Killing double bonds softly: the reduction of polymer-bound alkenes†
Daniel
Fürniss
a,
Ute
Schepers
b and
Stefan
Bräse
*ab
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: stefan.braese@kit.edu; Fax: +49 721 608-48581; Tel: +49 721 608-42903
bInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 19th September 2012
Abstract
Alkenes can be reduced through “transfer hydrogenation” with dimethylamine-borane adduct and Wilkinson's catalyst. This reaction can also be carried out by solid-phase synthesis as a heterogeneous reaction. Furthermore, the behaviour of various functional groups under hydrogenation conditions was tested.
Introduction
Boranes, especially amino-borane adducts, were long known as reducing agents for carbonyl compounds (aldehydes, ketones, carboxylic acids, esters, amides) as well as imines. Moreover, they find use in reductive aminations and in the reduction of a variety of functional groups such as highly polarized carbon–carbon double bonds, oximes, tosylhydrazones, indoles, iminium salts, enamines and nitriles.2 In the presence of transition metal catalysts amino-borane adducts also reduce aryl perfluorosulfonates, alkenes, quinoline, epoxides, alkynes, halides, aryl triflates and aryl nitro compounds.3,4N-Alloc deprotection, epoxide openings and cleavage of benzyl esters are also known.4,5 Berke et al. also showed that transfer hydrogenation of activated olefins with an ammonia-borane adduct is possible without a catalyst.6As far as solid phase methods are concerned, only a few protocols are available for the reduction of double bonds. Previous works on solid-phase hydrogenation point out that there are difficulties to adapt traditional heterogeneous catalysis due to poor kinetics. One approach to circumvent the kinetic problems is to apply homogeneous reagents (resin or catalysts).7 The most employed method is probably the diimide reduction with sulfonohydrazides.8 Other “hydrogen-free” reductions employ copper(I) hydride and titanocene reagents, respectively,9 or Grubbs' catalyst and triethylsilane.10
During our research on solid-phase polyamine synthesis, we observed a reduction of double bonds of immobilized substrates as a byproduct under certain hydroboration conditions (Scheme 1). The hydroboration reaction with only pinacolborane gave poor yields, so we tried to improve the conversion by adding Wilkinson's catalyst. In on-bead 13C NMR we could identify signals for both hydroborated and hydrogenated products (2 and 3).
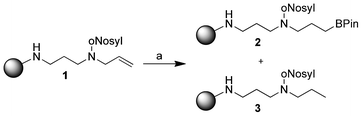 | ||
| Scheme 1 Initial observation of double bond reduction. (a) Pinacolborane (HBPin, 5.00 equiv.), Wilkinson's catalyst (0.10 equiv.), dry CH2Cl2, 15 h. | ||
Since a comparable method is only known in solution phase and not for solid-phase, we wanted to optimize our conditions towards hydrogenation and establish a mild reduction protocol.
For solution phase, Manners et al. developed a method for late transition metal-catalyzed dehydrocoupling of amine-borane adducts and thereby observed hydrogenation of COD (1,5-cyclooctadiene) while using rhodium catalysts. Cyclohexene was hydrogenated under the same conditions (Scheme 2).1
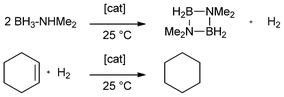 | ||
| Scheme 2 General equation for the catalytic dehydrocoupling of dimethylamine-borane adduct and hydrogenation of cyclohexene according to Manners et al.1 | ||
Herein, we now report a new method for reduction in solid-phase synthesis under mild conditions using dimethylamine-borane adduct and Wilkinson's catalyst (tris(triphenylphosphine)-rhodium(I) chloride). We immobilized a mono-protected diamine, which gave us ample opportunities to introduce different alkenes and other functional groups that we tested under these conditions via alkylation or acylation.
