DOI:
10.1039/C2RA22291D
(Paper)
RSC Adv., 2012,
2, 11306-11317
Triazine functionalized ordered mesoporous organosilica as a novel organocatalyst for the facile one-pot synthesis of 2-amino-4H-chromenes under solvent-free conditions†
Received
25th September 2012
, Accepted 25th September 2012
First published on 25th September 2012
Abstract
A highly ordered 2D-hexagonal mesoporous material functionalized with a triazine moiety has been synthesized via post-synthetic modification of mesoporous SBA-15 with thiols followed by a thiol–ene click reaction using 2,4,6-triallyloxy-1,3,5-triazine. A facile one-pot three-component condensation reaction of aromatic aldehyde, malononitrile and activated phenols for the synthesis of a diverse range of 2-amino-4H-chromenes has been efficiently catalyzed over this novel mesoporous metal-free heterogeneous organocatalyst under solvent-free reaction conditions. Further, this organocatalytic reaction is waste-free, easy to work-up and efficiently reused. The organic products have been isolated from the reaction mixture by using easily disposable solvents and can be easily purified by re-crystallization.
Introduction
During the last decade, organocatalytic processes have attracted increasing interest due to their huge potential in a wide range of value added organic transformations.1 The robustness, non-toxicity, the lack of metal-leaching possibility during the reaction, inertness towards moisture and oxygen, simple handling and storage are the main advantages of organocatalysts,2 which could largely overcome the major drawbacks traditionally associated with metal-based heterogeneous catalysts, like long reaction times, metal-leaching and structural stability of the catalyst for prolonged recycling experiments.3 In this context, sustainable chemistry has emerged as one of the key areas of research for the designing of chemical processes that cause minimal environmental impact and prevents pollution at its source.4 Existing chemical processes have been tuned by using solvent-free reaction media, aqueous medium, supercritical CO2, ionic liquids, etc., for the development of novel, environmentally benign processes and much improved catalytic performance.5 Thus, for developing sustainable and environmentally friendly chemical synthesis processes, the design of novel organocatalysts is highly desirable.6 Although, the success of organocatalysts in homogeneous reactions is hugely acknowledged, there are several drawbacks, like high catalyst loading, and the recovery and reuse of the expensive catalysts. In this context a successful strategy for the design of a heterogeneous organocatalyst is to heterogenize the active homogeneous counterpart via an immobilization technique. Ion exchange, physical adsorption, dipolar attraction and covalent attachment of the species containing the active sites to solid supports are well-known strategies for the immobilization of homogeneous organocatalysts. However, due to the improved catalyst recycling efficiency and minimum possibility of leaching of the organic fragment, the last approach has been considered as the most useful for immobilization of a homogeneous catalyst.7 Some organocatalysts have been successfully prepared through the immobilization of organic ligands, such as proline, diarylprolinol silyl ether, etc. Proline-based organocatalysts have been synthesized by anchoring proline into the organic polystyrene matrix8 or inorganic (zeolite, mesoporous silica) insoluble supports.9 Very recently, an organocatalyst based on a chitosan hydrogel has shown excellent catalytic activity.10 Due to the substantial swelling and stability problems, low surface area and also lower accessibility of the catalytic center, polymer supported organocatalysts often show slower reaction rates compared to their homogeneous counterparts.11
Mesoporous materials have gained increasing interest for drug delivery,12 adsorption/separation,13 sensing,14 catalysis,15etc., due to their exceptionally high surface area, thermal and mechanical stability. Thus, in this context, the design of organocatalysts based on mesoporous hybrid organosilica is a promising one.16 A mesoporous silica-based organocatalyst with a controlled loading of the active fragment, such as –NH2, –OH, –COOH, and –SH groups, on the support can be synthesized either by surfactant-assisted sol–gel synthesis or post-grafting into the mesoporous silica by employing some chemical reactions using the organosilane precursor.17 An active organic fragment of the organocatalyst in the mesoporous silica framework can be preserved due to the strong Si–C covalent bonding. The periodicity of nanoscale pores in these materials could favor the easy accessibility of the organic functions in addition to the possible diffusion of the solvent–soluble reactants within the insoluble solid matrix and is thus very attractive for liquid–solid phase catalytic reactions. Therefore, the covalent grafting of catalytically active organic centers onto the surface of functionalized mesoporous silica becomes of considerable technological interest in the field of molecular catalysis.16
2-Amino-chromenes are an important class of heterocyclic compounds, attracting a diverse array of research interests, ranging from natural products to biological and pharmacological activities, such as antimicrobial,18 antiviral,19 mutagenicity,20 antiproliferative,21 antitumor and central nervous system activity.22 These compounds are also hugely recognized in the field of cosmetics, pigments and potential biodegradable agrochemicals.23 Therefore, in recent times increasing attention has been paid for the synthesis of these heterocyclic compounds. These heterocyclic compounds can be synthesized by the one-pot three-component condensation reaction of aromatic aldehyde, malononitrile and activated phenol in a straightforward manner by using piperidine as a basic catalyst.24 The synthesis of 2-amino-chromene via condensation reactions has been conducted by employing many homogeneous catalysts such as NaOH,25 K2CO3,26 I2/K2CO3,27 TiCl4,28 InCl3,29 heteropolyacid30 and Et3N.31 Recently, Makerem et al. reported the electrochemically induced multicomponent condensation of resorcinol, malononitrile and aldehyde in propanol in an undivided cell in the presence of NaBr as an electrolyte.32 However, these homogeneous catalysts require long reaction times, toxic solvents and greater amounts of reagents than its stoichiometry. On the other hand, only a few heterogeneous catalysts, such as nano-sized magnesium oxide, Mg/Al hydrotalcite, and nano-structured diphosphate (Na2CaP2O7), have been developed for the synthesis of 2-amino-chromene.33 However, all of the heterogeneous catalysts are metal based, causing environmental hazards. So a successful approach for the development of an alternative, non-air sensitive, easily separable heterogeneous organocatalyst devoid of metal ions is highly desirable to address the industrial and environmental concerns.
Herein, we report the synthesis of a new robust and non-air sensitive metal-free triazine functionalized mesoporous organocatalyst TFMO-1 and its excellent catalytic activity in the one-pot three-component condensation reactions for the syntheses of 2-amino-chromene derivatives under solvent-free conditions. TFMO-1 with a 2D-hexagonal mesoporous structure has been designed via consecutive surface functionalization of SBA-15 with 3-mercaptopropyl triethoxysilane followed by a thiol–ene click reaction with 2,4,6-triallyloxy-1,3,5-triazine in the presence of azobisisobutylronitrile (AIBN) initiator. The organocatalyst has been thoroughly characterized by XRD, FT-IR, HRTEM, FE SEM, N2 sorption, 13C and 29Si MAS NMR studies. The increasing importance of metal-free catalysis has provoked us to use this organically modified triazine functionalized mesoporous silica as a catalyst for the synthesis of 2-amino-chromenes from a mixture of aromatic aldehyde, malononitrile and activated phenols under solvent-free conditions.
Experimental section
Organocatalyst preparation
Synthesis of mesoporous SBA-15.
In a typical synthesis, 2.0 g of P123 was dissolved in 60 mL of 2.0 M aqueous HCl and 15.0 g of distilled water under stirring at room temperature. Then 4.25 g of tetraethyl orthosilicate (TEOS) was added dropwise to the solution of P123 in acid media at 40 °C. After TEOS was added, the mixture was stirred for 24 h and then transferred into a Teflon-lined autoclave and kept at 100 °C for another 24 h. Finally the solid product was recovered by filtration, washed with distilled water, and air-dried. The template was removed by calcination of the sample at 500 °C in air for 5 h to get the final product SBA-15.
General procedure for the synthesis of –SH functionalized SBA-15.
0.1 g of calcined SBA-15 in 10 mL of dry toluene was placed in a 25 mL round bottom flask equipped with a condenser. A solution of 3-mercaptopropyltriethoxysilane (0.00081 mol, 0.158 g, Aldrich) in 5 mL of dry toluene with two drops of pyridine was added dropwise to the dispersed SBA-15 solution under continuous stirring. After complete addition of the organosilane precursor, the reaction mixture was kept under refluxing conditions for about 12 h under a nitrogen atmosphere. After completion of the reaction, the reaction mixture was allowed to cool at room temperature. It was filtered through suction and washed thoroughly with toluene (10 mL), dichloromethane (10 mL) and diethyl ether (10 mL), respectively. The white colored sample was dried at room temperature.
Synthesis of triazine functionalized SBA-15 (TFMO-1).
