The protein corona of dendrimers: PAMAM binds and activates complement proteins in human plasma in a generation dependent manner†
Anna
Åkesson
a,
Marité
Cárdenas
a,
Giuliano
Elia
b,
Marco P.
Monopoli
*bc and
Kenneth A.
Dawson
*c
aInstitute of Chemistry and Nano-Science Center, Copenhagen University, Copenhagen, DK 2100, Denmark
bMass Spectrometry Resource, Conway Institute for Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland
cCentre for BioNano Interactions, School of Chemistry and Chemical Biology and Conway Institute for Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: kenneth.a.dawson@cbni.ucd.ie; marco.monopoli@cbni.ucd.ie
First published on 21st September 2012
Abstract
Dendrimers are polymers with a strong role in nanomedicine. In the current work we have developed a platform for mapping out the biomolecule corona for polyamidoamine (PAMAM) dendrimers. Complement proteins including C3 and C4b were found for the high generation dendrimers, suggesting high affinities to the dendrimers and most importantly complement activation.
PAMAM dendrimers were synthesized and characterized for the first time in 1985 by Tomalia et al.1 and since then several applications in the medical field have been proposed including drug/gene delivery and Magnetic Resonance Imaging.2–9 Dendrimers are well defined highly branched polymers presenting low polydispersity. Their size and shape are tuneable since their synthesis requires the progressive addition of monomers to a multifunctional core and they are typically classified by their generation number, which represents the number of repeated branching cycles around the core. The multifunctional core is dominant for dendrimers of low generation number while as the generation number increases the branching units and surface groups become progressively more predominant. Finally, their ‘surface’ can be easily chemically modified to add varied functionality. Because of these properties, dendrimer use in nanomedicine is expected to continue to grow. For example dendrimers can chelate gadolinium molecules providing a new contrast agent nanocarrier or they can improve the solubility of hydrophobic drugs and under some circumstances increase blood circulation time and modify their biodistribution.8
The understanding of dendrimers, quantum dots and other very small nanoparticles (of several nanometres) in a biological context will require a clear characterization of the most long lived biomolecules (the hard protein corona) adsorbed to them from the bio-environment,10–15 as the protein corona is believed to provide the real nanomaterial identity in the biological environment. The surface properties of the nanomaterial (charge, surface curvature and so forth) and the nature of the biological fluid will strongly influence the proteins that are associated with the nanoparticle12–14,16–20 and hence also the cellular response, the biocompatibility of the material and other system level signalling responses.20–23
Dendrimers will interact with biomolecules when introduced into the blood circulation system with unknown molecular consequences. For example the protein corona is believed to strongly effect the dendrimers biodistribution as the binding to dysopsonin proteins, like serum albumin and apolipoproteins, will promote a longer circulation time. On the other hand, binding to opsonin proteins, like fibrinogen, immunoglobulin and complement proteins are most likely to promote recognition by macrophages and phagocytes and trigger an inflammation response.19,21,24,25
Dendrimers have been shown to bind via non-specific interactions (electrostatic/hydrophobic forces) with the lipid membrane26–30 and with soluble proteins,31–33 where dendrimer generation and chemical group modification will strongly influence the protein binding affinity.31,32
We have studied the corona formed on cationic PAMAM dendrimers that have shown strong generation dependent interactions with the cell membrane and cell toxicity.34 Interestingly the haemolytic ability of dendrimers was found to be significantly decreased when human serum albumin (HSA) was present in the cell medium.35 Incubation of these dendrimers with human blood induced platelet activation, with the degree of activation being generation and surface group dependent.36 Dendrimer exposure to serum blood has also been shown to cause the activation of complement pathways.37
Here we report the detailed analogue of the hard corona (high affinity complex) of PAMAM dendrimers of generation (G) 4 to 7 (that will have same surface modification but differ in size) and blood plasma proteins. Because PAMAM dendrimer sizes38,39 and densities are comparable to most blood proteins, the typical methodologies evolved for studying protein corona complexes for large nanoparticles (for examples, based on different sedimentation time for the coated nanomaterial from unbound plasma proteins under applied centrifugal forces13,16) are not appropriate. Nor is there an easy analogue (washing) to differentiate what is truly bound, and what is simply an impurity of a highly abundant protein. Here we have applied electrophoretic mobility techniques (often used to separate charged molecules and small nanoparticles that migrate though porous agarose gels40,41) followed by Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) to address these questions.
