The effect of the N-mesityl group in NHC-catalyzed reactions†
Jessada
Mahatthananchai
and
Jeffrey W.
Bode
*
Laboratory for Organic Chemistry, Swiss Federal Institute of Technology (ETH) Zürich, Switzerland. E-mail: bode@org.chem.ethz.ch; Fax: (+41)44-633-1235
First published on 18th August 2011
Abstract
The majority of N-heterocyclic carbene catalyzed reactions of α-functionalized aldehydes, including annulations, oxidations, and redox reactions, occur more rapidly with N-mesityl substituted NHCs. In many cases, no reaction occurs with NHCs lacking ortho-substituted aromatics. By careful competition studies, catalyst analogue synthesis, mechanistic investigations, and consideration of the elementary steps in NHC-catalyzed reactions of enals, we have determined that the effect of the N-mesityl group is to render the initial addition of the NHC to the aldehyde irreversible, thereby accelerating the formation of the Breslow intermediate. These studies rationalize the experimentally observed catalyst preference for all classes of NHC-catalyzed reactions of aldehydes and provide a roadmap for catalyst selection and design.
Introduction
Since the first reports in 2004 by our group,1 Rovis,2 and Glorius,3 more than 100 publications have documented the unique reactivity of the catalytically generated intermediates arisen from the combination of an α-functionalized aldehyde and an N-heterocyclic carbene catalyst.4 Multiple catalyst types including imidazolium, triazolium, and thiazolium-derived NHCs are employed but one clear trend emerges: all reactions of α,β-unsaturated aldehydes proceed more effectively with ortho,ortho′ disubstituted aromatic groups on the azolium ring. It is therefore no surprise that most practitioners in this field have adopted N-mesityl substituted catalysts, usually N-mesityl substituted triazolium derivatives,5 for NHC-catalyzed redox reactions, oxidations, and annulations.6 In contrast, the majority of “simple” aldehydes employed in NHC-catalyzed benzoin and Stetter-type reactions or redox reactions of α-heteroatomic aldehydes prefer N-pentafluorophenyl triazolium salts.7 Despite this repeated observation by multiple groups, no explanation for the marked superiority of N-mesityl substituted carbenes in NHC-catalyzed reactions of α,β-unsaturated aldehydes has appeared.8The effect of the N-mesityl substituent is not simply one of enhancing selectivity or stereochemistry; it is responsible for dramatically enhanced rates of reaction and different outcomes from otherwise identical substrates and conditions. A striking example of its activity is the redox esterification of cinnamaldehyde with methanol to give methyl cinnamate, a prototypical NHC-catalyzed reaction of enals (Scheme 1). When performed with N-mesityl substituted triazolium precatalyst 1, the reaction proceeds to completion even in the absence of added base.9 The corresponding N-C6F5 precatalyst 2 gives no conversion without base and slower reactions than 1 in the presence of Hünig's base. This is surprising; the N-C6F5 precatalyst is more acidic than the N-mesityl precatalyst. Indeed, an NMR investigation on the extent of deprotonation between the two catalysts revealed that precatalyst 2 became fully deprotonated (from the observation of protonated DBU) while the N-mesityl precatalyst 1 was only partially deprotonated (Fig. 1). This observation prompted the question of why the N-mesityl precatalyst 1, which generates less of the active NHC species, is the superior catalyst10 for reactions with enals?