Triazine functionalized SBA-15 (TFMO-1) has been prepared via a thiol–ene click reaction of thiol functionalized SBA-15 with 2,4,6-triallyloxy-1,3,5-triazine in the presence of AIBN. A solution of 2,4,6-triallyloxy-1,3,5-triazine (0.067 g, 0.00027 mol) and AIBN (10 mg, 6.3 × 10−2 mmol) were added to the as-prepared thiol functionalized SBA-15 dispersed in 15 mL of chloroform solution at room temperature. The mixture was heated to reflux at 60 °C for 10 h. Then the mixture was allowed to cool at room temperature. The off-white colored material was collected by filtration after washing several times with CHCl3 and MeOH mixture. This functionalized mesoporous material has been designated as TFMO-1. Elemental analysis of the mesoporous material gave C (20.91%); H (2.51%); N (4.06%). The outline for the synthesis of the organocatalyst TFMO-1 is shown in Scheme 1 .
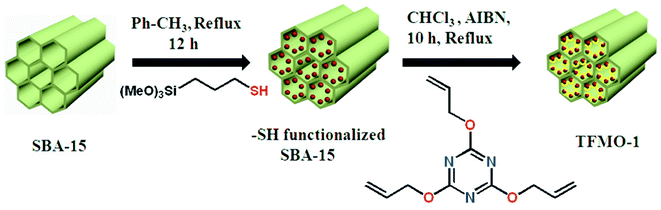 |
| | Scheme 1 Synthesis of triazine functionalized mesoporous organocatalyst TFMO-1 via thiol-ene click reaction. | |
General procedure for the synthesis of 2-amino-chromenes over TFMO-1.
A mixture of aromatic aldehyde 1 (1 mmol), malononitrile 2 (1 mmol, 66 mg), 1-naphthol 3 (1 mmol, 144 mg) and organocatalyst (0.04 g) was heated at 110 °C in an oil bath for an appropriate period of time under solvent-free conditions. After completion of the reaction (monitored by TLC), the reaction mixture was allowed to cool and 3 mL MeOH was poured into the reaction mixture. The resulting reaction mixture was stirred vigorously for about 2 h. The catalyst was separated from the reaction mixture by filtration and the catalyst was washed with acetone and ethyl acetate several times. The recovered catalyst was dried at 80 °C for 4 h and stored in a desiccator. Pure organic compound was afforded by evaporation of the solvent in a rotary evaporator and the residue was purified by crystallization using eco-compatible solvents (EtOH or EtOH/water) leading to 2-amino-chromene 4. The isolated pure product was characterized by 1H, 13C NMR. An outline of the condensation reaction over the organocatalyst is shown in Scheme 2.
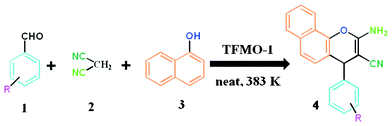 |
| | Scheme 2 General procedure for the synthesis of 2-amino-chromenes over TFMO-1. | |
Characterization techniques.
Powder X-ray diffraction patterns are recorded on a Bruker D-8 Advance SWAX diffractometer operated at a voltage of 40 kV and a current of 40 mA. The instrument was calibrated with a standard silicon sample, using Ni-filtered Cu-Kα (λ = 0.15406 nm) radiation. Nitrogen adsorption/desorption isotherms were obtained by using a Bel Japan Inc. Belsorp-HP at 77 K. Prior to gas adsorption, samples were degassed for 4 h at 393 K under high vacuum conditions. A JEOL JEM 6700F field emission scanning electron microscope was used for the determination of morphology of the particles. FT IR spectra of the samples were recorded using a Nicolet MAGNA-FT IR 750 Spectrometer Series II. High resolution transmittance electron microscopic (HR TEM) images were recorded in a JEOL JEM 2010 transmission electron microscope. Thermogravimetric analysis (TGA) was performed on a SBTQ600 Thermal Analyzer (USA) with a heating rate of 10 °C min−1 over a temperature range of 40–800 °C under flowing compressed N2 (100 mL min−1). 1H and 13C NMR (solution) experiments were carried out on a Bruker DPX-300 NMR spectrometer. The solid state MAS NMR spectra of the samples were taken in a Bruker MSL 500 spectrometer.
Results and discussion
The small angle powder X-ray diffraction (PXRD) pattern of the thiol-functionalized mesoporous SBA-15 is shown in the Fig. 1a. Three characteristic diffraction patterns in the 2θ region of 0.9 to 2.0 can be observed, which can be attributed to the 100 (strong), 110 (weak) and 200 (weak) reflections respectively, corresponding to the 2D-hexagonal mesostructure. When this thiol-functionalized mesoporous silica was subjected to the thiol–ene click reaction, for the synthesis of the triazine-functionalized organocatalyst, a considerable decrease in the intensity and a shift of the peaks are observed, however, the 2D-hexagonal ordering has been retained (Fig. 1b). The small shift in the XRD pattern of the triazine-functionalized organocatalyst could be attributed to the formation of a functional molecule layer via the click reaction of the surface –SH groups with the allyl groups.
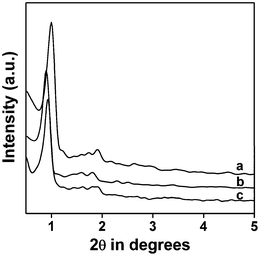 |
| | Fig. 1 Small angle powder XRD pattern of thiol-functionalized SBA-15 (a), triazine-functionalized organocatalyst TFMO-1 (b) and reused organocatalyst after the catalytic reaction (c). | |
N2 adsorption/desorption isotherms of the triazine-functionalized mesoporous organocatalyst at 77 K is shown in the Fig. 2. The sample shows a typical type IV isotherm with a very large hysteresis loop in the 0.5 to 0.8 P/P0 range. The isotherm accounts for the relatively ordered mesopores and the hysteresis loop, which is characteristic for the large tubular pores of SBA 15. The BET surface areas for the pure SBA-15 and TFMO-1 are 610 m2 g−1 and 405 m2 g−1 respectively. A considerable decrease in the BET surface area upon covalent grafting of the organic triazine groups through the thiol–ene click reaction at the surface of the thiol-functionalized SBA-15 material suggests that the organic groups have been anchored at the surface of the mesopores. The pore size distribution of the organocatalyst (TFMO-1) was calculated by employing non-local density functional theory (NLDFT), shown in the inset of (Fig. 2), which suggests a uniform distribution of large mesopores throughout the sample with pore dimensions of ca 6.7 nm. The calculated pore volume (0.517 ccg−1) and pore size of ca. 6.7 nm, for the organocatalyst (TFMO-1), is sufficient enough to allow the diffusion of large molecules, requiring only short reaction times for carrying out of the three-component condensation reactions. Further, the pore dimension for the SBA-15 material was 9.6 nm,16a which has been distinctly reduced to 6.7 nm after the thiol–ene click reaction. The considerable decrease in the pore dimension clearly suggests that the large triazine molecules have been anchored into the pore channels of the mesoporous SBA-15-SH material.
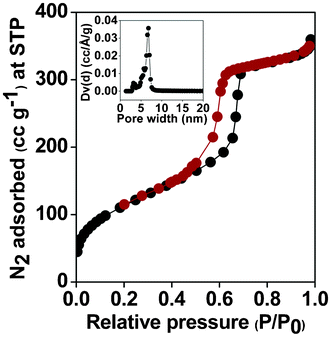 |
| | Fig. 2 N2 adsorption/desorption isotherm of the triazine-functionalized mesoporous organocatalyst (TFMO-1). Inset: Pore size distributions estimated through the NLDFT method. | |
The HR TEM image of mesoporous organocatalyst TFMO-1 (Fig. 3A) clearly suggests that uniformly ordered mesopores with dimensions of ca. 6.1–6.4 nm have been arranged in a honey-comb like hexagonal array throughout the sample. The FFT diffractogram shown in the inset of the Fig. 3A further suggests 2D-hexagonal pore channels. The TEM image (Fig. 3B, perpendicular to the pore axis) also illustrates that the channel directions are parallel to the thickness of the plate, and the 110 reflection plane of the sample is clearly viewed in this image. From these TEM images we can conclude that the mesoporous organocatalyst has ordered mesopores with a 2D-hexagonal structure, and that the hexagonally ordered mesoporous structure of SBA-15 is also preserved after functionalization.