Dendrimers of different generation, and thus of different sizes (G4, G5, G6 and G7) and with known surface proprieties (Table S.1, ESI†) were incubated for a fixed time (30 min) with low and high plasma concentration (representing protein concentration typically for the in vitro and in vivo environment) and the electrophoretic mobility was measured using 0.5% agarose gel under non denaturing conditions to preserve the dendrimer–plasma protein interactions (Fig. 1). Control samples of pristine dendrimers and human plasma were additionally run to measure their electrophoretic mobility independently. As expected, due to their positive charge, pristine dendrimers migrated towards the cathode with the degree of migration being size (generation) dependent (Fig. 1). Shorter migration times were found for G4 than G7 given their difference in size (G4 and G7 are 4 and 8 nm in diameter, respectively).38,39 After exposure to plasma, migration led to the separation of the dendrimer–plasma complexes from the excess of plasma. In low plasma concentration G4 dendrimers showed a dual distribution in which one population of dendrimers migrated towards the anode (associated with the plasma protein) while another migrated towards the cathode (free dendrimers). On the other hand, higher generation dendrimers migrated only towards the anode in the presence of plasma. Plasma control samples, where only plasma proteins were run in the gel, showed that most of the plasma proteins migrated to the anode end of the gel giving an intense band, while the other two intermediate bands were only visible at higher plasma concentration. No free plasma proteins had similar migration distances as the dendrimer–plasma protein complexes, indicated by the while arrows in Fig. 1. From the low plasma concentration in the agarose gel, it is evident that the amount of non-bound proteins decreased with increasing generation (black arrows in Fig. 1). Thus, higher generation dendrimers had a larger protein binding capacity.
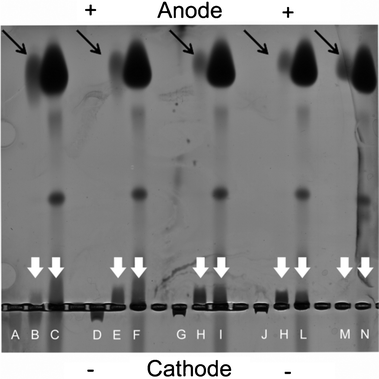 | ||
| Fig. 1 Electrophoretic separation of pristine dendrimers and protein–dendrimer complexes in low and high concentrations of plasma. Generation 4 (A–C), 5 (D–F), 6 (G–I) and 7 (J–L) dendrimer and control samples without dendrimer (M–N) were run. The first lane in each group contains pristine dendrimer, the second and third contains low and high plasma together with 0.5 mg ml−1 dendrimer. The white arrows indicate dendrimers plasma protein complexes while the black arrows indicate unbound proteins. | ||
In order to further isolate and characterize the protein–dendrimer complexes, those bands where the plasma–dendrimer mixtures differed from the plasma control (indicated by the white arrows in Fig. 1) were cut, mixed with SDS-PAGE loading buffer (a highly denaturing environment) and run on SDS-PAGE. By this method, and subsequent MS analysis, we could fully determine the hard corona proteins associated with the dendrimers. Spectral counting values, which represent the total number of the MS/MS spectra for all peptides attributed to a matched protein, have been used as a semi-quantitative analysis to calculate protein abundance (Table 1). SDS-PAGE gel and gel band densitometry are shown in Fig. 2 for high plasma conditions and in Fig. SI.1 in the ESI† for low plasma concentration. To investigate whether some plasma proteins had similar electrophoretic mobility as the dendrimer–protein complexes, the bands in the plasma control (Fig. 1, lane M and N) were also cut and run by SDS-PAGE.