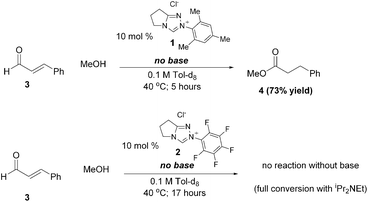 | ||
| Scheme 1 Redox esterification of enal in the absence of base. | ||
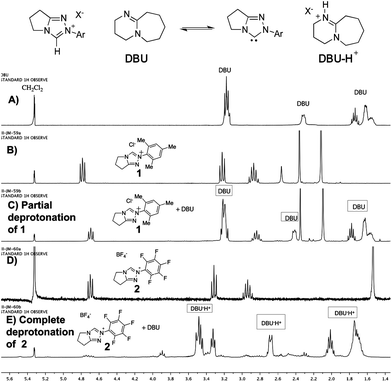 | ||
| Fig. 1 Extent of deprotonation of azolium salts 1 and 2. A) 1H NMR spectra of DBU in CD2Cl2 as reference; B) and D) 1H NMR spectra of triazolium salts 1 and 2; C) and E) 1H NMR spectra of triazolium salts following addition of 1.0 equiv. DBU. | ||
Results and discussion
We have previously shown that α,β-unsaturated Breslow intermediates from N-mesityl substituted triazoliums do not revert to aldehyde and NHC.11 We therefore hypothesized that the success of the N-mesityl substituted catalyst 1 lay in the irreversible formation of the Breslow intermediate, while that formed from the N-C6F5 substituted salt 2 was reversible. To test this, we examined numerous reactions, such as that of D-cinnamaldehyde (5, Scheme 2) in the presence of a proton source with both N-mesityl and N-C6F5 substituted triazoliums. In no case did we observe H/D exchange in the recovered aldehyde at any conversion. These results show that the formation of the Breslow intermediate from α,β-unsaturated aldehydes is irreversible12 with either catalyst and contrary to the generally accepted mechanism for azolium catalyzed benzoin-type reactions.13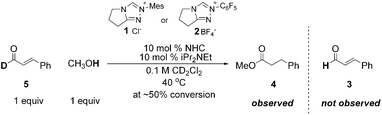 | ||
| Scheme 2 Irreversible formation of the Breslow intermediate. | ||
We revised our hypothesis to state that formation of the initial adduct between the aldehyde and the carbene is reversible for the N-C6F5 catalyst 2 and irreversible for the N-mesityl catalyst 1. The resulting adduct would then undergo a proton transfer sequence that would lead to the Breslow intermediate.14 This hypothesis would explain both why less acidic N-mesityl chloride 1 is able to catalyze reactions with only Cl− as a base – because any carbene generated would react irreversibly with the enals – as well as why the reactions with the N-mesityl catalyst are faster than with the more acidic N-C6F5 precatalyst 2. In order to investigate the origin of the observed reactivity, two additional catalysts were synthesized: the N-3,4,5-trimethoxy derivative 9, intended to be electronically similar to N-mesityl but sterically closer to N-C6F5 catalyst and N-2,4,6-trichloro triazolium 10 with the opposite profile.
We first needed to determine which of the elementary steps in NHC-catalyzed reactions of enals was responsible for the reactivity differences. To eliminate catalyst turnover from the acyl azolium intermediate as the origin of the catalyst divergency, we examined two NHC-catalyzed redox esterification reactions, one from cinnamaldehyde5a and the other from an α-chloroaldehyde (Scheme 3).15 These two reactions converge to the same acyl triazolium intermediate that serves as a catalytically generated activated carboxylate. We found that N-mesityl catalyst 1 is faster for cinnamaldehyde while the N-C6F5 catalyst is superior with α-chloroaldehyde 6. This discrepancy established that the rate acceleration for catalyst 1 in the reaction with enals must come from an elementary step prior to acyl azolium formation.
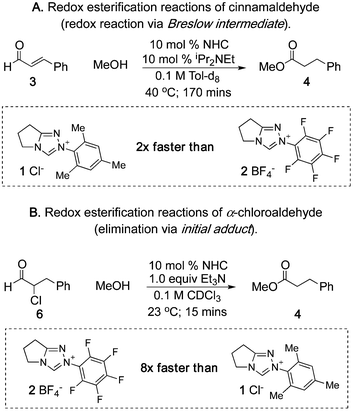 | ||
| Scheme 3 Redox esterifications of α-functionalized aldehydes. | ||
This left two possible origins for the divergent reactivity: 1) the β-protonation of the Breslow intermediate and 2) the formation of the Breslow intermediate from the initial adduct. To distinguish these, we examined the oxidative esterification reaction16 of cinnamaldehyde (Scheme 4A), a reaction that requires generation of the Breslow intermediate from an α,β-unsaturated aldehyde but not its protonation. The N-mesityl catalyst 1 proved to be an excellent catalyst for this reaction, followed by the N-2,4,6-trichloro 10, the N-C6F52, and the N-3,4,5-trimethoxy 9. The identical trend was observed in the redox esterification of ynal17 – a process that passes through the same α,β-unsaturated acyl azolium intermediate18 (Scheme 4B). The observation that the more sterically hindered catalysts initiate faster reactions is most dramatically illustrated by the oxidative esterification of α-hydroxyenone.11 Despite the fact that the requisite retro-benzoin reaction is very sensitive to steric hindrance, the bulkier catalysts were far superior (Scheme 4C). The absolute rates of all of these reactions are much faster than the redox esterification of cinnamaldehyde and together rule out the oxidation of the Breslow intermediate as the rate-limiting step. This leaves the formation of the Breslow intermediate as the origin of the reactivity difference.