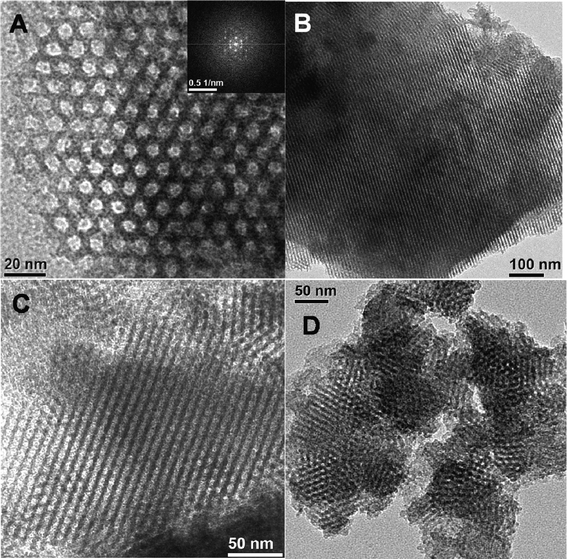 |
| | Fig. 3 HR TEM images (A, parallel to pore axis, FFT pattern is shown in the inset), (B, perpendicular to pore axis) of triazine functionalized mesoporous organocatalyst (TFMO-1). TEM images of the reused catalyst after the second (C) and sixth (D) catalytic cycles. | |
In Fig. 4, the FTIR spectra of the thiol-functionalized silica and TFMO-1 samples are shown. A weak absorbance at 1470 and 1420 cm−1 is observed with the introduction of 3-mercaptopropyltrimethoxysilane functionality in SBA-15. These peaks could be assigned to the bending vibrations of the methylene groups. Further, an absorbance in the range of 2860–2930 cm−1, corresponding to the methylene stretching vibrations of the propyl chain appeared, indicating the incorporation of the organic moiety in SBA-15. Before the thiol–ene click reaction, the –SH stretching is observed at 2567 cm−1 (inset of Fig. 4) with a very weak intensity. The disappearance of the –SH peak in TFMO-1 signifies that the –SH group takes part in the thiol–ene click reaction. In the FTIR spectrum of TFMO-1 (Fig. 4b) a strong band at 1570 cm−1 appears, which could be attributed to the ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration and an additional sharp band at 1415 cm−1 due to the stretching vibration of –CH2 group. Additionally, the characteristic mode around 814 cm−1 is further evidence of the presence of triazine units in this mesoporous organocatalyst. Further, the allylic double bond (–CC–) stretching of the 2,4,6-triallyloxy-1,3,5-triazine moiety at 1840 cm−1, has been disappeared (Fig. 4b), which clearly suggests that the CC bond of the triazine moiety undergoes the thiol–ene click reaction. The peak corresponding to the stretching vibration of C–N, at 1142 cm−1, cannot be observed due to the overlap with the absorbance of the Si–O–Si stretch in the 1000–1130 cm−1 range. In Fig. 5 the FE SEM image of the mesoporous TFMO-1 is shown. As seen from this image, very tiny particles with dimensions of ca. 40 nm have self-assembled to form uniform 3D-microspheres with diameters that vary from 1.84 μm to 1.98 μm. Microspheres are seen to adhere with each other at their surfaces. This could be due to the random thiol–ene click reaction of the allyl groups with the surface –SH groups of the material. During the initial period of this reaction, the triazine derivative is preferentially linked to the carbon nuclei due to its higher reactivity, implying that triallyloxy triazine acts as a cross-linker. The 3D-functional moiety of the triazine molecule could help the random thiol–ene click reaction in all directions leading to the 3D microsphere.
N stretching vibration and an additional sharp band at 1415 cm−1 due to the stretching vibration of –CH2 group. Additionally, the characteristic mode around 814 cm−1 is further evidence of the presence of triazine units in this mesoporous organocatalyst. Further, the allylic double bond (–CC–) stretching of the 2,4,6-triallyloxy-1,3,5-triazine moiety at 1840 cm−1, has been disappeared (Fig. 4b), which clearly suggests that the CC bond of the triazine moiety undergoes the thiol–ene click reaction. The peak corresponding to the stretching vibration of C–N, at 1142 cm−1, cannot be observed due to the overlap with the absorbance of the Si–O–Si stretch in the 1000–1130 cm−1 range. In Fig. 5 the FE SEM image of the mesoporous TFMO-1 is shown. As seen from this image, very tiny particles with dimensions of ca. 40 nm have self-assembled to form uniform 3D-microspheres with diameters that vary from 1.84 μm to 1.98 μm. Microspheres are seen to adhere with each other at their surfaces. This could be due to the random thiol–ene click reaction of the allyl groups with the surface –SH groups of the material. During the initial period of this reaction, the triazine derivative is preferentially linked to the carbon nuclei due to its higher reactivity, implying that triallyloxy triazine acts as a cross-linker. The 3D-functional moiety of the triazine molecule could help the random thiol–ene click reaction in all directions leading to the 3D microsphere.
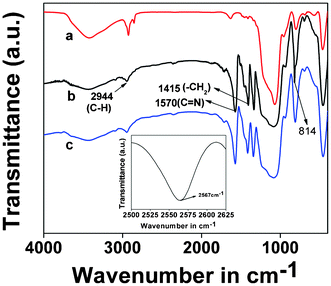 |
| | Fig. 4 FTIR spectra of the of the thiol-functionalized SBA-15 (a), triazine functionalized mesoporous organocatalyst TFMO-1 (b) and reused catalyst after the first cycle (c). Enlarged portion of thiol functionalized SBA-15 is shown in the inset. | |
 |
| | Fig. 5 FE SEM image of the mesoporous organocatalyst TFMO-1. | |
13C and 29Si MAS NMR results provide useful information regarding the chemical environment and the presence of an organic functional group in the organic–inorganic hybrid frameworks of the mesoporous organocatalyst. 29Si MAS NMR spectrum of TFMO-1 (Fig. 6A) exhibits two broad Tn and two Qn signals with the chemical shifts at –57.3, −67.3, −101.0 and −109.1 ppm, which could be attributed to the T2 [C–Si(OSi)2(OH)], T3 [(OSi)3Si–R], Q3 [Si(OSi)3(OH)] and Q4 [Si(OSi)4] species, respectively.16a The 13C CP MAS NMR spectrum (Fig. 6B) for the organocatalyst displays characteristic signals for the melamine bridging group at C1 (68.2 ppm), C2 (27.1 ppm), C3 (33.7 ppm), C4 (51.0 ppm), C5 (16.5 ppm) and C6 (10.5 ppm). A sharp signal with a maxima at 172.6 ppm can be attributed to the triazine-C of the melamine unit. The intensity of the sp2 hybridized carbon atoms of the allyl unit, which appear at 116 and 131 ppm, respectively, were substantially decreased (Fig. 6B). The decrease in peak intensities corresponding to the olefinic carbon atoms suggested that a predominant thiol–ene click reaction occurs with the surface –SH groups of the thiol-modified mesoporous silica. The radicals usually react with thiol groups in the thiol–ene reaction due to the difference in the kinetics between the thiol and alkene groups. It is possible that some amount of the unreacted alkene groups could remain at the surface of TFMO-1 due to incomplete thermal radical reaction in the presence of the initiator azobisisobutyronitrile (AIBN). These MAS NMR experimental results indicate that the functional group bearing the triazine moiety is covalently grafted inside the pore walls of the mesoporous catalyst. Elemental analysis of the organocatalyst gave C (20.91%); H (2.51%); N (4.06%). The organic group loading of the mesoporous organocatalyst (TFMO-1) is found to be 0.96 mmol g−1, as determined by the nitrogen content (4.06%) in the elemental analysis. Thermal stability of the triazine supported organocatalyst was investigated by using TGA analysis (ESI, Fig. S1†). The first one (ca. 5% weight loss) was found in the region 200 °C, which is attributed to the surface bound or the intercalated water absorbed in the internal pores or due to the loss of coordinated solvent molecules. The second weight loss (ca. 30%) for the organocatalyst in the temperature region between 200 °C and 400 °C corresponds to the decomposition of the organic fragments.
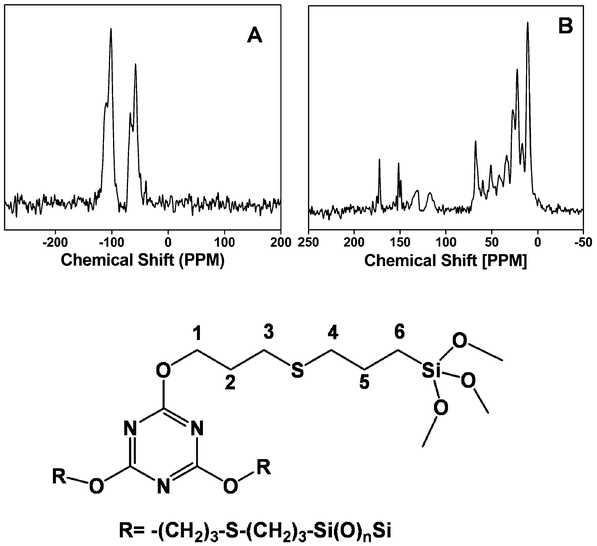 |
| | Fig. 6
29Si MAS NMR (A) and 13C MAS NMR (B) spectrum of the mesoporous organocatalyst TFMO-1. | |
Catalysis
Effect of catalyst dose.
