or 7,8-dihydro-8,14-dihydroxycodeinone (DHDHC, 14) and trans-10-hydroxythebaine (15) to be isolated.16 DHDHC is formed via the acid-catalysed aqueous hydrolysis of 5.3b,17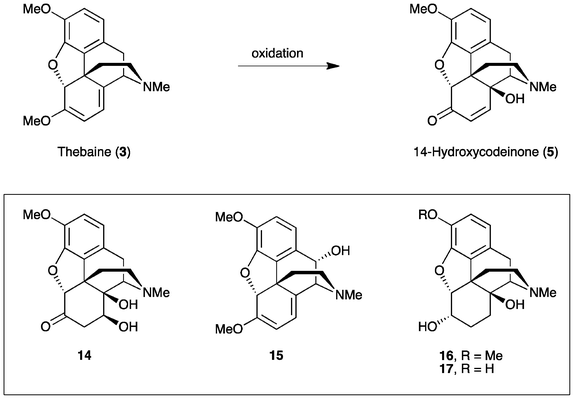
Oripavine (4) is reported to be more sensitive to the oxidation conditions than thebaine (3).4 For example, studies have shown the free phenolic OH to be a liability in the oxidative transformation of ring C atoms (vide supra).11 Others have reported that the double bond in 14-hydroxymorphinone (6) is even more susceptible to hydration than that in 14-hydroxycodeinone (5).18
According to the procedure described by Hauser et al., the synthesis of 14-hydroxycodeinone (5) from thebaine (3) was reproduced for up to 1 gram of the substrate.14 However, in our hands, the yields were variable for larger scale reactions. Oripavine (4) was found to be even more sensitive to these reaction conditions, resulting in diminished and varying yields of 14-hydroxymorphinone (6), particularly for reactions greater than one gram in scale (data not shown).
To avoid unnecessary complications due to competing N-oxide formation,15 oxidation using the hydrochloride salt of thebaine (3) and oripavine (4) was investigated. Rearrangement reactions of thebaine are known to occur in mineral acids, leading to the formation of compounds such as morphothebaine and thebenine.19 These rearrangements are particularly facile under more strongly acidic conditions and at higher temperatures. Notably, it was found that by carefully adding cold 10% HCl into a mixture of either thebaine (3) or oripavine (4) in ice-cold water until pH 2 was obtained, followed by a simple extraction with either CHCl3 or CHCl3/i-PrOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), side-products due to rearrangements were avoided. This procedure has consistently provided the respective hydrochloride salts of these opiates in near-quantitative yields.
:1), side-products due to rearrangements were avoided. This procedure has consistently provided the respective hydrochloride salts of these opiates in near-quantitative yields.
Subsequently, oxidation of the Δ6, Δ8-morphinane was performed using m-CPBA in 10% HOAc(aq). m-CPBA was added in portions to a stirred solution of thebaine hydrochloride in 10% HOAc(aq) at ambient temperature. When the oxidation was complete (via TLC analysis), removal of the m-CBA by-product was achieved via washing with CHCl3. Concentration of the aqueous solution gave 14-hydroxycodeinone hydrochloride (5·HCl) in a 98% yield. In a similar fashion, 14-hydroxymorphinone hydrochloride (6·HCl) was prepared from oripavine hydrochloride (4·HCl), again in an excellent yield (99%). Both compounds had purities of 95% and 94% respectively, according to HPLC analysis. Hence, simply employing the hydrochloride of the Δ6, Δ8-morphinane in the reaction rather than the more traditionally-used free base form has provided the α,β-unsaturated 14-hydroxyopiate in high yield and purity.
Reduction of the α,β-unsaturated ketone 5 or 6, is typically carried out via catalytic hydrogenation. For these substrates, this seemingly fundamental transformation has been reported to proceed in only ∼85% yields, although one early report involving the catalytic hydrogenation of the specific substrate (+)-14-hydroxycodeinone gave (+)-oxycodone in a 95% yield.20 Notably, an unusually high catalyst loading (1 g of catalyst for 2 g of substrate) was employed. For more typical reactions, depending upon the reaction conditions, a number of side-products are produced. For example, it is now common practice in industry to perform the two-step sequence in one-pot, without isolation of the intermediate α,β-unsaturated ketone. In such one-pot operations, the presence of any excess oxidising agent has been reported to result in the formation of 1,1′-dimer by-products, which are not easily removed.21 The problem may be alleviated by the addition of antioxidants such as ascorbic acid or sodium hydrosulfite.21 Additionally, incomplete reduction has also been an issue.22 The presence of small amounts of these α,β-unsaturated ketone precursors is of concern, as these compounds are potentially genotoxic. Small amounts of 14-hydroxycodeinone (5) can also be present in the final product since any DHDHC (14) produced during the oxidation step readily dehydrates into 5 during the conversion of the oxycodone base (1) into its hydrochloride salt.22 Some groups have attempted to remove residual levels of 5 through exposure of the final product to a further dissolving metal or a Clemmenson reduction with zinc or magnesium, to give mainly dihydrohydroxythebainone and a number of other products.22 Alternatively, residual amounts of the contaminant could be removed by treating the material with nucleophiles, such as thiols, giving more benign derivatives.22 It is apparent that the reduction of the α,β-unsaturated ketone via catalytic hydrogenation in aqueous acetic acid for an extended period can lead to the formation of more side-products, further compromising the yields. For the synthesis of 1, Frances and coworkers have also reported obtaining varying amounts of the over-reduced product, 14-hydroxydihydrocodeine (16) as a by-product.3c In this instance, various catalyst deactivating agents, such as amines and sulphur additives, were employed in an attempt to address the problem.
