Lipid-coated nanoscale coordination polymers for targeted delivery of antifolates to cancer cells†
Rachel C.
Huxford‡
,
Kathryn E.
deKrafft‡
,
William S.
Boyle
,
Demin
Liu
and
Wenbin
Lin
*
Department of Chemistry, CB#3290, University of North Carolina, Chapel Hill, NC 27599, USA. E-mail: wlin@unc.edu; Fax: +1-919-962-2388; Tel: +1-919-962-6320
First published on 20th October 2011
Abstract
Nanoscale coordination polymers (NCPs) have been demonstrated as an interesting platform for drug delivery, as they possess many advantages over small-molecule chemotherapeutics such as high payloads, lower systemic toxicity, tunability, and enhanced tumor uptake. Existing formulations for the delivery of methotrexate (MTX), an antifolate cancer drug, have very low drug loadings. Herein, we report the incorporation of MTX as a building block in an NCP formulation with exceptionally high drug loadings (up to 79.1 wt%) and the selective delivery of the NCP to cancer cells. Encapsulation of the NCP in a functionalized lipid bilayer allows for targeted delivery and controlled release to cancer cells. A phosphor can be doped into the NCPs for monitoring particle uptake by optical imaging. The lipid-coated and anisamide-targeted NCPs have superior in vitro efficacy against acute lymphoblastic leukemia cells when compared to the free drug.
Introduction
Coordination polymers, also known as metal–organic frameworks (MOFs), have recently emerged as an interesting class of hybrid materials comprised of organic bridging ligands coordinatively bound to metal-connecting points.1,2 The highly tunable nature of coordination polymers has allowed the design of numerous promising materials for a number of applications, including catalysis,3–8 nonlinear optics,9 light harvesting,10 selective gas adsorption,11,12 chemical sensing,13–16 and gas storage.17–22 When these materials are scaled down to the nano-regime to afford nanoscale coordination polymers (NCPs), they can be exploited for a number of biomedical applications.23–26 For example, we and others have demonstrated the potential utility of NCPs in magnetic resonance imaging,27–32 computed tomography,33 optical imaging,34,35 and biosensing.36 Whereas Horcajada and co-workers demonstrated the delivery of chemotherapeutics in the pores of the MIL family of NCPs,37,38 our group successfully delivered cisplatin prodrugs by either direct incorporation of the prodrug into an NCP or postsynthetic covalent attachment of a prodrug to a pre-synthesized NCP.39,40 In this work, we are utilizing a different direct incorporation strategy to deliver organic antifolate chemotherapeutics possessing functional groups that can bridge metal-connecting points in NCP formulations.41,42 Bulk scale coordination polymers have also been built from other biologically relevant molecules,43–45 pointing to the possibility of expanding the scope of their biological applications.Methotrexate (MTX) is a small molecule chemotherapeutic agent that works by inhibiting dehydrofolate reductase, thereby preventing DNA synthesis. While MTX is toxic to many cancer cells and is the first-line treatment for acute lymphoblastic leukemia (ALL), its efficacy is compromised by an array of drawbacks, including poor pharmacokinetics, low tolerated doses, and resistance.46 Large doses of MTX are required as a result of its non-specific distribution and rapid renal clearance, which can lead to systemic toxicity. Prolonged MTX treatment of ALL patients can result in numerous side effects such as mucositis, hematological toxicity, and secondary cancer. Drug delivery with nanoparticulate carriers can overcome many of the drawbacks of conventional chemotherapy, as nanoparticles can carry a large payload and significantly improve tumor uptake by taking advantage of the enhanced permeation and retention (EPR) effect.47 Drug encapsulation in liposomes has, for example, led to enhanced efficacy due to improved pharmacokinetics.48 Tumor uptake of nanotherapeutics can be further improved by surface conjugation of affinity molecules that bind to certain receptors overexpressed by cancer cells.47
Although several nanoparticle systems have been examined for MTX delivery, relatively low drug loadings were achieved in these studies.49–54 We hypothesized that MTX can be incorporated into NCPs as a bridging ligand since it contains two carboxylate groups which can coordinate to metal connecting points. Very high loadings of MTX can be achieved in such NCP formulations. Thus, we wish to report the synthesis and characterization of MTX-based NCPs, their stabilization and cancer targeting with a lipid bilayer, and in vitro efficacy against Jurkat ALL cells (Scheme 1). A carboxylic acid derivative of luminescent Ru(bpy)32+ (bpy = 2,2′-bipyridine) was also incorporated into the NCPs to allow the monitoring of MTX uptake by cancer cells via confocal microscopy.