Catalytic activity of the mesoporous organocatalyst TFMO-1 has been examined for the synthesis of a series of 2-amino-chromenes. It was found that the condensation reaction among aromatic aldehyde (1), malononitrile (2) and 1-naphthol (3) produces 2-amino-chromenes in good yields over the mesoporous organocatalyst TFMO-1 under solvent-free conditions (Scheme 2). All products were characterized by 1H and 13C NMR spectroscopy. To determine the best weight of the catalyst required for the condensation reaction, the synthesis was performed in the presence of varying amounts of catalyst (from 0.01 to 0.08 g) at 110 °C (Fig. 7). From Fig. 7, it has been found that 0.04 g of TFMO-1 is the optimum amount of catalyst required to carry out the reactions. Increasing the amount of catalyst does not improve the yield of the products any further, whereas decreasing the amount of catalyst leads to a decrease in the product yield. The optimal conditions for the 2-amino-chromene synthesis with a broad range of aromatic aldehydes and yield of the products are listed in the Table 2.34
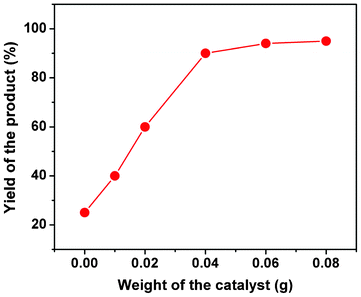 |
| | Fig. 7 Effect of the weight of organocatalyst for the three-component condensation reaction. | |
Table 1 Screening of different catalyst sources for the synthesis of 2-amino-4H-chromenesa
| Entry |
Catalyst |
Temperature (°C) |
Solvent |
Yield of 4cb (%) |
|
Reaction conditions: 4-chlorobenzaldehyde (1 mmol, 140 mg), 1-naphthol (1 mmol, 144 mg), malononitrile (1 mmol, 66 mg), solvent-free or solvent (CH3CN, 5 mL), catalyst (40 mg).
Isolated yield of pure product.
No reaction was observed, rt refers here to 25 °C.
|
| 1 |
No catalyst |
rt |
None |
NRc |
| 2 |
No catalyst |
rt |
CH3CN |
5 |
| 3 |
No catalyst |
40 |
CH3CN |
12 |
| 4 |
No catalyst |
70 |
None |
22 |
| 5 |
No catalyst |
110 |
None |
25 |
| 6 |
Piperidine |
70 |
None |
43 |
| 7 |
NEt3 |
70 |
None |
40 |
| 8 |
Pyridine |
70 |
None |
40 |
| 9 |
2,4,6-triallyloxy-1,3,5-triazine |
70 |
None |
58 |
| 10 |
Imidazole |
70 |
None |
52 |
| 11 |
MPTAT-1 |
110 |
None |
75 |
| 12 |
Mg-Al-hydrotalcite |
110 |
None |
65 |
| 13 |
SBA-15 |
110 |
None |
NRc |
| 14 |
SBA-15-SH |
110 |
None |
NRc |
| 15 |
TFMO-1 |
110 |
None |
90 |
| 16 |
2,4,6-triallyloxy-1,3,5-triazine |
110 |
None |
65 |
In order to confirm that the catalytic activity arises from the basic triazine moiety of the organocatalyst, a control experiment for the condensation reaction was performed by taking 4-chlorobenzaldehyde, malononitrile and 1-naphtol in the absence of any catalyst and solvent at room temperature (Table 1, entry 1). Under these conditions no reaction occurs, and the starting components remain unreacted. Then CH3CN was added into the reaction mixture and the mixture was allowed to stir at room temperature for 8 h, where the yield of the isolated product was about 5% (Table 1, entry 2). In order to improve the experimental results, the reaction was carried out at 40 °C without any catalyst in CH3CN. No appreciable increase in product yield was observed (Table 1, entry 3). The reaction was also performed under solvent-free conditions without any catalyst at 70 °C and 110 °C, providing yields of 22% and 25% respectively (Table 1, entries 4 and 5). It is found that the initial reaction starts but gives very poor yield to the final product (25%) at high temperatures. Triggered by these unsatisfactory results, we proceeded to perform the reaction by using a catalyst in order to increase the yield. Therefore, we surveyed some Lewis base catalysts (homogeneous and heterogeneous) for the condensation reaction to increase the product yield. The reaction was conducted at 70 °C under solvent-free conditions by employing several homogeneous Lewis base organocatalyst such as piperidine, triethylamine, pyridine and imidazole (Table 1, entries 6–8 and 10). The catalytic activity of the homogeneous catalyst 2,4,6-triallyloxy-1,3,5-triazine has been considered poorer than TFMO-1 (Table 1, entry 9). Very recently we have synthesized a mesoporous organic polymer MPTAT-1 bearing the backbone of the 2,4,6-triallyloxy-1,3,5-triazine fragment.35 When MPTAT-1 was used as a catalyst for this reaction, a 75% yield of the product 2-amino-chromene was obtained (Table 1, entry 11). The higher yield obtained by our TFMO-1 organocatalyst over MPTAT-1 could be attributed to the presence of sulfur atoms in the pore channels of the catalyst. Sulfur atoms increase the electrophilicity of the aldehyde group and also the cyanide group, which helps in the Michael addition and intra-molecular cyclization reaction, as a result, the condensation reaction could be more facile. Further, MPTAT-1 is not stable at higher temperatures, somehow the destruction of C–C bonds occur at high temperatures and thus some loss of the catalyst active sites occur during the course of the workup procedure. As a result, the TFMO-1 catalyst becomes more superior to the mesoporous polymer MPTAT-1. A control experiment over the well known solid base catalyst Mg-Al-hydrotalcite has been performed (Table 1, entry 12) for the condensation reaction. The yield of the condensation product 4c was 65%. However our TFMO-1 catalyst gives a 4c yield of 90%. When the mesoporous SBA-15 and SH-functionalized SBA-15 supported silica were used as an organocatalyst for the condensation reaction under solvent-free conditions no product was obtained (Table 1, entries 13 and 14). This data clearly signifies that the mesoporosity plays a significant role in this condensation reaction.
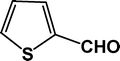
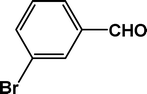
The condensation reaction for aldehydes with both electron-donating (Table 2, entries 4 and 5) and electron-withdrawing groups (Table 2, entries 1–3 and 7) proceeded smoothly with very good efficiency. Thus, the nature and position of the substitution in the aromatic ring did not have a great effect on the reactions. The meta-substituted aromatic aldehyde (Table 2, entry 7), as well as the α,β-unsaturated aromatic aldehyde (Table 2, entry 9), both undergo the condensation reaction without any difficulty. Several sensitive functional groups, such as –Cl, –Br, and –NO2, attached to the aromatic ring are also compatible in this reaction. A heterocyclic aldehyde, like thiophene-2-adehyde, participated in the condensation reaction with equal efficiency (Table 2, entry 6). The condensation reaction becomes quite facile for the bromo-substituted ortho-hydroxybenzaldehyde (Scheme 3), with a corresponding yield of 92%. The turn over number (TON) for different reactions varies from 24–22.6 for different aldehydes, suggesting high catalytic efficiency of TFMO-1 in these reactions. In contrast, the reaction at 298 K under identical conditions (Table 2, entry 10) was found to be very slow, yielding only a trace amount of the product, suggesting that the high temperature is more suitable for the one-pot three-component condensation reaction. In order to extend the scope of the reaction, the reaction was performed with various activated phenols (Table 3) using 4-chlorobenzaldehyde as the representative case for 4 h. It was found that 1-naphthol showed the best catalytic activity among the activated phenols in this condensation reaction. The condensation reaction was performed with different active methylene compounds, like malononitrile, ethyl cyanoacetate and diethyl malonate, under solvent-free conditions using 4-chlorobenzaldehyde as the representative aldehyde. However, only malononitrile showed a high product yield, suggesting that it was the best-suited active methylene compound for the synthesis of 2-amino-chromene derivatives over our mesoporous organocatalyst TFMO-1.
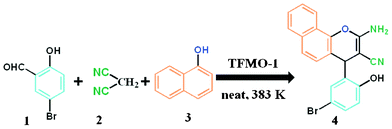 |
| | Scheme 3 Procedure for the synthesis of 2-amino-chromene of 5-bromosalicylaldehyde over TFMO-1. | |
Table 2 Synthesis of 2-amino-4H-chromenes over TFMO-1 under solvent-free conditionsa
| Entry |
Aromatic aldehyde |
Time (h) |
Yieldb (%) |
TONc |
Product |
|
Reaction conditions: aromatic aldehyde (1 mmol, 4-nitrobenzaldehyde, 151 mg, entry 1), 1-naphthol (1 mmol, 144 mg), malononitrile (1 mmol, 66 mg), solvent-free conditions, reaction temperature: 110 °C (oil bath), catalyst TFMO-1: 40 mg.