To investigate the reduction via catalytic hydrogenation, some initial experiments on a sample of purified 14-hydroxycodeinone free base (5) were conducted, using either 5% Pd/BaSO4 or 3% Pd/C as catalysts. 14-Hydroxycodeinone (5) was prepared according to the method reported by Hauser and co-workers.14 As a benchmark, 10% HOAc(aq) was chosen as the solvent as it is the most common medium used for this reaction. The reduction was allowed to proceed for a specified period at room temperature and aliquots were taken for LC-MS analysis. When complete, the reaction mixture was filtered and the filtrate was concentrated to dryness and analysed via1H NMR and LC-MS. The results and conditions are summarised in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Unless otherwise indicated, 10 mg of catalyst and 10 mL of solvent per 100 mg of substrate.a 20 mL of solvent per 100 mg of substrate.b Determined by 1H NMR (D2O); ratios were determined by comparison of the intensities of the H-6 proton of 16 or 17 and the corresponding aromatic protons.c Not detected. | |||||
| Entry | Substratea | Catalyst | Solvent | Time (h) | % 6-Hydroxy by-product (16/17)b |
| 1 | 5 | 5% Pd/BaSO4 | 10% HOAc(aq) | 1 | 10 |
| 2 | 5 | 5% Pd/BaSO4 | 10% HOAc(aq) | 1.3 | 12 |
| 3 | 5 | 3% Pd/C | 10% HOAc(aq) | 1 | 7 |
| 4 | 6·HCl | 5% Pd/BaSO4 | 10% HOAc(aq) | 1.3 | 10 |
| 5 | 6·HCl | 3% Pd/C | 10% HOAc(aq) | 1.3 | 11 |
| 6 | 6·HCl | 5% Pd/BaSO4 | 10% HOAc(aq) + NH4OH (pH 5) | 1.8 | 10 |
| 7 | 6·HCl | 5% Pd/BaSO4 | 10% HOAc(aq) + Et3N (pH 6) | 16 | 12 |
| 8 | 5 a | 5% Pd/BaSO4 | MeOH | 7 | n.d.c |
| 9 | 5·HCl | 5% Pd/BaSO4 | MeOH | 24 | n.d.c |
| 10 | 5·HCl | 3% Pd/C | MeOH | 1 | 5 |
| 11 | 6·HCl | 5% Pd/BaSO4 | MeOH | 8 | 3 |
| 12 | 6·HCl | 3% Pd/C | MeOH | 1 | 3 |
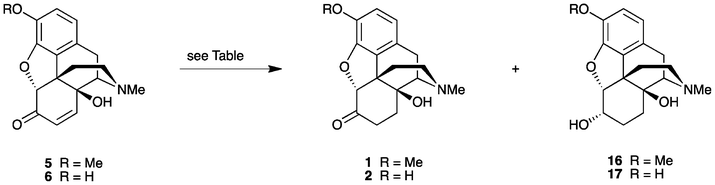
The reduction of 14-hydroxycodeinone (5) to oxycodone (1) was initially trialed using 5% Pd/BaSO4 and 3% Pd/C as hydrogenation catalysts in aqueous acetic acid (10% HOAc). In both cases, the mass spectral data showed essentially two products, one with an expected ion m/z of 316 [M + H]+ (100%) along with ions due to a main impurity at a m/z of 318. The impurity was postulated to be the corresponding 6-hydroxy analog, produced by the over-reduction of the desired product. This was later confirmed by comparison of the 1H NMR data of the respective crude reaction product with the published data for 14-hydroxydihydrocodeine (16).23 Subsequent experiments were conducted on the crude sample of 14-hydroxymorphinone hydrochloride (6·HCl), previously prepared via the oxidative transformation of oripavine hydrochloride (4·HCl). The reduction of 6·HCl also gave a major impurity with a m/z of 304 [M + H + 2]+. Once again, the contaminant was the corresponding 6-hydroxy compound, produced by the over-reduction of the desired product, as confirmed by comparison of the 1H NMR data of the respective crude reaction product with published data for 14-hydroxydihydromorphine (17).24 As further confirmation, a sample of 17 was prepared,25 as an approximate 5:1 ratio of the 6α:6β epimers, via NaBH4-reduction of 14-hydroxymorphinone (6).