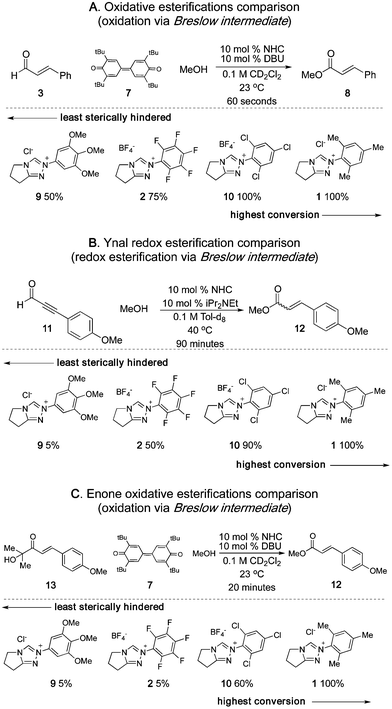 | ||
| Scheme 4 NHC catalyzed esterifications. | ||
The success of sterically hindered catalysts 1 and 10 originates from their rapid formation of the key Breslow intermediate II (kb, Fig. 2) – a consequence of the irreversible addition19 of the NHC to the electrophile when sterically hindered NHCs are used. In other words, the reverse reaction (k-a) has a higher activation barrier than the forward proton transfer steps (kb) that leads to the Breslow intermediate. The situation is the opposite with less-sterically demanding catalysts 2 and 9, which prefer to rest on the side of enal and catalyst – that is kb > k−a.
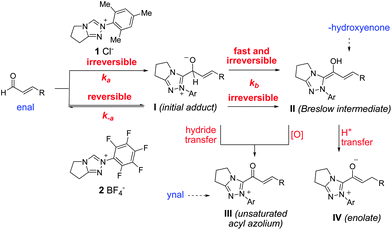 | ||
| Fig. 2 A proposed theory and mechanistic probes. | ||
This assertion predicts that the combination of N-mesityl catalyst 1 and cinnamaldehyde, in the absence of other reagents, should give an observable adduct. In contrast, N-C6F5 catalyst 2 should favor the starting materials: aldehyde and N-heterocyclic carbene should be observed. Indeed, the N-C6F5 triazolium 2 was completely deprotonated by one equiv. of DBU (by the detection of DBU-H+) but no adduct was observed upon the addition of cinnamaldehyde (Scheme 5). In contrast, the N-mesityl variant 1 was only partially deprotonated by DBU. Upon addition of cinnamaldehyde the extent of catalyst deprotonation increased, reaching full deprotonation only after two equiv. of aldehyde were added. A titration with cinnamaldehyde showed that in the absence of an oxidant, electrophile, or alcohol, the N-mesityl carbene forms a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct with cinnamaldehyde. This is a consequence of the irreversible formation of the initial adduct, which reacts with another equivalent of the starting aldehyde when no other forward pathway is available. Although in our case this gives a complex mixture, possibly the diastereomeric forms of the 2:1 NHC:aldehyde adduct recently described by Berkessel.20 Similar behavior was observed with triazolium 10 (see ESI†). The less hindered carbenes more rapidly revert to starting materials in the absence of a forward pathway.
:1 adduct with cinnamaldehyde. This is a consequence of the irreversible formation of the initial adduct, which reacts with another equivalent of the starting aldehyde when no other forward pathway is available. Although in our case this gives a complex mixture, possibly the diastereomeric forms of the 2:1 NHC:aldehyde adduct recently described by Berkessel.20 Similar behavior was observed with triazolium 10 (see ESI†). The less hindered carbenes more rapidly revert to starting materials in the absence of a forward pathway.