Isolated yield of the pure product.
Turn over number (TON) = moles of substrate converted per mole of active site.
Reaction conditions: 4-nitrobenzaldehyde (1 mmol, 151 mg), 1-naphthol (1 mmol, 144 mg), malononitrile (1 mmol, 66 mg), solvent-free conditions, reaction temperature: 25 °C (room temperature), catalyst TFMO-1: 40 mg.
|
| 1 |
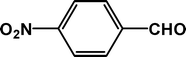
|
4 |
92 |
24.0 |
4a
|
| 2 |
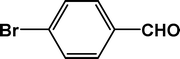
|
5 |
92 |
24.0 |
4b
|
| 3 |
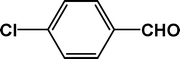
|
4 |
90 |
23.4 |
4c
|
| 4 |
|
4 |
90 |
23.4 |
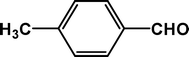
4d
|
| 5 |
|
6 |
88 |
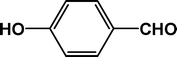
23.0 |
4e
|
| 6 |
|
5 |
86 |
22.3 |
4f
|
| 7 |
|
5 |
86 |
22.3 |
4g
|
| 8 |
|
6 |
88 |
23.0 |
4h
|
| 9 |
|
7 |
87 |
22.6 |
4i
|
| 10d |
|
10 |
— |
— |
4j
|
Table 3 Synthesis of 2-amino-4H-chromene derivatives catalyzed by TFMO-1a
| Activated phenol |
Reaction time (h) |
Yieldb (%) |
|
Reaction conditions: 4-chlorobenzaldehyde (1 mmol, 140 mg) activated phenolic compounds (1-naphthol,1 mmol, 144 mg), malononitrile (1 mmol, 66 mg), solvent-free, reaction temperature: 110 °C (oil bath), catalyst TFMO-1: 40 mg.
Isolated yield of pure product.
Reaction conditions: 4-chlorobenzaldehyde (1 mmol, 140 mg) 2-naphthol (1 mmol, 144 mg), malononitrile (1 mmol, 66 mg), solvent free, reaction temperature: 110 °C (oil bath), catalyst TFMO-1: 40 mg and complete consumption of reactants not observed on TLC for this reaction.
|
| 1-Naphthol |
4 |
90 |
| 2-Naphtholc |
4 |
45 |
| Resorcinol |
4 |
64 |
Effect of reaction temperature.
The temperature played a crucial role in determining the activity of the organocatalyst for the synthesis of the chromene derivatives. The effect of reaction temperature on the catalyst TFMO-1 was investigated by performing the condensation of 4-chlorobenzaldehyde, malononitrile and 1-naphtol under solvent-free conditions at different temperatures (Fig. 8). When the reaction was performed at room temperature (25 °C) under solvent-free conditions, only a trace amount of product (5%) was obtained. As the temperature of the reaction was increased to 40 °C, the product yield increased to 30%. As the product yield increases with the increase of temperature, we further performed the reaction at three other higher temperatures like 60 °C, 80 °C and 110 °C. From Fig. 8 it is clear that the temperature of 110 °C is the optimum temperature for completion of this reaction (Table 1, entry 15, yield 90%). Beyond 110 °C, no increase in product yield was observed. In order to compare the catalytic activity of the homogeneous phase and heterogeneous phase at the same reaction temperature and to clarify the role of the mesoporous organosilica we have performed the catalytic reaction of 4-chlorobenzaldehyde, 1-naphthol and malononitrile in the presence of 2,4,6-triallyloxy-1,3,5-triazine as the homogeneous phase at 110 °C under solvent-free conditions (Table 1, entry 16, yield 65%). This data clearly suggests that the mesoporosity plays a crucial role in this condensation reaction. Sulfur atoms are grafted with the mesoporous silica via the thiol–ene click reaction. These sulfur atoms increase the electrophilicity of the –CHO and –CN groups, which promotes the Michael addition and intra-molecular cyclization reaction. With the increase of electrophilicity of the –CN groups, the addition reaction as well as the nucleophilic attack by the phenoxide groups, become quite facile to provide the 2-amino chromene derivatives. These functionalities, as well as the surface area are absent in the homogeneous catalyst of 2,4,6-triallyloxy-1,3,5-triazine. Thus, the yield of the condensation product over our mesoporous organocatalyst TFMO-1 is much higher than the homogeneous 2,4,6-triallyloxy-1,3,5-triazine form.
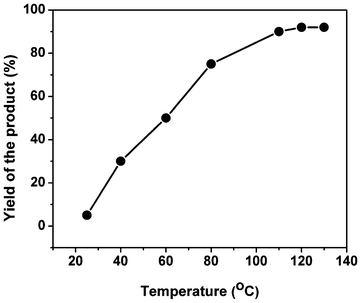 |
| | Fig. 8 Effect of reaction temperature for the three-component condensation reaction. | |
Reusability of the catalyst.
In order to establish the recyclability of the catalyst (TFMO-1), the condensation reaction under similar conditions, between 4-chlorobenzaldehyde, malononitrile and 1-naphtol, was taken as the representative case. After the reaction was over, the catalyst was separated from the reaction mixture by filtration, washed with dichloromethane followed by diethyl ether. Then the recovered catalyst was dried at room temperature for 6 h and used for a further six additional reaction cycles. In all cases, the silica-supported organocatalyst TFMO-1 exhibited consistent catalytic activity, establishing the recycling and reusability of the catalyst without any significant loss of catalytic activity (Table 4). Only a very small drop in the product yield in each catalytic cycle is observed. It is clear from Table 4 that the TON of the catalyst is retained from fresh to the sixth reaction cycle, suggesting the high catalytic efficiency of TFMO-1.
Table 4 Recycling potential of the mesoporous organocatalyst (TFMO-1)
| No. of cyclesa |
Fresh |
Run 1 |
Run 2 |
Run 3 |
Run 4 |
Run 5 |
Run 6 |
|
Reaction conditions: 4-chlorobenzaldehyde (1 mmol, 140 mg) 1-naphthol (1 mmol, 144 mg), malononitrile (1 mmol, 66 mg), solvent-free, reaction temperature: 110 °C (oil bath), catalyst TFMO-1: 40 mg.
Isolated yield of pure product.
Turn over number (TON) = moles of substrate converted per mole of active site.
|
| Yieldb(%) |
90 |
90 |
90 |
88 |
88 |
85 |
84 |
| Time (h) |
4 |
4 |
4 |
4 |
4 |
4 |
4 |
| TONc |
23.4 |
23.4 |
23.4 |
23.0 |
23.0 |
22.1 |
21.8 |
Leaching test.
Furthermore, the leaching of the organic groups from the catalyst support can be checked by performing an in situ filtration technique. In such a technique, the supported catalyst TFMO-1 was stirred at room temperature with dichloromethane for 24 h. The catalyst was then separated by filtration, and the condensation reaction of 4-chlorobenzaldehyde, malononitrile and 1-naphtol was conducted with the filtrate for 24 h at room temperature. No yield of the product 2-amino-chromene has been observed, establishing the fact that no leaching of the organic group occurs during the course of the reaction. This result confirms the heterogeneous nature of the mesoporous organocatalyst and the organic moiety was covalently anchored inside the pore wall of the catalyst. The absence of the organic triazine moiety in the filtrate was further confirmed by investigating the UV-vis spectra of the filtrate. The UV-vis spectra did not show any absorption peaks corresponding to the triazine moiety, indicating that no leaching of the active component 2,4,6-triallyloxy-1,3,5-triazine from the backbone of mesoporous silica occurs during the course of the reaction. Further, to confirm clearly that no leaching occurs during the course of the reaction, the leaching test was also performed using the standard reaction (hot filtration conditions) of 4-chlorobenaldehyde, malononitrile and 1-naphthol under optimized reaction conditions at 110 °C. After 2 h of reaction (as this reaction takes 4 h to complete, so 2 h is the mid-way of the reaction) the reaction was stopped. Then the reaction mixture was cooled and stirred with 2 mL of MeOH for 6 h at room temperature. Then the solid catalyst was filtered from the mixture. MeOH was evaporated to dryness to provide the crude product with 50.4% yield. The NMR data suggests that the reactants and product are also present in the mixture and product yield is 50%, however, the NMR peaks corresponding to the triazine moiety were absent. Then this crude product (product with the reactant mixture) was allowed to heat at 110 °C for another 2 h, after which the reaction mixture was worked-up as described previously. Absolutely no increase in the product yield in the reaction mixture was observed (yield 50.2%). If the triazine moiety, which is responsible for the catalysis, had leached from the silica backbone unit and was present in the reaction mixture (as the triazine moiety is soluble in MeOH), it would have facilitated the reaction further, improving the product yield. The UV-visible spectrum of the filtrate did not show bands corresponding to the triazine moiety. Thus, we can conclude that the triazine moiety has been covalently grafted with the silica framework and no leaching of the organic fragments occur during the course of the reaction under the optimized reaction conditions.