It has been found that the carbonyl moieties in oxycodone·HCl (1·HCl) and oxymorphone·HCl (2·HCl) are more prone to the formation of enolic derivatives than conventional ketones.26,27 One possible explanation for this is that the resultant Δ6 bond for the enol form permits flattening of the C ring, thus relieving torsional ring strain.27 The rather significant amount of the 6-hydroxy contaminant obtained is consistent with the reported facile enolate formation in these 14-hydroxydihydromorphinane systems.
A number of factors may influence the keto–enol tautomeric equilibrium, including the pH or temperature at which the reduction is conducted. Since the oxidation and reduction protocol can be conducted in one-pot3 and as 10% HOAc(aq) is an efficient medium for the oxidation step, we were interested in investigating whether varying the pH for the subsequent reduction step via the addition of concentrated aqueous ammonia or Et3N would be beneficial. These reactions were conducted on 6·HCl as the substrate. The progress of the reductions was monitored using LC-MS after 1 h and thereafter at intervals until complete. As the results in Table 1 (entries 6 and 7) show, there appears to be little change in the amount of 17 formed. We have found that the reduction of the α,β-unsaturated ketones 5 and 6·HCl and oxycodone/oxymorphone are competing reactions, albeit at a slower rate for the latter systems (via LC-MS analysis).
It is also well-known that different solvents influence the keto–enol tautomeric equilibria in a complex way.27 In particular, the prevailing form can be greatly dependent on the protic nature and polarity of the solvent.28 For example, the equilibrium can be shifted via the addition of organic solvent into the aqueous media. There have been numerous studies conducted in an attempt to improve the yield and reduce the various side-product formation associated with the reduction of these α,β-unsaturated keto-morphinans via catalytic hydrogenation. Surprisingly, solvent effects have not previously been investigated to the best of our knowledge. To this end, we investigated the hydrogenation of 14-hydroxycodeinone (5) as the free base in a non-aqueous medium. Methanol was the solvent of choice since both the free base and the hydrochloride salt of the α,β-unsaturated ketones 5 and 6 are soluble in this medium. Thus, when employing 5% Pd/BaSO4 as the catalyst, the reduction of the 14-hydroxycodeinone free base (5) was complete after 7 h at ambient temperature (Table 1, entry 8). After filtration, the filtrate was concentrated to afford the product in a near-quantitative yield (99%). Methanol proved to be an ideal solvent choice as a cursory examination of the 1H NMR data for the crude product revealed no over-reduced 14-hydroxydihydrocodeine (16) by-product. The purity of the oxycodone (1) obtained was 98% (via HPLC). Reduction using methanol was subsequently repeated on the crude hydrochloride salt of 14-hydroxycodeinone (5·HCl), using either 5% Pd/BaSO4 or 3% Pd/C (Table 1, entries 9 and 10). Also, as entry 9 shows, hydrogenation in methanol over 5% Pd/BaSO4, even for an extended period of time, did not result in any detectable amount of 16 being formed (via1H NMR). The yield of the crude oxycodone·HCl (1·HCl) prepared from the crude 5·HCl was 98% (93% purity via HPLC analysis). On the other hand, methanol offered no significant benefit as a solvent when 3% Pd/C was employed as the catalyst (cf. entries 3 and 10).
Similar results were obtained for the reduction of 14-hydroxymorphinone·HCl (6·HCl) (Table 1, entries 11 and 12), with the reduction in MeOH over 5% Pd/BaSO4 resulting in a greatly reduced amount of 17 being formed. After filtration, the filtrate was concentrated to furnish oxymorphone·HCl (2·HCl) in a 98% yield and a purity of 94% by HPLC. Notably, this was achieved from oripavine (4) without the requirement of any column chromatography or purification of intermediates. The reaction conditions, as summarised in entry 11 on a 100 mg scale, were also amenable to scale-up. Hence, when starting with 2 grams of 4, 2·HCl was obtained in a 96% yield over 3-steps (95% purity via HPLC).