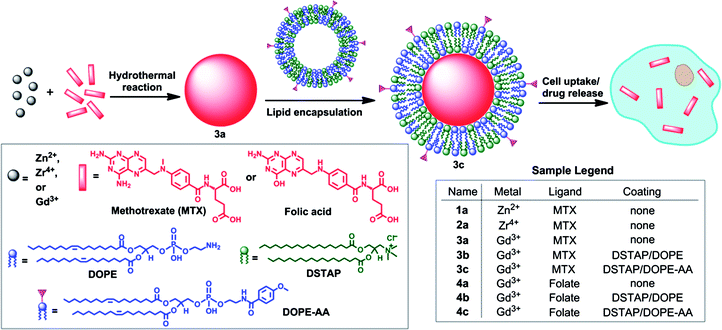 | ||
| Scheme 1 Synthesis of NCPs and functionalization of MTX-containing NCPs with a lipid bilayer and targeting moiety. | ||
Experimental section
Synthesis of Zn-MTX NCP (1)
NCPs of 1 were synthesized by a high-temperature surfactant-assisted method. Two microemulsions with W = 15 were prepared by the addition of 270 μL of an aqueous solution of MTX dimethylammonium salt (0.10 M, pH = 10.6) and 270 μL of an aqueous solution of Zn(NO3)2·6H2O (0.11 M) to separate 10 mL aliquots of a 0.1 M CTAB/0.5 M 1-hexanol/isooctane mixture. The separate microemulsions were stirred vigorously for 10 min at room temperature, then the two microemulsions were combined, and the resultant 20 mL microemulsion with W = 15 was transferred to a sealed microwave vessel. The reaction was rapidly heated to 120 °C and held at this temperature for 10 min with stirring. After cooling, the nanoparticles were isolated by centrifugation at 13,000 rpm for 10 min. After the removal of the supernatant, the particles were washed twice, using 10 mL of ethanol each time. For each wash, the particles were redispersed by sonication and then recovered by centrifugation at 13,000 rpm for 10 min. Yield: 11.8 mg (84.2%).Synthesis of Zr-MTX NCP (2)
ZrCl4 (6.99 mg, 0.03 mmol) was dissolved in 6 mL N,N-dimethylformamide (DMF), followed by 13.67 mg (0.03 mmol) methotrexate. The clear yellow solution was placed in a sealed microwave vessel and heated at 60 °C for 5 min (300 W) without stirring. The product was isolated from the resulting yellow dispersion by centrifugation at 13,000 rpm for 15 min, washed by sonication and centrifugation, with H2O then EtOH, and dispersed in EtOH. Yield: 8.88 mg (54.4%).Synthesis of Gd-MTX NCP (3a)
Both Gd(NO3)3·6H2O (13.54 mg, 0.0300 mmol) and MTX (13.63 mg, 0.0300 mmol) were dissolved in 6 mL DMF and the solution was heated in a sealed microwave vessel at 80 °C for 5 min without stirring. The product was isolated from the resulting yellow dispersion by centrifugation at 13,000 rpm for 15 min, washed by sonication and centrifugation with DMF then EtOH, and dispersed in EtOH. Yield: 15.6 mg (85%).Synthesis of Gd-folate NCP (4a)
22.5 mg (0.0498 mmol) Gd(NO3)3·6H2O was dissolved in 3 mL DMF, and 22.2 mg (0.0503 mmol) folic acid was dissolved in 7 mL DMF. The solutions were combined, and the heating procedure was similar to that of 3a. Yield: 7.5 mg (25.2%).Synthesis of [Ru(5,5′-CO2-bpy)(bpy)2]-doped analogs of 3a–c and 4a–c
10 mol% [Ru(5,5′-CO2-bpy)(bpy)2]-doped 3a was synthesized by the same procedure as 3a, but with the addition of 2.02 mg (0.00308 mmol, 10 mol%) [Ru(5,5′-CO2H-bpy)(bpy)2](PF6)2 to the DMF solution. Particles were isolated in a yield of 20.9 mg (72%). 10 mol% [Ru(5,5′-CO2-bpy)(bpy)2]-doped 4a was synthesized by the same procedure as 4a, but with the addition of 3.3 mg (0.0050 mmol, 10 mol%) Ru(bpy)2(bby-CO2H) to the DMF solution. Particles were isolated in a yield of 9.45 mg (29%). Synthesis of lipid-coated versions of doped 3a and 4a were carried out by the same methods for 3b, 3c, 4b, and 4c.Synthesis of lipid-coated NCPs.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (by mol) DSTAP/DOPE liposomes in 1 mM aq. KCl were heated to 50 °C and mixed together. The dispersion was vortexed and allowed to sit at room temperature for 30 min. Empty liposomes were removed by centrifugation at 6,000 rpm for 10 min, and the resulting particles of 3b were redispersed in 1 mM aq. KCl by sonication.