Characterization of the reused catalyst.
The reused organocatalyst was further characterized by small angle powder XRD, TEM, FT-IR studies in order to clarify if any change occur in the catalyst after catalysis. In the small angle powder XRD pattern of the reused organocatalyst (Fig. 1c) characteristic mesophase was obviously observed, which suggests that the hexagonal mesostructure of the reused catalyst are well preserved during the course of the catalytic reaction. FT-IR studies of the reused catalyst after the sixth catalytic cycle (Fig. 4c) were recorded, where the two strong absorption bands at 1570 cm−1 and 1415 cm−1, corresponding to the CN stretching and the stretching vibration of the –CH2 groups, were obviously observed. The IR spectra of the reused catalyst reveals that no obvious change occurs in the structure of the organocatalyst. The TEM images of the reused organocatalyst after the second (Fig. 3C) and sixth (Fig. 3D) catalytic cycle suggests that the ordered mesostructure of the reused catalyst remains unaffected after the reaction. The above results revealed that the mesoporous organocatalyst was very stable during the catalysis and could withstand these conditions.
Plausible reaction pathway.
The possible mechanistic pathway for the one-pot three-component condensation reaction over TFMO-1, containing basic sites, is shown in the Fig. 9. A Knoevenagel reaction, Michael addition and intra-molecular cyclization are involved simultaneously in the synthesis of 2-amino-4H-chromene derivatives.36 The one-pot three-component condensation reaction could proceed as follows. In the first step, the aromatic aldehyde undergoes a Knoevenagel condensation reaction with malononitrile to afford α-cyanocinnamonitrile derivative (A). The 1-naphthol endures a Michael addition reaction with the A to afford the product (B). Here, the sulfur atoms of the catalyst increases the electrophilicity of the –CN group followed by abstraction of the proton from the –OH group by the basic N-sites of the catalyst. The Michael addition product (B) undergoes intra-molecular cyclization through nucleophilic attack of the phenoxide group on the cyano moiety and is subsequently tautomerized to yield the 2-amino-chromene derivatives (C).37
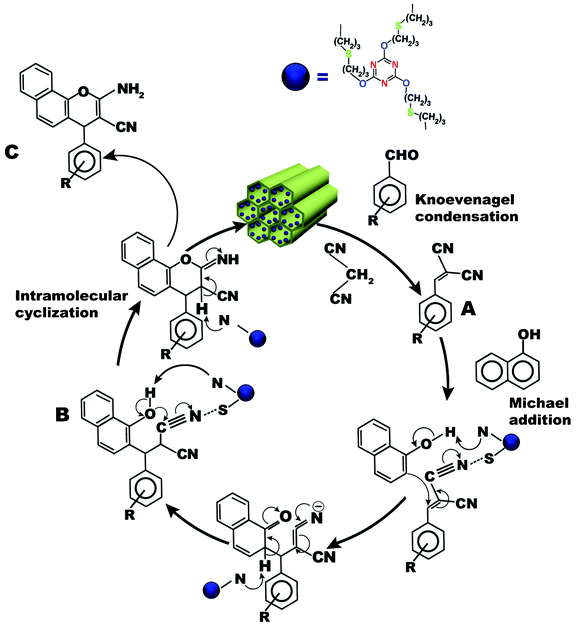 |
| | Fig. 9 Plausible reaction pathway for condensation reaction of aromatic aldehyde, 1-naphthol and malononitrile in the presence of TFMO-1. | |
Conclusion
In conclusion, we have developed a novel metal-free mesoporous organocatalyst possessing a highly ordered 2D-hexagonal nanostructure via the thiol–ene click reaction of 3-mercaptopropyl grafted SBA-15 with 2,4,6-triallyloxy-1,3,5-triazine. This mesoporous organocatalyst can be used for the one-pot three-component condensation reaction of aromatic aldehyde, malononitrile and 1-naphthol under solvent-free conditions for the synthesis of 2-amino-4H-chromenes. The organic 2-amino-4H-chromene derivative was isolated from the mixture by using the eco-compatible and easily disposable solvent MeOH, and was then reused several times without any significant loss of catalytic activity. An efficient organocatalytic pathway is described herein for the synthesis of 2-amino-4H-chromene under solvent-free conditions, which may contribute significantly to designing value added therapeutic molecules.
Acknowledgements
JM and AM thank the CSIR, New Delhi for their respective senior research fellowships. This work is partly funded by the DST Unit on Nanoscience.
References
-
(a) B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS;
(b) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS;
(c) R. Sebesta, I. Kmentova and S. Toma, Green Chem., 2008, 10, 484–496 RSC.
-
(a) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS;
(b) A. M. Marcet, X. Cattoën, D. A. Alonso, C. Nájera, M. W. C. Man and R. Pleixats, Green Chem., 2012, 14, 1601–1610 RSC.
- B. Das, B. Ravikanth, R. Ramu, K. Laxminarayana and B. V. Rao, J. Mol. Catal. A: Chem., 2006, 255, 74–77 CrossRef CAS.
-
(a) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3691 CrossRef CAS;
(b) A. Corma, Nature, 2009, 461, 182–183 CrossRef CAS;
(c) R. E. Galian and J. Perez-Prieto, Energy Environ. Sci., 2010, 3, 1488–1498 RSC;
(d) Y. Wang, X. Wang and M. Antonietti, Angew. Chem. Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS.
-
(a) T. Ishida and M. Haruta, Angew. Chem. Int. Ed., 2007, 46, 7154–7156 CrossRef CAS;
(b) X. Huang, X. M. Liu, Q. Luo, J. Q. Liu and J. C. Shen, Chem. Soc. Rev., 2011, 40, 1171–1184 RSC.
-
(a) T. Ray, S. F. Mapolie and J. Darkwa, J. Mol. Catal. A: Chem., 2007, 267, 143–148 CrossRef;
(b) T. Luts, W. Suprun, D. Hofmann, O. Klepel and H. Papp, J. Mol. Catal. A: Chem., 2007, 261, 16–23 CrossRef CAS.
-
(a) D. Font, C. Jimeno and M. A. Pericàs, Org. Lett., 2006, 8, 4653–4655 CrossRef CAS;
(b) M. Gruttadauria, A. M. P. Salvo, F. Giacalone, P. Agrigento and R. Noto, Eur. J. Org. Chem., 2009, 5437–5444 CrossRef CAS;
(c) A. Bañón-Caballero, G. Guillena and C. Nájera, Green Chem., 2010, 12, 1599–1606 RSC.
-
(a) A. Fuerte, A. Corma and F. Sánchez, Catal. Today, 2005, 107–108, 404–409 CrossRef CAS;
(b) E. G. Doyagüez, F. Calderón, F. Sánchez and A. Fernández-Mayoralas, J. Org. Chem., 2007, 72, 9353–9356 CrossRef;
(c) E. A. Prasetyanto, S. C. Lee, S. M. Jeong and S. E. Park, Chem. Commun., 2008, 1995–1997 RSC;
(d) J. Gao, J. Liu, D. Jiang, B. Xiao and Q. Yang, J. Mol. Catal. A: Chem., 2009, 313, 79–87 CrossRef CAS.
- D. Kuehbeck, G. Saidulu, K. R. Reddy and D. D. Diaz, Green Chem., 2012, 14, 378–392 RSC.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
-
(a) M. Vallet-Regi, Dalton Trans., 2006, 5211–5220 RSC;
(b) A. M. Sauer, A. Schlossbauer, N. Ruthardt, V. Cauda, T. Bein and C. Braeuchle, Nano Lett., 2010, 10, 3684–3691 CrossRef CAS;
(c) Y. S. Lin, N. Abadeer, K. R. Hurley and C. L. Haynes, J. Am. Chem. Soc., 2011, 133, 20444–20457 CrossRef CAS;
(d) S. K. Das, M. K. Bhunia, D. Chakraborty, A. R. Khuda-Bukhsh and A. Bhaumik, Chem. Commun., 2012, 48, 2891–2893 RSC.
-
(a) S. S. Park, J. H. Shin, D. Y. Zhao and C. S. Ha, J. Mater. Chem., 2010, 20, 7854–7858 RSC;
(b) Z. Zhu, X. Yang, L.-N. He and W. Li, RSC Adv., 2012, 2, 1088–1095 RSC.