The improved oxidation–reduction protocol was then investigated for the direct synthesis of N-noroxymorphone (9) from N-nororipavine (10). Oxidation of N-nororipavine·HCl (10·HCl) using m-CPBA in 10% HOAc(aq) at ambient temperature afforded 14-hydroxy-N-normorphinone·HCl (13·HCl) in a 99% yield (purity 96% via HPLC). Washing the reaction mixture with CHCl3 once again allowed for the facile removal of the m-CBA by-product. The crude material was used in the subsequent reduction step without further purification. The conditions and results for the reduction experiments are summarised in Table 2. Once again, 5% Pd/BaSO4 and MeOH performed the best (Table 2, entry 3), and furnished (after filtration and concentration to remove volatiles), N-noroxymorphone·HCl (9·HCl) in a 97% yield with a purity of 95% via HPLC. Interestingly, although the reduction was significantly slower when conducted with MeOH compared to 10% HOAc(aq), the extended reaction time did not lead to more 14-hydroxydihydro-N-normorphine·HCl (18·HCl) being formed. The identity of the 6-hydroxy contaminant 18 in these experiments was confirmed via NaBH4-reduction of a sample of 9. The ratio of the product 9 and contaminant 18 was established by comparison of the intensities of the multiplet at Δ4.27 ppm due to the H-6 proton of 18 and the aromatic protons of the crude product in D2O.
|
|
||||
|---|---|---|---|---|
| Unless otherwise indicated, 10 mg of catalyst and 10 mL of solvent was used for 100 mg of substrate.a Determined by 1H NMR.b The additional amount of catalyst was added at intervals. | ||||
| Entry | Catalyst | Solvent | Time (h) | % 14-hydroxydihydro-N-normorphine·HCl (18·HCl)a |
| 1 | 5% Pd/BaSO4 | 10% HOAc(aq) | 2 | 12 |
| 2 | 3% Pd/C | 10% HOAc(aq) | 1 | 11 |
| 3 | 5% Pd/BaSO4 (15 mg + 20 mg)b | MeOH (20 mL) | 72 | 2 |
| 4 | 3% Pd/C (15 mg) | MeOH (20 mL) | 24 | 6 |
Finally, the sample of crude N-noroxymorphone·HCl (9·HCl) obtained from Table 2, entry 3, was subjected to N-alkylation using established conditions for the synthesis of naltrexone (7) from N-noroxymorphone (9).8 Treatment of the crude N-noroxymorphone·HCl (9·HCl) with (bromomethyl)cyclopropane and NaHCO3 in DMA–H2O at 60 °C provided, after a standard workup, crude naltrexone in a 95% yield and with a purity of 92% via HPLC. Subsequent column chromatography furnished naltrexone in an 82% yield. This represents an overall yield of 79% over three steps from N-nororipavine (10).
Conclusion
Oxycodone (1) is typically synthesised from thebaine (3) in two steps. Herein, we have demonstrated improved conditions for the initial oxidation step that involves the use of the hydrochloride salt of thebaine (3) rather than the free base form. The subsequent reduction step was also significantly improved by using methanol as the solvent rather than an aqueous medium. These conditions also work well for the conversion of oripavine (4) into oxymorphone (2), which was achieved in over a 90% yield over two steps.In combination with new methodology, we have recently reported the N-demethylation of oripavine (4).12,13 This approach also provides an efficient route for the preparation of noroxymorphone (9), a key intermediate in the synthesis of a number of opiate pharmaceuticals such as naltrexone. N-Noroxymorphone (9) was prepared in a 78% yield from oripavine (4) following N-demethylation, oxidation to introduce the 14-hydroxy group and alkene reduction. N-Noroxymorphone (9) was subsequently alkylated with (bromomethyl)cyclopropane to exemplify the synthesis of the known pharmaceutical opiate naltrexone (7) (in a 64% overall yield from oripavine).
Experimental
General