Results and discussion
Our testing system core was mono-nosyl protected diaminopropane 5 which was immobilized on 2-chlorotrityl chloride resin (4). In a substitution reaction the protected amine and N,N-diisopropylethylamine (DIPEA) were added to the resin 4. Through Fukuyama-alkylation with various allylic halides and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in N,N-dimethyl-formamide (DMF) we obtained the resin 6 (see Experimental part). After these alkylation reactions, the nosyl-protecting group was removed using 2-mercaptoethanol and DBU to give resin 8. Subsequently, the immobilized alkenes were hydrogenated using our method, which is highlighted in detail below, to yield resin 9. The product 10 was cleaved from the resin in a final step using trifluoroacetic acid (TFA, Scheme 3).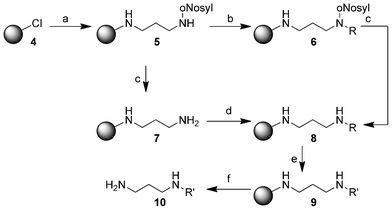 | ||
| Scheme 3 Synthesis of the substituted diamines 10 on 2-chlorotrityl chloride resin (4). (a) N-(3-Aminopropyl)-2-nitrobenzenesulfonamide (3.00 equiv.), DIPEA (3.00 equiv.), dry CH2Cl2, 15 h; (b) alkyl halide (6.00 equiv.), DBU (6.00 equiv.), DMF, 15 h; (c) 2-mercaptoethanol (10.0 equiv.), DBU (5.00 equiv.), DMF, 2 h; (d) carboxylic acid (5.00 equiv.), HOBt (5.00 equiv.), DIC (5.00 equiv.), DMF, 15 h; (e) dimethylamine-borane adduct (5.00 equiv.), Wilkinson's catalyst (0.10 equiv.), dry CH2Cl2, 15 h; (f) 5% TFA in CH2Cl2, 15 h. All reactions were conducted at room temperature. For R, R′ see Table 1 and Table 2. | ||
Immobilization of mono-nosyl protected diaminopropane on 2-chlorotrityl chloride resin (4), the following Fukuyama alkylation, nosyl deprotection and final cleavage from the resin were performed according to the established protocols from our group11 and slightly adapted protocols, respectively.
The reaction that really interested us was the hydrogenation step, which is now discussed in detail. The resins 8a–e were subjected to slightly modified dehydrocoupling/hydrogenation conditions, compared to the ones Manners et al.1 applied, using dimethylamine-borane adduct and Wilkinson's catalyst in dry dichloromethane at room temperature (Scheme 3). Under these conditions, no hydroboration was observed and even without the addition of Wilkinson's catalyst and under elevated temperature no hydroboration occurred. Possible hydroboration was ruled out since afterwards there was no reaction with hydrogen peroxide/sodium hydroxide.
However, we achieved hydrogenation of the carbon–carbon double bonds. Not only terminal double bonds (Entry 1), but also substituted and internal double bonds (Entries 2–4) as well as alkynes (8e, Entry 5) gave good results. For analysis and quantification, the resulting compounds had to be cleaved from the resin and purified by HPLC. After five steps (immobilization, alkylation, deprotection, reduction and cleavage) we obtained our desired compounds in 60–96% overall yield (Table 1). Although we did not quantify every single step—most steps were monitored by NMR—overall yields for our five-step procedure show that every reaction proceeds in good to very good yields, including the hydrogenation step. In addition, there was no olefin observed after the final step, so the conversion should be complete.
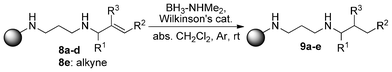 | ||
| Scheme 4 Hydrogenation of alkene and alkyne substrates. | ||
As shown in Table 1, hindered as well as unhindered alkenes can be reduced with our method. The reduction of the alkyne 8e cleanly resulted in the formation of the alkane derivative 9e.