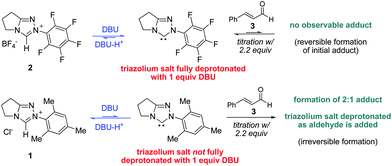 | ||
| Scheme 5 Titration of azoliums with DBU and cinnamaldehyde. | ||
These findings leave the question: why does the N-mesityl catalyst form essentially irreversible adducts with enals while the addition of the N-C6F5 catalyst to the aldehyde is reversible? We initially believe this to be due to electronic differences. The more electron deficient N-C6F5 carbene should be a better leaving group, thereby facilitating the reverse reactions (k−a, Fig. 2). The fact that the N-trimethoxy catalyst 9 behaves similarly to the N-C6F52 and the N-trichloro catalyst 10 more like the N-mesityl catalyst 1 argues against this electronic effect and implicates steric argument as the key determinant. This may be the consequence of ground state destabilization of the initial adduct I (Fig. 2); rapid formation of the Breslow intermediate II provides relief from this sterically congested initial adduct. The initial adduct I bearing N-C6F5 moiety has a relative higher activation barrier for Breslow intermediate formation than the reverse reaction to enal and catalyst – an observation consistent with Rovis' recent findings concerning the Stetter reaction.21
This insight also explains one reaction of enals in which the N-C6F5 triazolium salt is superior to the N-mesityl variant. In 2009, Csáky reported an NHC-catalyzed oxidative esterification of aldehydes with 1,4-naphthoquinone as the stoichiometric oxidant.22 Electron-deficient catalysts 2 and 10 proved to be viable catalysts while electron-rich catalysts 1 and 9 did not lead to the desired product (Scheme 6). The proposed mechanism is not oxidation of the Breslow intermediate but rather hydride transfer from the initial adduct I. This reaction is mechanistically related23 to the Cannizzaro reaction and follows similar electronic preference for faster hydride transfer with an electron-withdrawing-aryl group.24 Because this reaction does not occur from electron-transfer oxidation25via the intermediacy of the Breslow intermediate II, it does not follow the steric preference outlined previously. We therefore present this observation as “the exception that proves the rule.”
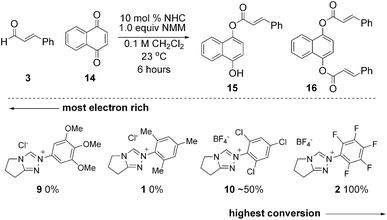 | ||
| Scheme 6 Hydride transfer oxidative esterification comparison (reaction via initial adduct). | ||
Conclusion
The conclusion that the effect of the N-mesityl group in NHC-catalyzed reactions is to enhance the rate of formation of the Breslow intermediate is consistent with known NHC-catalyzed reactions of aldehydes.10 Less hindered azolium salts, such as the N-C6F5 derivatives, are preferred for benzoin and Stetter type reactions (i.e.Scheme 7A); the use of the N-mesityl catalyst is likely to change the rate-limiting step from Breslow intermediate formation to the attack of the acyl anion equivalent onto the electrophile. Indeed, Rovis very recently reported an elegant enantioselective intermolecular Stetter reaction between α,β-unsaturated aldehydes and nitroalkenes using a chiral analog of 2.21b The N-C6F5 triazolium catalyst is also preferred for redox esterification reactions of α-heteroatomic and α,β-epoxy aldehydes.26 These reactions do not require formation of the Breslow intermediate (Fig. 2),1a,27 and catalyst turnover is most likely the turnover-limiting step – a process facilitated by more electron deficient carbenes, which are better leaving groups. The relative ease of C–C bond cleavage during catalyst turnover of N-C6F5 catalysts is apparent in a reaction between ynal 22 and kojic acid derivative 23 in which N-C6F5 catalyst 2′ exclusively afforded the ester product 25 while N-mesityl catalyst 1′ gave only the Coates-Claisen rearrangment9 product 24 (Scheme 7D).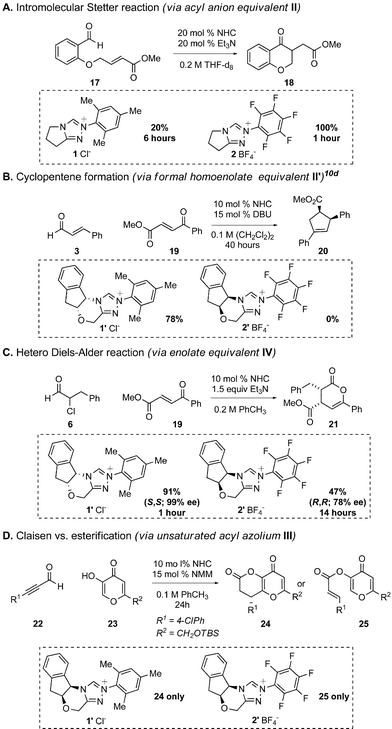 | ||