m-Chloroperbenzoic acid, (bromomethyl)cyclopropane, 5% Pd/BaSO4 and 3% Pd/C were purchased from Sigma-Aldrich. Solvents (Merck) were used as supplied. Thin layer chromatography (TLC) was performed with 0.25 μm TLC silica gel 60 F aluminium plates with a 254 nm fluorescent indicator and the plates were visualized using both UV light and a molybdate stain. 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively. Chemical shifts (δ (ppm)) were referenced with solvent residual peaks. Coupling constants are given in Hertz. High resolution mass spectra were obtained using a Waters LCT Premier XE time-of-flight mass spectrometer fitted with an electrospray (ESI) ion source and controlled with MassLynx software version 4.5. 14-Hydroxycodeinone free base (5) was prepared from thebaine, in 75–80% yields, according to literature14 methods. N-Nororipavine hydrochloride (10·HCl) was prepared via N-demethylation of oripavine-N-oxide hydrochloride using a combination of Fe(0) and FeCl3·6H2O, according to published13 procedures.14-Hydroxycodeinone·HCl (5·HCl)
Ice-cold 10% HCl was added dropwise to a stirred slurry of thebaine (3) (1.00 g, 3.21 mmol) in ice-cold H2O (mL) until the pH of the resulting solution was around 2. The solution was extracted with CHCl3 (10 mL × 4), the extracts combined, dried (Na2SO4), filtered and concentrated to give thebaine hydrochloride (3·HCl) as a white solid. m-CPBA (0.58 g of a max. 77% reagent, 3.37 mmol) was added portionwise to a stirred solution of 3·HCl in 10% HOAc(aq) (20 mL) over 2 h at RT. After stirring for a further 30 min, the mixture was washed with CHCl3 (10 mL × 3) and concentrated to dryness to afford the title compound as an off-white solid, 1.10 g (98%). This material was used in the subsequent step without further purification; 1H NMR (D2O) δ 7.03 (d, J 6.0, 1H), 6.98 (d, J 8.4, 1H), 6.90 (d, J 8.4, 1H), 6.27 (d, J 6.0, 1H), 5.07 (s, 1H), 3.99 (d, J 6.0, 1H), 3.85 (s, 3H), 3.58 (d, J 20.4, 1H), 3.65 (dd, J 4.8 and 13.2, 1H), 3.16 (dd, J 6.0 and 20.4, 1H), 3.01 (s, 3H), 2.95 (ddd, J 4.0, 12.8 and 12.8, 1H), 2.77 (ddd, J 5.2, 13.2 and 13.2, 1H), 1.97 (dd, J 3.2 and 13.2, 1H); 13C NMR (D2O) δ 196.3, 147.3, 143.4, 142.5, 133.1, 128.3, 122.8, 121.1, 115.5, 85.9, 67.4, 65.0, 56.5, 47.2, 45.5, 40.8, 26.2, 23.0; HRMS C18H20NO4 calcd for [M + H]+ 314.1387, found 314.1377.14-Hydroxymorphinone·HCl (6·HCl)
The above reaction was repeated using oripavine (4) (2.00 g, 6.72 mmol) instead of thebaine. This provided 14-hydroxymorphinone·HCl as an off-white solid, 2.24 g (99%). This material was used in the subsequent step without further purification; 1H NMR (D2O) δ 7.04 (d, J 10.4 Hz, 1 H), 6.84 (d, J 8.4, 1H), 6.81 (d, J 8.4, 1H), 6.26 (d, J 8.4, 1H), 5.05 (s 1H), 3.97 (d, J 6.4 1H), 3.55 (d, J 20.4, 1H), 3.36 (dd, J 4.8 and 13.2, 1H), 3.13 (dd, J 6.4 and 20.4, 1H), 3.00 (s, 3H), 2.96 (ddd, J 4.0, 13.2 and 13.2, 1H), 2.77 (dd, J 4.8 and 13.2, 1H); 13C NMR (D2O) δ 196.7, 147.3, 142.5, 138.6, 133.0, 128.5, 122.0, 121.1, 118.8, 85.7, 67.4, 65.0, 47.3, 45.5, 40.8, 26.1, 23.0; HRMS C17H18NO4 calcd for [M + H]+ 300.1230, found 300.1241.14-Hydroxy-N-normorphinone·HCl (13·HCl)
m-CPBA (291 mg of a 77% max reagent, approx. 1.30 mmol) portionwise was added to a stirred solution of N-nororipavine hydrochloride (10·HCl) (397 mg, 1.24 mmol) in 10% HOAc(aq) (15 mL) over 10 min. The reaction mixture was stirred at RT for an additional 10 min and then heated at 60 °C for 30 min. When cooled, the mixture was washed with CHCl3 (20 mL × 3) and concentrated to dryness to afford the title compound as an off-white foam (395 mg, 99%). This material was used in the subsequent step without further purification; 1H NMR (D2O) δ 7.07 (d, J 8.8, 1H), 6.88 (d, J 8.4, 1H), 6.84 (d, J 8.4, 1H), 6.29 (d, J 8.8, 1H), 5.06 (s, 1H), 4.12 (d, J 4.8, 1H), 3.40–3.28 (m, 3H), 3.04 (ddd, J 4.0, 13.2 and 13.2, 1H), 2.76 (ddd, J 4.8, 13.6 and 13.6, 1H), 1.98 (dd, J 4.0 and 13.6, 1H); 13C NMR (D2O) δ 196.7, 147.8, 142.6, 138.6, 132.9, 128.8, 122.3, 121.1, 118.7, 86.0, 66.4, 56.4, 46.1, 37.1, 27.4, 25.0; HRMS C16H16NO4 calcd for [M + H]+ 286.1074, found 286.1085.General procedure for the reduction of 5, 5·HCl, 6·HCl and 13·HCl via catalytic hydrogenation