With these results in hand we wanted to investigate the scope of the reaction by testing different functionalized substrates in the same manner (Table 2). Substrates that were available as benzyl halides were conjugated as described above. For carboxylic acids the sequence needed to be adapted (Scheme 3). The only difference was that resin 5 was first deprotected with 2-mercaptoethanol and DBU to give resin 7 and afterwards acylated with a carboxylic acid, 1-hydroxybenzotriazole (HOBt) and N,N′-diisopropylcarbodiimide (DIC) to yield resin 8. After this step the syntheses were the same.
| Entry |
|
Resin 9 | Isol. yield of amine 10a/Average yield per step [%] |
|---|---|---|---|
| a Total yield over five steps. b Total yield over six steps. c Traces of amine/alcohol were observed in ESI-MS. d Equivalents of reagents were doubled. | |||
| 6 |
|
|
30/78 |
|
24/75 | ||
| 7 |
 d
d
|
|
49/87 |
|
51/87 | ||
| 8 |
|
|
47/86 |
| 9 |
|
|
62/91 |
| 10 |

|
|
56/89 |
| 11 |
|
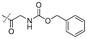
|
66/92 |
| 12 |
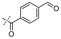
|
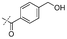
|
74/94 |
| 13 |
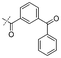
|
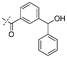
|
62/91 |
| 14 |
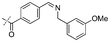
|
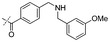
|
66b/93 |
| 15 |
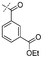
|
|
38/83c |
| 16 |
|
|
89/98 |
| 17 |
|
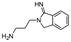
|
95/99c |
| 18 |
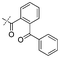
|
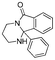
|
47/86 |
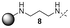
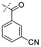
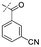
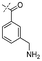
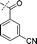
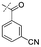
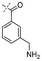
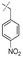
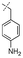
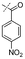
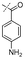
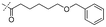
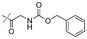
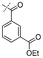
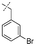
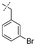
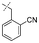
In our study we tested aldehydes (Entry 12), ketones (Entries 13 and 18), imines (Entry 14), arylesters (Entry 15) and -halides (Entry 16) as well as nitrile- (Entries 6, 7 and 17), nitro- (Entries 8 and 9), benzylether- (Entry 10) and benzyloxycarbonyl (Cbz)- protecting (Entry 11) groups. We found that benzylethers (Entry 10), halides (Entry 16), arylesters (Entry 15) and the Cbz-group (Entry 11) were stable under the reducing conditions and gave good overall yields for the unreduced product. However, after reduction of arylester (Entry 15), traces of the corresponding alcohol could be found by ESI-MS. Again, the overall yields were consulted for conclusions. All steps proceed in good to excellent yields and the reduction step does not lead to any serious degradation.
Aldehydes (Entry 12), ketones (Entry 13), imines (Entry 14) and nitro-compounds (Entries 8 and 9) were readily reduced to the corresponding products in good overall yields. Nitriles were only partially reduced. Still, the overall yields considering both products were very good (Entries 6 and 7). After receiving preliminary results that showed incomplete conversion for the nitriles (Entry 6), we tried again with double the equivalents of the reducing agent (Entry 7). Again, the conversion was not complete and we obtained a mixture of nitrile and amine, but the ratio was shifted towards the amine. Longer reaction times or repetition of the reduction step might lead to complete reduction in this case, too.
It is also noteworthy that amides were not reduced during the reaction. In Table 2 the results for the reduction are mapped including the yields that were obtained after cleavage of the amine 10 from the resin.
Entries 17 and 18 in Table 2 are showcases to demonstrate that ketones and nitrile react only slowly and were not reduced in the presence of an ortho-amine group: they reacted intramolecularly to form aminals or amidates, respectively.
In summary we showed that our method not only hydrogenates C–C double bonds, but also reduces some functional groups whereas other functional groups are stable under the used conditions.
Conclusion
In conclusion, we were able to establish a new protocol for the reduction of alkenes and alkynes on a solid-phase. Aldehydes, ketones, imines and nitro-groups are also reduced under these conditions. In our test halides, arylesters, benzylethers, amides and Cbz-groups were stable. Only nitriles were found to be partially instable with our method.Experimental
Attachment of monoprotected diamine to the resin
13C NMR (Gel) (100 MHz, CDCl3): δ = 147.7 (CAr–NO2), 133.3 (CAr), 132.5 (CAr), 130.7 (CAr), 127.6 (CAr), 125.1 (CAr), 42.3 (CH2NH), 40.1 (CH2NH), 30.5 (CH2CH2) ppm.