| Scheme 7 Comparison of reactions catalyzed by 1, 1′, 2, and 2′. | ||
The N-mesityl triazoliums are preferred in the NHC-catalyzed hetero-Diels–Alder reaction of α-halo aldehydes (Scheme 7C) due to the increase in electron density of the enolate IV, which deterred tautomerization to the activated carboxylate. Similarly, annulation cascade via a formal homoenolate addition prefers catalyst 1′ due to increased electron density of the Breslow intermediate (Scheme 7B).10b–d This explanation is also consistent with recent reports from Scheidt,28 You,29 and Chi30 in their applications of ortho,ortho′ disubstituted aromatic NHCs in formal homoenolate annulations, and Glorius on NHC-catalyzed hydroacylation reactions of olefins – a process that requires formation of the Breslow intermediate and proceeds more efficiently with N-mesityl substituted carbenes.31 These experiments, coupled with the ever-growing body of literature on NHC-catalyzed reactions, allows us to make predictions about the reactivity of the intermediates generated during NHC-catalyzed reactions of aldehydes. Fig. 3 provides a roadmap for selecting the most likely azolium salt for reactions with the desired mode of reactivity. In certain cases, such as γ-lactone formation annulations of enals and aldehydes1b,3 and air oxidation of aldehydes,32 more electron rich imidazolium or thiazolium carbenes are preferred but the choice of N-aryl substituent remains the same.33 NHC-catalyzed reactions of substrates other than aldehydes, such as esters34 and ketenes,35 may not follow these generalizations. For the majority of NHC-catalyzed reactions, however, either the N-mesityl or N-C6F5 will be the most competent catalyst.
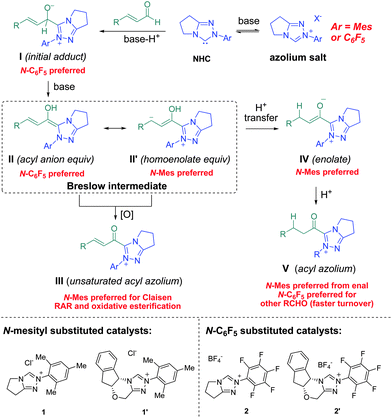 | ||
| Fig. 3 Roadmap for triazolium selection for the reaction modes available from NHC-catalyzed reactions of aldehydes. | ||
Acknowledgements
This work was supported by NIH GM-079339 and ETH-Zürich. J.M. is grateful for a predoctoral fellowship from Novartis. The authors thank Michael Rommel and Pinguan Zheng for preliminary investigations, and Juthanat Kaeobamrung and Pei-Chen Chiang for helpful discussions. We also thank Prof. Tomislav Rovis (Colorado State University) for his suggestion of catalyst 10.Notes and References
- (a) K. Y.-K. Chow and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 8126–8127 CrossRef CAS; (b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS.
- N. T. Reynolds, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2004, 126, 9518–9519 CrossRef CAS.
- C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS.
- For recent reviews on NHC catalysis, see (a) P.-C. Chiang and J. W. Bode, in N-Heterocyclic Carbenes, The Royal Society of Chemistry, 2011, 399–435 Search PubMed; (b) J. L. Moore and T. Rovis, Top. Curr. Chem., 2009, 291, 77–144; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC; (d) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS.