A stirred solution of the α,β-unsaturated ketone 5, 5·HCl, 6·HCl or 13·HCl (100 mg) in 10% HOAc(aq) (10 mL) was evacuated and then backfilled with nitrogen. 5% Pd/BaSO4 or 3% Pd/C (10 mg) was added and the reaction vessel was evacuated and backfilled with hydrogen. This latter process was repeated twice. The progress of the reduction was monitored at intervals via LC-MS. When complete, the solids were removed by filtration and the filtrate was concentrated to dryness to afford respectively, oxycodone, oxycodone·HCl, oxymorphone·HCl or N-noroxymorphone·HCl. The free base form of these compounds was obtained via treatment of the hydrochloride salt with concentrated ammonia. The above reaction was repeated using 3% Pd/C and replacing 10% HOAc(aq) with MeOH (see Table 1 and 2 for details).Oxycodone (1)
White solid: mp 215–217 °C (lit.29 mp 218–219 °C); 1H NMR (CDCl3) δ 6.71 (d, J 8.0, 1H), 6.54 (d, J 8.0, 1H), 5.02–4.90 (br s, 1H), 4.67 (s, 1H), 3.91 (s, 3H), 3.16 (d, J 14.4, 1H), 3.03 (ddd, J 4.8, 14.4 and 14.4, 1H), 2.88 (d, J 6.0, 1H), 2.57 (dd, J 6.0 and 18.8, 1H), 2.52–2.36 (m, 5H), 2.30 (ddd, J 3.2, 3.2 and 14.4, 1H), 2.22–2.13 (m, 1H), 1.88 (ddd, J 2.8, 4.8 and 13.2, 1H), 1.68–1.55 (m, 2H); 13C NMR (CDCl3) δ 208.4, 144.8, 142.7, 129.2, 124.8, 119.3, 114.8, 90.2, 64.4, 56.6, 50.0, 45.1, 42.5, 35.9, 31.2, 30.3, 21.7; HRMS C18H22NO4 calcd for [M + H]+ 316.1543, found 316.1531.Oxymorphone (2)
White solid: mp 244–247 °C (lit.30 mp 248–249 °C); 1H NMR (CDCl3) δ 6.73 (d, J 8.0, 1H), 6.62 (d, J 8.0, 1H), 5.70–5.20 (br s, 2H), 4.69 (s, 1H), 3.16 (d, J 18.4, 1H), 3.05 (ddd, J 5.2, 14.4 and 14.4, 1H), 2.89 (d, J 5.6, 1H), 2.56 (dd, J 6.0 and 18.8, 1H), 2.53–2.39 (m, 5H), 2.32 (ddd, J 2.8, 2.8 and 14.4, 1H), 2.24–2.17 (m, 1H), 1.89 (ddd, J 3.2, 5.2 and 13.6, 1H), 1.69–1.58 (m, 2 H); 13C NMR (DMSO-d6) δ 208.8, 143.5, 139.5, 129.3, 119.1, 117.3, 89.4, 70.0, 63.9, 49.6, 45.2, 42.3, 39.9, 35.8, 31.1, 29.9, 21.6; HRMS C17H20NO4 calcd for [M + H]+ 302.1387, found 302.1387.N-Noroxymorphone·HCl (9·HCl)
Off-white solid: mp > 250 °C; 1H NMR (D2O) δ 6.87 (d, J 8.0, 1H), 6.83 (d, J 8.0, 1H), 5.05 (s, 1H), 3.91 (d, J 4.0, 1H), 3.33–3.20 (m, 3H), 3.03 (ddd, J 4.2, 14.8 and 14.8, 1H), 2.89 (ddd, J 4.4, 13.2 and 13.2, 1H), 2.69 (ddd, J 4.2, 13.6 and 13.6, 1H), 2.35 (ddd, J 2.8, 2.8 and 14.8, 1H), 2.07 (ddd, J 3.2, 5.2 and 14.4, 1H), 1.82–1.71 (m, 2H); 13C NMR for 9 (DMSO-d6) δ 208.7, 143.5, 139.5, 129.3, 123.5, 119.1, 117.3, 89.4, 69.5, 56.8, 50.2, 37.3, 35.7, 31.3, 30.9, 29.1; HRMS C16H18NO4 calcd for [M + H]+ 288.1230, found 288.1226.14-Hydroxydihydromorphine (17)
A slurry of NaBH4 (54 mg, 1.43 mmol) in MeOH (2 mL) was added in a dropwise fashion to a stirred solution of oxymorphone (120 mg, 0.398 mmol) in MeOH (5 mL). The reaction mixture was stirred for 15 min and then concentrated to dryness. MeOH (5 mL) was added to the remaining white solid and the resulting solution concentrated to dryness. The process of adding MeOH and concentrating was repeated twice. The remaining residue was dissolved in H2O (5 mL), the pH of the solution was adjusted to 8.5 with NH4OH and extracted with 3:1 CHCl3/i-PrOH (5 mL × 3). The extracts were combined, dried (Na2SO4), filtered and concentrated to give a 5:1 mixture of 17 and its 6-β epimer (iso-14-hydroxydihydromorphine) as a white solid, 105 mg (87%). 14-Hydroxydihydromorphine (17): selected 1H NMR (CDCl3) δ 6.73(d, J 8.4, 1H), 6.56 (d, J 8.4, 1H), 4.67 (d, J 4.4, 1H), 4.25 (ddd, J 4.4, 14.4 and 11.2, 1H), 3.23, d, J 18.4, 1H), 2.78 (d, J 6.4, 1H), 2.58 (dd, J 6.4 and 18.4, 1H), 2.48–2.38 (m, 1H), 2.36 (s, 3H), 1.20–1.08 (m, 1H). Iso-14-Hydroxydihydromorphine: selected 1H NMR (CDCl3) δ 4.50 (d, J 6.0, 1H), 3.57 (ddd, J 4.8, 5.6 and 10.8, 1H); HRMS C17H22NO4 calcd for [M + H]+ 304.1543, found 304.1542.