Fukuyama-alkylation
13C NMR (Gel) (100 MHz, CDCl3): δ = 147.7 (CAr–NO2), 133.3 (CAr), 132.7 (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 131.5 (CAr), 130.6 (CAr), 127.6 (CAr), 124.0 (CAr), 119.1 (CHCH2), 49.8 (CH2CHCH2), 45.1 (CH2N), 41.7 (CH2N), 29.1 (CH2(CH2)2) ppm.
CH2), 131.5 (CAr), 130.6 (CAr), 127.6 (CAr), 124.0 (CAr), 119.1 (CHCH2), 49.8 (CH2CHCH2), 45.1 (CH2N), 41.7 (CH2N), 29.1 (CH2(CH2)2) ppm.
13C NMR (Gel) (75 MHz, CDCl3): δ = 147.8 (CAr–NO2), 136.6 (CCHCH3), 133.3 (CAr), 131.5 (CAr), 130.6 (CAr), 127.7 (CAr), 125.1 (CAr), 123.9 (CCHCH3), 49.1 (CCHCH2N), 44.7 (NHCH2CH2CH2N), 41.7 (CH2NH), 40.3 (CH2NH), 28.5 (CH2(CH2)2), 17.7 (trans-CH3), 12.8 (cis-CH3) ppm.
13C NMR (Gel) (75 MHz, CDCl3): δ = 147.7 (CAr–NO2), 134.2 (CAr), 133.4 (CAr), 131.5 (PhCCH), 130.7 (CAr), 128.5 (CAr), 127.9 (CAr), 126.4 (PhCCH), 124.0 (CAr), 49.4 (CCHCH2N), 45.2 (CH2N), 40.2 (CH2N), 29.3 (CH2(CH2)2) ppm.
13C NMR (Gel) (75 MHz, CDCl3): δ = 147.9 (CAr–NO2), 133.8 (CAr), 133.2 (CAr), 132.1 (CAr), 131.4 (NCHCHCH), 130.6 (CAr), 125.7 (CAr), 123.9 (NCHCHCH), 42.9 (CH2N), 40.5 (CH2N), 32.8 (CH2(CH2)2), 28.7 (NCHCH2), 24.3 (CHCHCH2), 21.6 (CHCHCH2CH2) ppm.
Nosyl-deprotection
13C NMR (Gel) (100 MHz, CDCl3): δ = 136.9 (CHCH2), 115.6 (CHCH2), 47.6 (CH2CHCH2), 42.7 (CH2N), 40.3 (CH2N), 31.1 (CH2(CH2)2) ppm.
CHCH3), 127.6 (cis-CCHCH3), 126.9 (trans-CCHCH3), 125.8 (cis-CCHCH3), 51.7 (CCHCH2N), 47.6 (NHCH2CH2CH2N), 46.0 (CH2NH), 42.7 (CH2NH), 31.2 (CH2(CH2)2), 17.7 (trans-CH3), 13.0 (cis-CH3) ppm.
CH), 128.4 (CAr), 127.2 (CAr), 126.1 (PhCCH), 124.0 (CAr), 51.9 (CCHCH2N), 47.7 (CH2N), 42.8 (CH2N), 31.2 (CH2(CH2)2) ppm.
CH), 128.5 (NCHCHCH), 45.2 (CH2N), 42.8 (CH2N), 31.5 (CH2(CH2)2), 29.5 (NCHCH2), 25.2 (CHCHCH2), 20.2 (CHCHCH2CH2) ppm.
Reduction
13C NMR (Gel) (100 MHz, CDCl3): δ = 22.2 (CH2), 11.7 (CH3) ppm.