- (a) For the first report of a N-mesityl substituted triazolium precatalyst, see: S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873–3876 Search PubMed; (b) For the first chiral variant, see: M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418–8420 Search PubMed; (c) For the synthesis of N-mesityl triazolium salts, see: J. R. Struble and J. W. Bode, Org. Synth., 2010, 87, 362–376 Search PubMed.
- P.-C. Chiang and J. W. Bode, TCI MAIL, 2011, 149, 2–17 Search PubMed.
- (a) The N-C6F5triazolium catalyst 2 was first reported in, M. S. Kerr, J. Read de Alaniz and T. Rovis, J. Org. Chem., 2005, 70, 5725–5728 Search PubMed; (b) The chiral amino alcohol-derived N-C6F5 precatalysts were first reported in, M. S. Kerr and T. Rovis, J. Am. Chem. Soc., 2004, 126, 8876–8877 Search PubMed; (c) For the synthesis of N-C6F5 triazolium salts, see: H. U. Vora, S. P. Lathrop, N. T. Reynolds, M. S. Kerr, J. Read de Alaniz and T. Rovis, Org. Synth., 2010, 87, 350–361 Search PubMed.
- For a discussion regarding the effect of different aryl substitution on triazolium salts in the context of intramolecular Stetter reactions see: T. Rovis, Chem. Lett., 2008, 37, 2–7 Search PubMed.
- We have reported that the weak base Cl− counterion deprotonates the triazolium to generate the NHC species: J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810–8812 Search PubMed.
- Examples include: (a) J. Kaeobamrung, M. C. Kozlowski and J. W. Bode, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20661–20665 CrossRef CAS; (b) J. Kaeobamrung and J. W. Bode, Org. Lett., 2009, 11, 677–680 CrossRef CAS; (c) M. He and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 418–419 CrossRef CAS; (d) P.-C. Chiang, J. Kaeobamrung and J. W. Bode, J. Am. Chem. Soc., 2007, 129, 3520–3521 CrossRef CAS.
- (a) P.-C. Chiang, M. Rommel and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 8714–8718 CrossRef CAS; (b) P.-C. Chiang, Y. Kim and J. W. Bode, Chem. Commun., 2009, 4566–4568 RSC.
- We noted an exception to this observation in the case of α-methyl cinnamaldehyde, in which H/D exchange is observed for both catalysts 1 and 2 (with 2 being faster at exchanging than 1, see ESI†). We attribute this to the reduced stability of the more sterically hindered Breslow intermediate formed in this case. This is similar to the Breslow intermediates derived from simple aldehyde such as benzaldehyde, which appeared to be formed reversibly. For example:
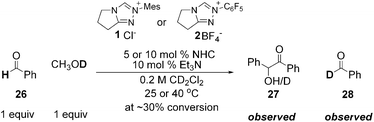 .
. - (a) R. Breslow and R. Kim, Tetrahedron Lett., 1994, 35, 699–702 CrossRef CAS; (b) J. H. Teles, J. P. Melder, K. Ebel, R. Schneider, E. Gehrer, W. Harder, S. Brode, D. Enders, K. Breuer and G. Raabe, Helv. Chim. Acta, 1996, 79, 61–83 CrossRef CAS; (c) M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124–5131 CrossRef CAS.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- N. T. Reynolds and T. Rovis, J. Am. Chem. Soc., 2005, 127, 16406–16407 CrossRef.
- (a) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS; (b) S. De Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef CAS.
- K. Zeitler, Org. Lett., 2006, 8, 637–640 CrossRef CAS.
- J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 1673–1677 CrossRef CAS.
- While this manuscript was under review, Mayr reported an elegant, detailed study on reaction of NHCs with various electrophiles and concluded that the observed reactivity of NHC catalysts originate from their high Lewis basicity, not their nucleophilicity. They concluded the attack of NHCs to the carbonyl group of α,β-unsaturated aldehydes occurs under kinetic control and “has a lower degree of reversibility.” Additionally, using Methyl Cation Affinity calculation, they found an N-mesityl substituted triazolium NHC to be more Lewis basic than the N-phenyl counterpart. B. Maji, M. Breugst and H. Mayr, Angew. Chem., Int. Ed., 2011, 50, 6915–6919 Search PubMed.