14-Hydroxydihydro-N-normorphine hydrochloride (18·HCl)
According to the procedure for the preparation of 14-hydroxydihydromorphine (17), N-noroxymorphone (200 mg, 0.696 mmol) was treated with NaBH4 (95 mg, 2.51 mmol). When the reduction was complete (via TLC analysis), the reaction mixture was concentrated, MeOH was added and the solution was evaporated to dryness. The latter process of adding MeOH and the concentration of the resulting solution was repeated twice. H2O (2 mL) was added to the white solid and the slurry was filtered. The solid was washed with H2O (2 mL × 2) then dissolved in 10% HCl(aq) and the solution was concentrated to dryness to afford a ∼14:1 mixture of the title compound and its 6-β epimer (iso-14-hydroxydihydro-N-normorphine hydrochloride) as a white solid, 212 mg (93%). 14-Hydroxydihydro-N-normorphine hydrochloride (18·HCl): 1H NMR (D2O) δ 6.86 (d, J 8.0, 1H),6.77 (d, J 8.0, 1H), 4.77 (d, J 5.2, 1H), 4.27 (ddd, J 4.0, 4.0 and 8.8, 1H), 3.79 (d, J 6.0, 1H), 3.31 (dd, J 6.4 and 19.6, 1H), 3.22–3.12 (m, 2H), 2.99 (ddd, J 4.4, 13.2 and 13.2, 1H), 2.47 (ddd, J 5.2, 13.6 and 13.6, 1H), 1.88–1.55 (m, 4H), 1.36 (ddd, J 6.8, 6.8 and 15.2, 1H). Iso-14-Hydroxydihydro-N-normorphine hydrochloride: selected 1H NMR (D2O) δ 6.88 (d, J 8.0, 1H), 6.81 (d, J 8.0, 1H), 4.55 (d, J 6.8, 1H), 3.51 (ddd, J 5.2, 6.4 and 12.0, 1H); HRMS C16H18NO4 calcd for [M + H]+ 288.1230, found 288.1220.
Naltrexone (7)
NaHCO3 (88 mg, 1.05 mmol) and (bromomethyl)cyclopropane (40 μL, 0.400 mmol) was added to a stirred solution of crude N-noroxymorphone·HCl (prepared via catalytic hydrogenation of a solution of 13·HCl in MeOH with 5% Pd/BaSO4; 87 mg, 0.269 mmol) in DMA (1 mL) and H2O (0.1 mL).8 The mixture was heated at 60 °C. When complete (via LC-MS), H2O (5 mL) was added and the solution was extracted with DCM (5 mL × 4). The extracts were combined, dried (Na2SO4), filtered and concentrated. The residue was purified via column chromatography on silica gel, eluting with a gradient of DCM–MeOH (50:1–20:1), which afforded naltrexone as a white solid, 75 mg, 82%; mp 174–175 °C (lit.31 mp 175.7 °C); 1H NMR (CDCl3) δ 6.72 (d, J 8.0, 1H), 6.60 (d, J 8.0, 1H), 5.50–5.05 (br s, 1H), 5.31 (s, 1H), 4.70 (s, 1H), 3.20 (d, J 6.0, 1H), 3.10–3.11 (m, 2H), 2.71 (dd, J 4.8 and 12.0, 1H), 2.58 (dd, J 6.0 and 18.4, 1H), 2.50–2.39 (m, 3H), 2.33 (ddd, J 3.2, 3.2 and 14.4, 1H), 2.17 (ddd, J 3.6, 12.0 and 12.0, 1H), 1.90 (ddd, J 2.8, 5.2 and 13.2, 1H), 1.65 (ddd, J 3.2, 13.2 and 14.4, 1H), 1.62–1.55 (m, 1H), 0.93–0.82 (m, 1H), 0.62–0.53 (m, 2H), 0.20–0.12 (m, 2H); 13C NMR (CDCl3) δ 210.0, 143.5, 138.8, 129.0, 124.0, 119.8, 117.9, 90.5, 70.4, 61.9, 59.1, 51.0, 43.5, 36.1, 31.3, 30.5, 22.6, 9.3, 3.9, 3.7; HRMS C20H24NO4 calcd for [M + H]+ 342.1700, found 342.1709.
Acknowledgements
The authors wish to thank the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology for financial support.References
- R. J. Bryant, Chem. Ind., 1988, 146–153 CAS
.