Cleavage
1H NMR (400 MHz, MeOH-d4): δ = 3.11 (t, J = 7.8 Hz, 2 H, CH2NH2), 3.05 (t, J = 7.6 Hz, 2 H, CH2NH), 2.98 (t, J = 7.8 Hz, 2 H, CH2NH), 2.07 (tt, J = 7.8 Hz, J = 7.6 Hz, 2 H, CH2CH2CH2), 1.72 (tq, J = 7.8 Hz, J = 7.4 Hz, 2 H, CH2CH3), 1.02 (t, J = 7.4 Hz, 3 H, CH3) ppm. – 13C NMR (100 MHz, MeOH-d4): δ = 50.7 (−, CH2NH), 45.8 (−, CH2NH), 37.9 (−, CH2NH2), 25.4 (−, CH2CH2CH2), 20.7 (−, CH2CH3), 11.2 (+, CH3) ppm. – MS (ESI): m/z: 117.1 [M++H]. – MS (FAB): m/z: 117.1 [M++H]. – HRMS (C6H16N2): calc. 116.1313; found 116.1312. – IR (ATR): ṽ = 3039 (w), 2974 (w), 2809 (w), 2558 (w), 2246 (vw), 1665 (s), 1608 (m), 1531 (w), 1485 (m), 1428 (m), 1349 (vw), 1195 (s), 1177 (s), 1124 (vs), 1004 (w), 962 (vw), 834 (m), 796 (m), 771 (w), 722 (s), 600 (w), 519 (w), 442 (w), 414 (w) cm−1.
1H NMR (400 MHz, MeOH-d4): δ = 3.11 (t, J = 7.8 Hz, 2 H, CH2NH2), 3.08–2.99 (m, 4 H, CH2NH), 2.07 (tt, J = 7.8 Hz, 2 H, CH2CH2CH2), 1.67 (tt, J = 7.8 Hz, 2 H, CH2CH2CH2), 1.43 (tq, J = 7.8 Hz, J = 7.3 Hz, 2 H, CH2CH3), 0.98 (t, J = 7.3 Hz, 3 H, CH3) ppm. – 13C NMR (100 MHz, MeOH-d4): δ = 48.9 (−, CH2NH), 45.8 (−, CH2NH), 37.9 (−, CH2NH2), 29.2 (−, CH2CH2CH2), 25.4 (−, CH2CH2CH2), 20.8 (−, CH2CH3), 13.9 (+, CH3) ppm. – MS (ESI): m/z: 131.2 [M++H].– MS (FAB): m/z: 131.1 [M++H]. – HRMS (C7H18N2): calc. 130.1470; found 130.1475. – IR (ATR): ṽ = 3038 (w), 2870 (w), 2230 (vw), 1665 (s), 1606 (m), 1531 (w), 1485 (m), 1429 (m), 1349 (vw), 1177 (s), 1124 (s), 969 (w), 920 (vw), 834 (m), 796 (m), 770 (w), 721 (s), 600 (w), 519 (w), 442 (w), 412 (w) cm−1. – mp: 149.4 °C.
1H NMR (400 MHz, MeOH-d4): δ = 7.32–7.17 (m, 5 H, HAr), 3.10 (t, J = 7.8 Hz, 2 H, CH2NH2), 3.07–2.99 (m, 4 H, CH2NH), 2.72 (t, J = 7.8 Hz, 2 H, CH2CAr), 2.06 (tt, J = 7.8 Hz, 2 H, CH2CH2CH2), 2.01 (tt, J = 7.8 Hz, 2 H, CH2CH2CH2) ppm. – 13C NMR (100 MHz, MeOH-d4): δ = 141.6 (Cquart), 129.7 (+, 2 × CHAr), 129.4 (+,2 × CHAr), 127.5 (+, CHAr), 48.7 (−, CH2NH), 45.8 (−, CH2NH), 37.8 (−, CH2NH2), 33.5 (−, CH2CAr), 29.0 (−, CH2CH2), 25.4 (−, CH2CH2) ppm. – MS (ESI): m/z: 193.2 [M++H]. – MS (FAB): m/z: 193.2 [M++H]. –HRMS (C12H21N2): calc. 193.1705; found 193.1704. – IR (ATR): ṽ = 3028 (w), 2944 (w), 2797 (w), 2231 (vw), 1663 (s), 1536 (w), 1496 (w) 1484 (w), 1454 (w), 1429 (w), 1326 (vw), 1177 (s), 1130 (s), 906 (vw), 836 (m), 796 (m), 768 (w), 751 (m), 723 (s), 694 (m), 600 (w), 570 (w), 519 (w), 493 (w), 461 (w), 441 (w), 411 (w) cm−1. – mp: 157.9 °C.