-
(a) A. Wada, S. Tamaru and M. Ikeda, J. Am. Chem. Soc., 2009, 131, 5321–5330 CrossRef CAS;
(b) K. Sarkar, K. Dhara, M. Nandi, P. Roy, A. Bhaumik and P. Banerjee, Adv. Funct. Mater., 2009, 19, 223–234 CrossRef CAS.
-
(a) Y. Kubota, H. Yamaguchi, T. Yamada, S. Inagaki, Y. Sugi and T. Tatsumi, Top. Catal., 2010, 3, 492–499 CrossRef;
(b) S. Verma, M. Nandi, A. Modak, S. L. Jain and A. Bhaumik, Adv. Synth. Catal., 2011, 353, 1897–1902 CrossRef CAS;
(c) J. Mondal, A. Modak, A. Dutta and A. Bhaumik, Dalton Trans., 2011, 40, 5228–5235 RSC;
(d) J. G. Hu, Q. Y. Wu, W. Li, L. Ma, F. Su, Y. H. Guo and Y. Q. Qiu, ChemSusChem, 2011, 4, 1813–1822 CrossRef CAS;
(e) N. Salam, P. Mondal, J. Mondal, A. S. Roy, A. Bhaumik and S. M. Islam, RSC Adv., 2012, 2, 6464–6477 RSC.
-
(a) M. Nandi, J. Mondal, K. Sarkar, Y. Yamauchi and A. Bhaumik, Chem. Commun., 2011, 47, 6677–6679 RSC;
(b) S. L. Jain, A. Modak and A. Bhaumik, Green Chem., 2011, 13, 586–590 RSC.
-
(a) S. Fujita and S. Inagaki, Chem. Mater., 2008, 20, 891–908 CrossRef CAS;
(b) J. Y. Shi, C. A. Wang, Z. J. Li, Q. Wang, Y. Zhang and W. Wang, Chem. Eur. J., 2011, 17, 6206–6213 CrossRef CAS;
(c) Q. Chen, C. Xin, L. L. Lou, K. Yu, F. Ding and S. G. Liu, Catal. Lett., 2011, 141, 1378–1383 CrossRef CAS.
- M. M. Khafagy, A. H. F. A. El-Wahas, F. A. Eid and A. M. El-Agrody, Farmaco, 2002, 57, 715–722 CrossRef CAS.
-
(a) A. G. Martinez and L. J. Marco, Bioorg. Med. Chem. Lett., 1997, 7, 3165–3170 CrossRef;
(b) W. P. Smith, L. S. Sollis, D. P. Howes, C. P. Cherry, D. I. Starkey and N. K. Cobley, J. Med. Chem., 1998, 41, 787–797 CrossRef.
- K. Hiramoto, A. Nasuhara, K. Michiloshi, T. Kato and K. Kikugawa, Mutat. Res., 1997, 395, 47–56 CrossRef CAS.
- C. P. Dell and C. W. Smith, European Patent Appl., EP
537949, Chem. Abstr., 1993, 119, 139102d Search PubMed.
-
(a) S. J. Mohr, M. A. Chirigos, F. S. Fuhrman and J. W. Pryor, Cancer Res., 1975, 35, 3750–3754 CAS;
(b) F. Eiden and F. Denk, Arch. Pharm. Weinhein Ger (Arch. Pharm.), 1991, 324, 353 CAS.
-
(a)
G. P. Ellis, A. In Weissberger and E. C. Taylor ed. in Chemistry of Heterocyclic Compounds Chromenes, Chromanes and Chromones; John Wiley: New York, 1977; Chapter II, pp 11–139 Search PubMed;
(b) E. A. Hafez, M. H. Elnagdi, A. G. A. Elagemey and F. M. A. A. El-Taweel, Heterocycles, 1987, 26, 903 CrossRef CAS.
-
(a) A. G. A. Elagemey, F. M. A. A. El-Taweel, M. N. M. Khodeir and M. H. Elnagdi, Bull. Chem. Soc. Jpn., 1993, 66, 464–468 CrossRef;
(b) J. Bloxham, C. P. Dell and C. W. Smith, Heterocycles, 1994, 38, 399–408 CrossRef CAS.
- A.-Q. Zhang, M. Zhang, H.-H. Chen, J. Chen and H.-Y. Chen, Synth. Commun., 2007, 37, 231–235 CrossRef CAS.
- M. Kidwai, S. Saxena, M. K. Rahman Khan and S. S. Thukral, Bioorg. Med. Chem. Lett., 2005, 15, 4295–4298 CrossRef CAS.
- Y. Ren and C. Cai, Catal. Commun., 2008, 9, 1017–1020 CrossRef CAS.
- B. S. Kumar, N. Srinivasulu, R. H. Udupi, B. Rajitha, Y. T. Reddy, P. N. Reddy and P. S. Kumar, J. Heterocycl. Chem., 2006, 43, 1691–1693 CrossRef CAS.
- G. Shanthi and P. T. Perumal, Tetrahedron Lett., 2007, 48, 6785–6789 CrossRef CAS.
- M. M. Heravi, K. Bakhtiari, V. Zadsirjan, F. F. Bamoharram and O. M. Heravi, Bioorg. Med. Chem. Lett., 2007, 17, 4262–4265 CrossRef CAS.
- A. Shaabani, R. Ghadari, S. Ghasemi, M. Pedarpour, A. H. Rezayan, A. Sarvary and S. Weng Ng, J. Comb. Chem., 2009, 11, 956–959 CrossRef CAS.
- S. Makarem, A. A. Mohammadi and A. R. Fakhari, Tetrahedron Lett., 2008, 49, 7194–7196 CrossRef CAS.
-
(a) D. Kumar, V. B. Reddy, B. G. Mishra, R. K. Rana, M. N. Nadagaouda and R. S. Varma, Tetrahedron, 2007, 63, 3093–3097 CrossRef CAS;
(b) M. P. Surpur, S. Kshirsagar and S. D. Samant, Tetrahedron Lett., 2009, 50, 719–722 CrossRef CAS;
(c) A. Solhy, A. Elmakssoudi, R. Tahir, M. Karkouri, M. Larzek, M. Bousmina and M. Zahouily, Green Chem., 2010, 12, 2261–2267 RSC.
-
1H and 13C NMR chemical shifts for different 2-amino chromene products reported in Table 2. 2-Amino-3-cyano-4-(4-nitrophenyl)-4H-benzo[h]-chromene (Table 2, entry 1), (4a), Yellow solid:1H NMR (300 MHz, DMSO-d6) δ 5.13 (s, 1H), 7.08–7.10 (d, 1H, J = 6 Hz), 7.31 (s, 2H), 7.51 (d, 2H), 7.54–7.79 (3H, m), 7.87–7.90 (1H, d, J = 9 Hz), 8.17–8.19 (2H, d, J = 6 Hz), 8.24 (1H, d); 13C NMR (300 MHz, DMSO-d6) δ 55.2 (s, C-4), 68.1 (s, C-3), 116.5 (s, CN), 120.2 (s, C-4a), 120.8 (s, C-10), 122.7 (s, C-10a), 124.5 (s, C-3′, 5′), 125.50 (s, C-6), 125.9 (s, C-5), 126.7 (s, C-8), 127.7 (s, C-9), 128.9 (s, C-7), 132.2 (s, C-2′, 6′), 132.9 (s, C-6a), 134.4 (s, C-4′), 146.4 (s, C-1′), 152.9 (C-10b), 160.3 (C-2). 2-Amino-3-cyano-4-(4-bromophenyl)-4H-benzo[h]-chromene (Table 2, entry 2), (4b), Yellow solid:1H NMR (300 MHz, DMSO-d6) δ 5.20 (s, 1H), 7.09–7.14 (d, 1H, J = 15 Hz), 7.34 (s, 2H), 7.51 (d, 2H), 7.53–7.79 (3H, m), 7.79–7.82 (2H, d, J = 9 Hz), 8.07–8.12 (1H, d, J = 15 Hz), 8.13–8.19 (1H, d, J = 18 Hz); 13C NMR (300 MHz, DMSO-d6) δ 55.8 (s, C-4), 68.1 (s, C-3), 117.2 (s, CN), 120.2 (s, C-4a), 120.6 (s, C-10), 122.7 (s, C-10a), 124.5 (s, C-3′, 5′), 125.1 (s, C-6), 126.0 (s, C-5), 126.8 (s, C-8), 127.6 (s, C-9), 129.9 (s, C-7), 132.2 (s, C-2′, 6′), 132.7 (s, C-6a), 134.3 (s, C-4′), 145.0 (s, C-1′), 153.0 (C-10b), 160.1 (C-2). 2-Amino-3-cyano-4-(4-chlorophenyl)-4H-benzo[h]-chromene (Table 2, entry 3), (4c), Yellow