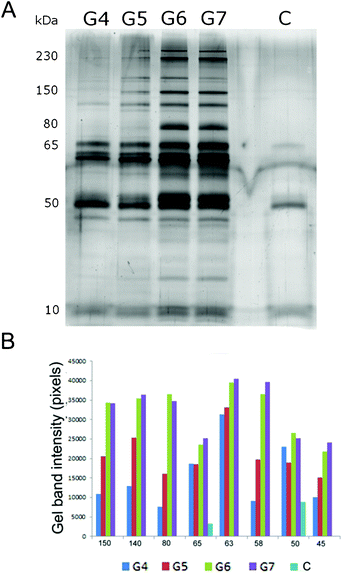 | ||
| Fig. 2 (A) SDS-PAGE of human plasma proteins, in high plasma concentration, associated with dendrimers of different generations (G4–G6) and plasma control (C). (B) Gel band densitometry of the most relevant SDS-PAGE gel bands. | ||
| Acc. nra | Protein identity | Mw (Da) | G4b | G5b | G6b | G7b | Cb |
|---|---|---|---|---|---|---|---|
| a Uniprot accession number. b Spectral counting for proteins associated with G4, G5, G6, G7 dendrimers and the control sample respectively. | |||||||
| P04114 | Apolipoprotein B100 | 515![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 283 283 |
1 | 19 | 63 | 50 | 3 |
| P01024 | Complement C3 | 187029 |
84 | 110 | 152 | 160 | 13 |
| P00450 | Ceruloplasmin | 122127 |
— | 35 | 75 | 72 | — |
| P00734 | Prothrombin | 69992 |
7 | 23 | 43 | 34 | — |
| P04003 | C4b-binding protein | 66989 |
13 | 12 | 24 | 23 | — |
| P04004 | Vitronectin | 54271 |
1 | 4 | 12 | 12 | — |
| P01871 | Ig mu chain | 49275 |
14 | 14 | 24 | 20 | 11 |
| P01857 | Ig gamma-1 chain C | 36083 |
9 | 10 | 14 | 13 | 14 |
From Fig. 2 it is clear that the lower dendrimer generations bound significantly less proteins compared to G6 and G7 even though similar protein patterns (in terms of protein molecular masses) were observed for all dendrimers. Some exceptions occurred for the gel bands of 230 kDa, 80 kDa and ∼20 kDa (identified as complement C3 and C4A, fibrinogen alpha chain and IgG (immunoglobulin G) kappa chain, see Table 1 and Table SI.2 in the ESI†), which appeared only for the largest dendrimers (G6 and G7). This indicates the selective affinity of these proteins towards high generation dendrimers only. Consistent duplet bands detected at 50 kDa were present for all dendrimers although they became stronger for higher generations. These bands were identified by MS analysis as fibrinogen beta and gamma chain (Table 1).
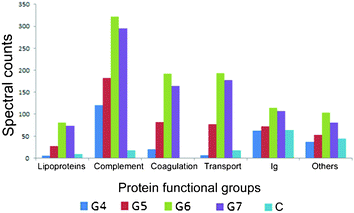 | ||
| Fig. 3 The functional classification of human plasma proteins, in high plasma concentrations, associated with dendrimers of different generations. | ||
Functional classification on the identified proteins associated on the dendrimers after incubation with high plasma concentration (Fig. 3, Table 1 and Table SI.3 in the ESI†) and low plasma concentration (Fig. SI.2 and Table SI.3 in the ESI,† respectively), showed that complement proteins are progressively enriched in higher generation dendrimers with complement C3 being the most abundant protein associated with these dendrimers based on the spectral counting values. Complement proteins C4A and C5B, derived from the enzymatic cleavage of full length C4 proteins were also associated with the dendrimer complexes. This suggests not only high affinities to the dendrimers but most importantly complement activation as result of exposure to these dendrimers. MS analysis also identified several key proteins of the complement cascade associated with the dendrimer–protein complexes for both G6 and G7, including IgG, complement C1q, complement C4 and complement C3. The proteins involved in the coagulation cascade, like fibrinogen and prothrombin, were also associated with the dendrimer–protein complexes and their abundance was significantly higher with G6 and G7 as shown in Table SI.2 in the ESI.† Similarly, a few proteins involved in the transport of nutrients and biomolecules in the blood stream, like serum albumin, ceruloplasmin and transthyretin are also associated with all the dendrimers but to a greater extent with the higher generation dendrimers.