1H NMR (400 MHz, MeOH-d4): δ = 3.21–3.02 (m, 5 H, CH2NH2, CH2NH, CHNH), 2.16–2.02 (m, 4 H, CH2CH2N + 2 × cHexH), 1.93–1.85 (m, 2 H, cHexH), 1.75–1.68 (m, 1 H, cHexH), 1.43–1.32 (m, 4 H, cHexH), 1.29–1.16 (m, 1 H, cHexH) ppm. – 13C NMR (100 MHz, MeOH-d4): δ = 58.7 (+, CHNH), 42.7 (−, CH2NH), 37.9 (−, CH2NH2), 30.3 (−, NH2CH2CH2), 26.1 (−, 2 × CHCH2), 25.6 (−, CHCH2CH2CH2), 25.5 (−, 2 × CHCH2CH2) ppm. – MS (ESI): m/z: 157.2 [M++H]. – MS (FAB): m/z: 157.2 [M++H]. – HRMS (C9H21N2): calc. 157.1705; found 157.1704. – IR (ATR): ṽ = 3034 (m), 2934 (m), 2857 (m), 1665 (s), 1529 (w), 1500 (w), 1431 (m), 1394 (w), 1172 (s), 1120 (s), 1069 (m), 1053 (m), 1032 (w), 972 (vw), 942 (vw), 897 (vw), 838 (m), 797 (s), 765 (w), 721 (s), 674 (vw), 598 (w), 581 (vw), 517 (w), 447 (w), 414 (w) cm−1. – mp: 153.7 °C.
1H NMR (400 MHz, MeOH-d4): δ = 3.11 (t, J = 7.8 Hz, 2 H, CH2NH2), 3.05 (t, J = 7.6 Hz, 2 H, CH2NH), 2.98 (t, J = 7.8 Hz, 2 H, CH2NH), 2.07 (tt, J = 7.8 Hz, 2 H, CH2CH2CH2), 1.72 (tq, J = 7.6 Hz, J = 7.4 Hz, 2 H, CH2CH3), 1.02 (t, J = 7.4 Hz, 3 H, CH3) ppm. – 13C NMR (100 MHz, MeOH-d4): δ = 50.7 (−, CH2NH), 45.8 (−, CH2NH), 37.9 (−, CH2NH2), 25.4 (−, CH2CH2CH2), 20.7 (−, CH2CH3), 11.2 (+, CH3) ppm. [Impurity: 2-mercaptoethanol <10%]. – MS (ESI): m/z: 117.1 [M++H]. – MS (FAB): m/z: 117.1 [M++H]. – HRMS (C6H16N2): calc. 116.1313; found 116.1314. – IR (ATR): ṽ = 2794 (w), 1666 (s), 1475 (w), 1427 (w), 1193 (s), 1126 (s), 835 (m), 798 (m), 758 (w), 721 (m), 598 (w), 518 (w), 438 (w), 411 (w) cm−1.
Acknowledgements
This work was supported by the Carl Zeiss Stiftung (fellowship for D. F.).References
- (a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS; (b) C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 2698–2699 CrossRef CAS; (c) M. E. Sloan, A. Staubitz, K. Lee and I. Manners, Eur. J. Org. Chem., 2011, 672–675 CrossRef CAS.