-
W. L. Way, E. L. Way and H. L. Fields, Opioid analgesics and antagonistsIn Basic and clinical pharmacology; 6th ed, ed. B. G. Katzung, Appleton and Lange: Norwalk, CT, 1995 Search PubMed
-
(a) See for example B. Weber and S. Sahli, Preparation of Low Impurity Opiates in a Continuous Flow Reactor, 2011 Search PubMed
-
(a) M. Freund and E. Speyer, Angew. Chem., 1914, 27, 350–351 Search PubMed
- R. E. Lutz and L. Small, J. Org. Chem., 1939, 4, 220–233 CrossRef CAS
-
(a) See for example P. X. Wang, T. Jiang, G. L. Cantrell and D. W. Berberich, Novel Opiate Reduction Utilising Catalytic Hydrogen Transfer Reaction, 2008 Search PubMed
-
A. F. Casy, R. T. Parfitt, Opioid analgesics, Plenum, New York, 1986 Search PubMed
- See for example P. X. Wang, T. Jiang, D. W. Berberich, Processes for the Synthesis of Tertiary Amines, US Pat. Appl. 2010081819, 2010 Search PubMed
-
K. C. Rice, A. M. Newman, Total Synthesis of Northebaine, Normorphine, Noroxymorphone Enantiomers and Derivatives via N-Nor Intermediates, US 5668285, 1997 Search PubMed
- B-S. Huang, Process for Preparing Oxymorphone, 2008 Search PubMed
- A. Sipos, S. Berényi and S. Antus, Helv. Chim. Acta, 2009, 92, 1359–1365 CrossRef CAS
- G. B. Kok and P. J. Scammells, Org. Biomol. Chem., 2011, 9, 1008–1011 CAS
- G. B. Kok and P. J. Scammells, Aust. J. Chem., 2011, 64, 1515–1521 CrossRef CAS
- F. M. Hauser, T-K. Chen and F. I. Carroll, J. Med. Chem., 1974, 17, 1117 CrossRef CAS
-
(a) R. Krassnig, C. Hederer and H. Schmidhammer, Arch. Pharm., 1996, 329, 325–326 CrossRef CAS
- B. Proksa, Arch. Pharm., 1999, 369–370 CrossRef CAS
-
(a) S. P. Findlay and L. F. Small, J. Am. Chem. Soc., 1950, 72, 3247–3249 CrossRef CAS
- U. Weiss, J. Org. Chem., 1957, 22, 1505–1508 CrossRef CAS
- F. E. Granchelli, C. N. Filer, A. H. Soloway and J. L. Neumeyer, J. Org. Chem., 1980, 45, 2275–2278 CrossRef CAS
- I. Iijima, J. Minamikawa, A. E. Jacobson, A. Brossi and K. C. Rice, J. Med. Chem., 1978, 21, 398–400 CrossRef CAS
-
P. X. Wang, T. Jiang, G. L. Cantrell and D. W. Berberich, Improved Preparation of Oxymorphone from Oripavine, WO 2008/118654 A1 Search PubMed
-
D. P. Cox, Y. Zhang, W-C. Zhang and K. E. James, Process for Preparing Oxycodone having Reduced Levels of 14-Hydroxycodeinone, WO 2008/070658 A1 Search PubMed
- V. Chaudhary, H. Leisch, A. Moudra, B. Allen, V. DeLuca, D. P. Cox and T. Hudlicky, Collect. Czech. Chem. Commun., 2009, 74(7–8), 1179–1193 CrossRef CAS
- R. C. Crouch, A. V. Bhatia and O. W. Lever Jr., J. Heterocycl. Chem., 1990, 27, 385–389 CrossRef CAS
- M. T. Long, A. M. Hailes, G. W. Kirby and N. C. Bruce, Appl. Environ. Microbiol., 1995, 61, 3645–3649 CAS
- H. Nagase, A. Abe and P. S. Portoghese, J. Org. Chem., 1989, 54, 4120–4125 CrossRef CAS
- See for example E. Ferrari, M. Saladini, F. Pignedoli, F. Spagnolo and R. Benassi, New J. Chem., 2011, 35, 2840–2847 RSC
- H. Watarai and N. J. Suzuki, J. Inorg. Nucl. Chem., 1974, 36, 1815–1820 CrossRef CAS
- A. C. Currie, J. Chem. Soc., 1960, 773–781 RSC
-
J. D. Andre, J. R. Dormoy and A. Heymes, Preparation of Morphinan Derivatives as Analgesics, EP 359647 A1 1990 Search PubMed
- M. O. Hamad, P. K. Kiptoo, A. L. Stinchcomb and P. A. Crooks, Bioorg. Med. Chem., 2006, 14, 7051–7061 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and IR spectra of all products. See DOI: 10.1039/c2ra21693k |
| This journal is © The Royal Society of Chemistry 2012 |