Conclusions
In conclusion, we have reported a new method to isolate and determine the “protein corona” associated with dendrimers (and potentially other small particles), which have similar sizes and densities to plasma proteins. Agarose gel electrophoresis showed increased plasma protein interactions with G6 and G7 dendrimers compared to lower generations as unbound protein intensities were detected in the gel (Fig. 1). The SDS-PAGE of the dendrimer–protein complexes showed a higher number and intensity of protein bands associated with the G6 and G7 dendrimers (Fig. 2) and a similarly higher protein abundance has also been detected by MS semi-quantitative analysis based on spectral counting (Fig. 3 and Table 1). Since higher generation dendrimers contain higher surface charge densities (see Table SI.1 in the ESI†), the increasing amount of protein associated with the higher generation dendrimers is likely to be the result of an increase of the electrostatic interactions,33 additionally, there is an increase in entropy of the system due to counterion release, as commonly found for polyelectrolyte–surfactant systems.42 Functional classification showed that complement proteins were strongly associated strictly to G6 and G7, and activated forms of the complements C4A and C5B were also found in the protein corona complexes. As activation emerged from a selective and direct complement interaction and it was particularly enhanced with the large generation dendrimers, it allows us to predict the likely outcome of these polymers in the blood stream: in this case an immunological response should be expected for dendrimers G6 or larger. In addition, we obtained a complete mapping out of all the biomolecules associated with the dendrimers.In essence we consider that by using this simple methodology it may be possible to shed light on the biological identity of dendrimers, quantum dots, and other small nanoparticles in the same way in which we have been able to map out the protein corona for larger particles. This could have considerable utility in screening them for nanosafety and nanomedicine applications and we comment briefly on them here. Firstly, as the dendrimer generation increases and more potential binding sites become available for molecules from the environment, the dendrimer–biomolecule complexes may grow in a manner (depending on the structure of the dendrimer) that is markedly different than for the surface of larger spherical nanoparticles. As with other systems and provided that there is a sufficient concentration of biomolecules in the environment,43 the biomolecules will occupy the available binding sites on the nanoparticle surface since there is a beneficial free energy of binding (derived in part from the chemical structure of the dendrimer). However, the whole process in a dense crowded biopolymer environment in biological fluids is expected to be different from that on a simple spherical particle surface. Thus, both in the nature of the kinetics of the approach to the dendrimer, and the competition in the binding and displacement process between the different species is quite different from a simple surface, and one may expect cases in which even similar polymeric backbones lead to entirely different outcomes depending on the structure of the dendrimer and its generation. Potentially the most significant difference is the selective binding of biomolecules and dendrimer type may be used to make further contact with coronas of other dendrimer generations if those interactions confer some particular benefit. Thus, our capacity to chemically tailor dendrimer structure potentially makes it feasible to form protein coronas that have highly complex (for example, potentially functional biologically) assembled structures turning the dendrimers into scaffolds to lower the entropic barrier to assembly formation in a highly complex environment. Some sign of this already occurs in the present (complement) case, but the motif is potentially of very considerable interest for future study. Needless to say, this paradigm has significance not just for the study and extraction of such complexes, but could have significance for the potential to target medicines in the future.
Acknowledgements
The use and access of the UCD Conway Mass Spectrometry Resource is gratefully acknowledged. This work was conducted under the INSPIRE programme, funded by the Irish Government's Programme for Research in Third Level Institutions, Cycle 4, National Development Plan 2007–2013. Funding from EU FP7 Large Collaborative project NAMDIATREAM (NMP4-LA-2010-246479) is also acknowledged. Additional funding from the European Science Foundation EpitopeMap project and the Helmholtz Virtual Institute Nano Tracking project supported by the Helmholtz Initiative and Networking fund are gratefully acknowledged. M.C. gratefully acknowledges the financial support from the “Centre for Synthetic Biology” at Copenhagen University funded by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation.References
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS.
- R. Duncan and R. Gaspar, Mol. Pharmaceutics, 2011, 8, 2101–2141 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS.
- A. Asthana, A. S. Chauhan, P. V. Diwan and N. K. Jain, AAPS PharmSciTech, 2005, 6, E536–542 CrossRef.
- S. K. Choi, T. Thomas, M. H. Li, A. Kotlyar, A. Desai and J. R. Baker, Jr., Chem. Commun., 2010, 46, 2632–2634 RSC.
- Z. Dong, H. Katsumi, T. Sakane and A. Yamamoto, Int. J. Pharm., 2010, 393, 245–252 CrossRef.
- J. Huang, F. Gao, X. X. Tang, J. H. Yu, D. X. Wang, S. Y. Liu and Y. P. Li, Polym. Int., 2010, 59, 1390–1396 CrossRef CAS.
- N. K. Jain and U. Gupta, Expert Opin. Drug Metab. Toxicol., 2008, 4, 1035–1052 CrossRef CAS.
- H. Kobayashi, N. Sato, S. Kawamoto, T. Saga, A. Hiraga, T. L. Haque, T. Ishimori, J. Konishi, K. Togashi and M. W. Brechbiel, Bioconjugate Chem., 2001, 12, 100–107 CrossRef CAS.