- (a) R. P. Barnes, J. H. Graham and M. D. Taylor, J. Org. Chem., 1958, 23, 1561–1562 CrossRef; (b) A. Pelter, R. M. Rosser and S. Mills, J. Chem. Soc., Perkin Trans. 1, 1984, 717–720 RSC; (c) M. D. Bomann, I. C. Guch and M. DiMare, J. Org. Chem., 1995, 60, 5995–5996 CrossRef CAS; (d) H. C. Brown, J. V. Bhaskar Kanth and M. Zaidlewicz, J. Org. Chem., 1998, 63, 5154–5163 CrossRef CAS; (e) C. F. Lane, Aldrichimica Acta, 1973, 6, 51–58 CAS; (f) R. O. Hutchins, K. Learn, B. Nazer, D. Pytlewski and A. Pelter, Org. Prep. Proced. Int., 1984, 16, 335–372 CrossRef CAS.
- B. H. Lipshutz, D. J. Buzard and R. W. Vivian, Tetrahedron Lett., 1999, 40, 6871–6874 CrossRef CAS.
- (a) M. Couturier, B. M. Andresen, J. L. Tucker, P. Dubé, S. J. Brenek and J. T. Negri, Tetrahedron Lett., 2001, 42, 2763–2766 CrossRef CAS; (b) M. Couturier, J. L. Tucker, B. M. Andresen, P. Dubé, S. J. Brenek and J. T. Negri, Tetrahedron Lett., 2001, 42, 2285–2288 CrossRef CAS; (c) M. Couturier, J. L. Tucker, B. M. Andresen, P. Dubé and J. T. Negri, Org. Lett., 2001, 3, 465–467 CrossRef CAS; (d) Y. Jiang and H. Berke, Chem. Commun., 2007, 3571–3753 RSC; (e) N. Blaquiere, S. Diallo-Garcia, S. I. Gorelsky, D. A. Black and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 14034–14035 CrossRef CAS.
- (a) P. Gomez-Martinez, M. Dessolin, F. Guibé and F. Albericio, J. Chem. Soc., Perkin Trans. 1, 1999, 2871–2874 RSC; (b) H. David, L. Dupuis, M.-G. Guillerez and F. Guibé, Tetrahedron Lett., 2000, 41, 3335–3338 CrossRef CAS.
- X. Yang, T. Fox and H. Berke, Chem. Commun., 2011, 47, 2053–2055 RSC.
- (a) S. Chen and K. D. Janda, J. Am. Chem. Soc., 1997, 119, 8724–8725 CrossRef CAS; (b) A. N. Whelan, J. Elaridi, M. Harte, S. V. Smith, W. R. Jackson and A. J .Robinson, Tetrahedron Lett., 2004, 45, 9545–9547 CrossRef CAS; (c) S. Nad, S. Roller, R. Haag and R. Breinbauer, Org. Lett., 2006, 8, 403–406 CrossRef CAS; (d) A. M. Harned, H. S. He, P. H. Toy, D. L. Flynn and P. R. Hanson, J. Am. Chem. Soc., 2005, 127, 52–53 CrossRef CAS.
- (a) P. Lacombe, B. Castagner, Y. Gareau and R. Ruel, Tetrahedron Lett., 1998, 39, 6785–6786 CrossRef CAS; (b) K. R. Buszek and N. Brown, J. Org. Chem., 2007, 72, 3125–3128 CrossRef CAS; (c) M. I. Garcia-Aranda, R. Gonzalez-Muniz, M. T. Garica-Lopez and M. J. Perez de Vega, Eur. J. Org. Chem., 2009, 4149–4157 CrossRef CAS; (d) V. Mamane, A. B. Garcia, J. D. Umarye, T. Lessmann, S. Sommer and H. Waldmann, Tetrahedron, 2007, 63, 5754–5767 CrossRef CAS.
- D. P. Dickson, C. Toh, M. Lunda, M. V. Yermolina, D. J. Wardrop and C. L. Landrie, J. Org. Chem., 2009, 74, 9535–9538 CrossRef CAS.
- A. A. Poeylaut-Palena, S. A. Testero and E. G. Mata, Chem. Commun., 2011, 47, 1565–1567 RSC.
- F. Hahn and U. Schepers, J. Comb. Chem., 2008, 10, 267–273 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental details and NMR spectra. See DOI: 10.1039/c2ra22189f |
| This journal is © The Royal Society of Chemistry 2012 |