solid:1H NMR (300 MHz, DMSO-d6) δ 4.95 (s, 1H), 7.12 (d, 1H), 7.22 (s, 2H), 7.28-7.31 (d, 2H, J = 9 Hz), 7.35–7.42 (d, 2H, J = 21 Hz), 7.56–7.65 (m, 3H), 7.88–7.90 (d, 1H, J = 6 Hz), 8.23–8.24 (d, 1H, J = 3 Hz); 13C NMR (300 MHz, DMSO-d6) δ 55.8 (s, C-4), 68.1 (s, C-3), 117.3 (s, CN), 120.2 (s, C-4a), 120.6 (s, C-10), 122.7 (s, C-10a), 123.9 (s, C-3′, 5′), 126.0 (s, C-6), 126.6 (s, C-5), 126.8 (s, C-8), 127.6 (s, C-9), 128.6 (s, C-7), 129.5 (s, C-2′, 6′), 131.4 (s, C-6a), 132.7 (s, C-4′), 142.7 (s, C-1′), 144.5 (C-10b), 160.1 (C-2). 2-Amino-3-cyano-4-(4-methylphenyl)-4H-benzo[h]-chromene (Table 2, entry 4), (4d), Yellow solid:1H NMR (300 MHz, DMSO-d6) δ 2.29 (S, 3H), 4.84 (s, 1H), 6.48–6.57 (d, 1H, J = 27 Hz), 7.03–7.06 (d, 1H, J = 9 Hz), 7.14 (s, 2H), 7.17–7.23 (d, 2H, J = 18 Hz), 7.35–7.46 (d, 2H), 7.54–7.64 (m, 2H), 7.86–7.88 (d, 1H, J = 6 Hz), 8.20–8.24 (d, 1H, J = 12 Hz); 13C NMR (300 MHz, DMSO-d6) δ 21.2 (–CH3), 41.1 (s, C-4), 57.0 (s, C-3), 118.7 (s, CN), 121.1 (s, C-4a), 121.3 (s, C-10), 123.3 (s, C-10a), 124.4 (s, C-3′, 5′), 128.8 (s, C-6), 127.0 (s, C-5), 127.2 (s, C-8), 127.7 (s, C-9), 128.8 (s, C-7), 131.3 (s, C-2′, 6′), 133.2 (s, C-6a), 136.6 (s, C-4′), 144.5 (s, C-1′), 157.4 (C-10b), 160.3 (C-2). 2-Amino-3-cyano-4-(4-hydroxyphenyl)-4H-benzo[h]-chromene (Table 2, entry 5), (4e), Yellow solid:1H NMR (300 MHz, DMSO-d6) δ 4.87 (s, 1H), 7.07–7.13 (m, 4H), 7.43–7.46 (d, 2H, J = 9 Hz), 7.55–7.59 (m, 2H), 7.61–7.63 (d, 1H, J = 6 Hz), 7.85–7.88 (d, 2H, J = 9 Hz), 8.22–8.24 (d, 1H, J = 6 Hz), 8.47 (s, 1H); 13C NMR (300 MHz, DMSO-d6) δ 57.0 (s, C-4), 68.7 (s, C-3), 118.6 (s, CN), 121.0 (s, C-4a), 121.3 (s, C-10), 122.5 (s, C-10a), 124.4 (s, C-3′, 5′), 125.1 (s, C-6), 126.6 (s, C-5), 126.9 (s, C-8), 127.9 (s, C-9), 129.3 (s, C-7), 131.2 (s, C-2′, 6′), 132.9 (s, C-6a), 136.6 (s, C-4′), 143.3 (s, C-1′), 146.2 (C-10b), 160.9 (C-2). 2-Amino-3-cyano-4-(thiophenyl)-4H-benzo[h]-chromene (Table 2, entry 6), (4f), Yellow solid:1H NMR (300 MHz, DMSO-d6) δ 4.78 (s, 1H), 6.85–6.84 (d, 1H, J = 3 Hz), 7.09 (s, 2H), 7.41–7.38 (m, 3H), 7.96–7.94 (d, 3H, J = 6 Hz), 8.31–8.29 (d, 2H, J = 6 Hz). 2-Amino-3-cyano-4-(3-bromophenyl)-4H-benzo[h]-chromene (Table 2, entry 7), (4g), Yellow solid:1H NMR (300 MHz, DMSO-d6) δ 4.95 (s, 1H), 7.10–7.30 (m, 4H), 7.35–7.52 (m, 6H), 7.57–7.61 (d, 1H, J = 12 Hz), 8.22–8.26 (d, 2H, J = 12 Hz); 13C NMR (300 MHz, DMSO-d6) δ 41.6 (s, C-4), 55.6 (s, C-3), 117.2 (s, CN), 120.7 (s, C-4a), 121.9 (s, C-10), 122.9 (s, C-9), 124.1 (s, C-6), 126.0 (s, C-10a), 126.7 (s, C-2′), 126.9 (s, C-4′), 127.7 (s, C-7), 128.2 (s, C-5), 129.9 (s, C-8), 130.2 (s, C-6′), 131.0 (C-6a), 132.7 (s, C-5′), 142.7 (C-1′), 148.4 (s, C-3′), 149.0 (s, C-10b), 160.2 (C-2). 2-Amino-3-cyano-4-(2-nitrophenyl)-4H-benzo[h]-chromene (Table 2, entry 8), (4h), Pale yellow solid: 1H NMR (500 MHz, DMSO-d6) δ 4.94 (s, 1H), 7.09 (d, 1H, J = 7.9 Hz), 7.19 (s, 2H), 7.21–7.23 (d, 2H, J = 7.6 Hz), 7.51 (d, 2H, J = 7.33 Hz), 7.59–7.65 (m, 3H), 7.89 (d, 1H, J = 7.7 Hz), 8.23 (d, 1H, J = 7.9 Hz); 13C NMR (500 MHz, DMSO-d6) δ 56.6, 118.1, 120.9, 121.2, 121.5, 123.59, 124.8, 126.9, 127.6, 127.7, 128.5, 130.8, 132.4, 133.6, 143.6, 145.9, 161.0. 2-Amino-3-cyano-4-(cinnamyl)-4H-benzo[h]-chromene (Table 2, entry 9), (4i), Yellow solid:1H NMR (500 MHz, DMSO-d6) δ 7.24–7.31 (m, 3H), 7.42–7.50 (m, 6H), 7.59–7.62 (d, 2H, J = 9 Hz), 7.75–7.80 (d, 4H, J = 15 Hz), 8.27–8.30 (d, 2H, J = 9 Hz); 13C NMR (500 MHz, DMSO-d6) δ 80.9, 108.0, 112.1, 114.1, 118.2, 121.9, 122.5, 124.4, 126.0, 126.6, 127.3, 12.1, 129.2, 131.8, 134.1, 151.1, 162.0. 2-Amino-3-cyano-4-(2-hydroxy-5-bromophenyl)-4H-benzo[h]-chromene (Scheme-3), Yellow solid:1H NMR (500 MHz, DMSO-d6) δ 5.19 (s, 1H), 6.79–6.78 (d, 1H, J = 5 Hz), 7.11–7.22 (dt, 5H), 7.55–7.64 (m, 3H), 7.89–7.87 (d, 1H, J = 10 Hz), 8.23–8.22 (d, 1H, J = 5 Hz), 10.004 (s, 1H); 13C NMR (500 MHz, DMSO-d6) δ 54.6 (s, C-4), 64.9 (s, C-3), 117.5 (s, CN), 117.8 (s, C-3′), 120.4 (s, C-4a), 120.6 (s, C-6), 122.6 (s, C-10), 123.7 (s, C-1′), 125.6 (s, C-8, 5), 126.5 (s, C-9), 126.6 (s, C-10a), 127.5 (s, C-7), 130.5 (s, C-4′), 131.3 (s, C-5′), 132.6 (s, C-6a), 134.1 (s, C-6′), 142.8 (s, C-10b), 154.0 (s, C-2′), 160.7 (C-.
- A. Modak, J. Mondal, M. Sasidharan and A. Bhaumik, Green Chem., 2011, 13, 1317–1331 RSC.
-
(a) D. B. Ramachary, M. Kishor and Y. V. Reddy, Eur. J. Org. Chem., 2008, 975–993 CrossRef CAS;
(b) Q. Ren, W. Y. Siau, Z. Y. Du, K. Zhang and J. Wang, Chem. Eur. J., 2011, 17, 7781–7785 CrossRef CAS.
-
(a) R. Ballini, G. Bosica, M. L. Conforti, R. Maggi, A. Mazzacani, P. Righi and G. Sartori, Tetrahedron, 2001, 57, 1395–1398 CrossRef CAS;
(b) M. M. Heravi, B. Baghernejad and H. A. Oskooie, J. Chin. Chem. Soc., 2008, 55, 659–662 CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra22291d |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.