- A. Berkessel, S. Elfert, K. Etzenbach-Effers and J. H. Teles, Angew. Chem., Int. Ed., 2010, 49, 7120–7124 CrossRef CAS . In the case of α,β-unsaturated aldehydes these adducts appear to be irreversible but chemically unstable and could not yet be conclusively identified. For the N-mesityl catalyst, a 2:1 titration is observed (50% conversion at 1 equiv of cinnamaldehyde)..
- (a) J. L. Moore, A. P. Silvestri, J. R. de Alaniz, D. A. DiRocco and T. Rovis, Org. Lett., 2011, 13, 1742–1745 CrossRef CAS; (b) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402–10405 CrossRef CAS.
- M. T. Molina, C. Navarro, A. Moreno and A. G. Csáky, J. Org. Chem., 2009, 74, 9573–9575 CrossRef CAS.
- A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2006, 128, 4558–4559 CrossRef CAS.
- S. E. Hazlet and D. A. Stauffer, J. Org. Chem., 1962, 27, 2021–2024 CrossRef CAS.
- (a) J. Castells, H. Llitjos and M. Moreno-Mañas, Tetrahedron Lett., 1977, 18, 205–206 CrossRef; (b) H. Inoue and K. Higashiura, J. Chem. Soc., Chem. Commun., 1980, 549–550 RSC.
- (a) H. U. Vora, J. R. Moncecchi, O. Epstein and T. Rovis, J. Org. Chem., 2008, 73, 9727–9731 CrossRef CAS; (b) K. Thai, L. Wang, T. Dudding, F.o. Bilodeau and M. Gravel, Org. Lett., 2010, 12, 5708–5711 CrossRef CAS; (c) H. Jiang, B. Gschwend, L. Albrecht and K. Anker Jorgensen, Org. Lett., 2010, 12, 5052–5055 CrossRef CAS; (d) L. Deiana, P. Dziedzic, G.-L. Zhao, J. Vesely, I. Ibrahem, R. Rios, J. Sun and A. Córdova, Chem.–Eur. J., 2011, 17, 7904–7917 CrossRef CAS.
- R. M. Nowak, J. Org. Chem., 1963, 28, 1182–1187 CrossRef CAS.
- (a) D. E. A Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766–771 CrossRef CAS; (b) D. T. Cohen, B. Cardinal-David and K. A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 1678–1682 CrossRef CAS.
- Y. Li, Z.-A. Zhao, H. He and S.-L. You, Adv. Synth. Catal., 2008, 350, 1885–1890 CrossRef CAS.
- X. Fang, K. Jiang, C. Xing, L. Hao and Y. R. Chi, Angew. Chem., Int. Ed., 2011, 50, 1910–1913 CrossRef CAS.
- (a) X. Bugaut, F. Liu and F. Glorius, J. Am. Chem. Soc., 2011, 133, 8130–8133 CrossRef CAS; (b) I. Piel, M. Steinmetz, K. Hirano, R. Fröhlich, S. Grimme and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 4983–4987 CrossRef CAS; (c) K. Hirano, A. T. Biju, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 14190–14191 CrossRef CAS.
- (a) P.-C. Chiang and J. W. Bode, Org. Lett., 2011, 13, 2422–2425 CrossRef CAS; (b) B. Maji, S. Vedachalan, X. Ge, S. Cai and X.-W. Liu, J. Org. Chem., 2011, 76, 3016–3023 CrossRef CAS.
- For a side-by-side comparison of NHC-catalyzed reactions with N-mesityl substituted chiral triazolium and imidazolium precatalysts, see: J. R. Struble, J. Kaeobamrung and J. W. Bode, Org. Lett., 2008, 10, 957–960 Search PubMed.
- S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176–14177 CrossRef CAS.
- (a) C. Concellon, N. Duguet and A. D. Smith, Adv. Synth. Catal., 2009, 351, 3001–3009 CrossRef CAS; (b) X.-N. Wang, H. Lv, X.-L. Huang and S. Ye, Org. Biomol. Chem., 2009, 7, 346–350 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization for all compounds. See DOI: 10.1039/c1sc00397f |
| This journal is © The Royal Society of Chemistry 2012 |