- X. R. Xia, N. A. Monteiro-Riviere and J. E. Riviere, Nat. Nanotechnol., 2010, 5, 671–675 CrossRef CAS.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Radler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS.
- A. S. Pitek, D. O'Connell, E. Mahon, M. P. Monopoli, F. Baldelli Bombelli and K. A. Dawson, PLoS One, 2012, 7, e40685 CAS.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS.
- J. Sund, H. Alenius, M. Vippola, K. Savolainen and A. Puustinen, ACS Nano, 2011, 5, 4300–4309 CrossRef CAS.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS.
- A. Elsaesser and C. V. Howard, Adv. Drug Delivery Rev., 2012, 64, 129–37 CrossRef CAS.
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS.
- P. Decuzzi, B. Godin, T. Tanaka, S. Y. Lee, C. Chiappini, X. Liu and M. Ferrari, J. Controlled Release, 2010, 141, 320–327 CrossRef CAS.
- I. Hamad, O. Al-Hanbali, A. C. Hunter, K. J. Rutt, T. L. Andresen and S. M. Moghimi, ACS Nano, 2010, 4, 6629–6638 CrossRef CAS.
- C. V. Kelly, M. G. Liroff, L. D. Triplett, P. R. Leroueil, D. G. Mullen, J. M. Wallace, S. Meshinchi, J. R. Baker, B. G. Orr and M. M. B. Holl, ACS Nano, 2009, 3, 1886–1896 CrossRef CAS.
- V. Tiriveedhi, K. M. Kitchens, K. J. Nevels, H. Ghandehari and P. Butko, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 209–218 CrossRef CAS.
- S. Parimi, T. J. Barnes and C. A. Prestidge, Langmuir, 2008, 24, 13532–13539 CrossRef CAS.
- A. Mecke, I. J. Majoros, A. K. Patri, J. R. Baker, M. M. B. Holl and B. G. Orr, Langmuir, 2005, 21, 10348–10354 CrossRef CAS.
- A. Akesson, K. M. Bendtsen, M. A. Beherens, J. S. Pedersen, V. Alfredsson and M. C. Gomez, Phys. Chem. Chem. Phys., 2010, 12, 12267–12272 RSC.
- E. Gabelleri, G. B. Strambini, D. Shcharbin, B. Klajnert and M. Bryszewska, Biochim. Biophys. Acta, Biomembr., 2006, 1764, 1750–1756 CrossRef.
- J. Giri, M. S. Diallo, A. J. Simpson, Y. Liu, W. A. Goddard, R. Kumar and G. C. Woods, ACS Nano, 2011, 5, 3456–3468 CrossRef CAS.
- L. Giehm, C. Christensen, U. Boas, P. A. H. Heegaard and D. E. Otzen, Biopolymers, 2008, 89, 522–529 CrossRef CAS.
- S. P. Mukherjee, M. Davoren and H. J. Byrne, Toxicol. in Vitro, 2010, 24, 169–177 CrossRef CAS.
- B. Klajnert, S. Pikala and M. Bryszewska, Proc. R. Soc. London, Ser. A, 2010, 466, 1527 CrossRef CAS.
- M. A. Dobrovolskaia, A. K. Patri, J. Simak, J. B. Hall, J. Semberova, S. H. De Paoli Lacerda and S. E. McNeil, Mol. Pharmaceutics, 2012, 9, 382–393 CrossRef CAS.
- C. Plank, K. Mechtler, F. C. Szoka, Jr. and E. Wagner, Hum. Gene Ther., 1996, 7, 1437–1446 CrossRef CAS.
- P. K. Maiti and R. Messina, Macromolecules, 2008, 41, 5002–5006 CrossRef CAS.
- Y. Liu, V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Am. Chem. Soc., 2009, 131, 2798–2799 CrossRef CAS.
- S. Park, N. Sinha and K. Hamad-Schifferli, Langmuir, 2010, 26, 13071–13075 CrossRef CAS.
- W. J. Parak, T. Pellegrino, C. M. Micheel, D. Gerion, S. C. Williams and A. P. Alivisatos, Nano Lett., 2002, 3, 33–36 CrossRef.
- C. Wang and K. C. Tam, Langmuir, 2002, 18, 6484–6490 CrossRef CAS.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra21866f |
| This journal is © The Royal Society of Chemistry 2012 |