:1 (by mol) DSTAP/DOPE liposomes, incorporating 10 mol% DOPE-AA.
:1 (by mol) DSTAP/DOPE liposomes in 1 mM aq. KCl were heated to 50 °C and mixed together. The dispersion was vortexed and allowed to sit at room temperature for 30 min. Empty liposomes were removed by centrifugation at 6,000 rpm for 10 min, and the resulting particles of 3b were redispersed in 1 mM aq. KCl by sonication.
:1 (by mol) DSTAP/DOPE liposomes, incorporating 10 mol% DOPE-AA.
Cytotoxicity assay of 3a, 3b, and 3c against Jurkat cells
Confluent Jurkat ALL cells were counted from the culture flask using a hemocytometer. Cells were plated in 6-well plates at a cell density of 5 × 104 cells per well in 1.5 mL RPMI-1640 complete growth medium. The cells were incubated at 37 °C and 5% CO2 overnight. MTX and particle dispersions of 3a, 3b, and 3c (16 μM) in RPMI-1640 media and additional media were added to wells, resulting in MTX concentrations (μM) of 0, 0.25, 0.5, 1, 2, and 4. Cells were incubated (37 °C, 5% CO2) with free MTX or particles for 48 h or 72 h. Viability was determined by the trypan blue exclusion assay.Viability control assays with Jurkat cells
This assay was conducted to show that MTX is the only contributor to cytotoxicity and that Gd3+ has no bearing on the cytotoxicity. Gd(NO3)3, 4a, 4b, and 4c were tested against Jurkat cells. Confluent Jurkat cells were counted from the culture flask using a hemocytometer. Cells were plated in 6-well plates at a cell density of 5 × 104 cells per well in 1.5 mL RPMI-1640 complete growth medium. The cells were incubated at 37 °C and 5% CO2 overnight. Free Gd3+ and particle dispersions (200 μM) in RPMI-1640 media and additional media were added to wells, resulting in folate concentrations (μM) of 0, 0.5, 1, 2.5, 5, and 10. Cells were incubated (37 °C, 5% CO2) with free Gd3+ or particles for 48 h. Viability was determined by the trypan blue exclusion assay.Confocal microscopy of [Ru(5,5′-CO2-bpy)(bpy)2]-doped analogs of 3a–c and 4a–c
Wells in 6-well plates were seeded with 500,000 cells and 2 mL total of RPMI-1640 media (10% FBS, 2% penicillin-streptomycin). The plates were incubated for 24 h at 37 °C and 5% CO2. Particle dispersions were prepared in RPMI-1640 medium, and aliquots of particle dispersions/additional media were added to the wells, resulting in a concentration of 0.04 mg per well for each particle. The cells were incubated with particles for 1 h, and the cell suspensions were centrifuged. Cell pellets were re-suspended in PBS, 15 μL Annexin V FITC conjugate was added, and the cells were allowed to sit at room temperature for 10 min. The cells were centrifuged and redispersed in 20μL of media. The entire cell suspension was placed on glass coverslips, adhered on glass slides with antifade mounting medium, and imaged at the UNC–CH Microscope and Imaging Facility.Results and discussion
Synthesis and characterization of MTX-containing NCPs
Three new NCPs were synthesized using MTX as the bridging ligand and either Zn2+, Zr4+, or Gd3+ ions as the metal-connecting points. The properties of the NCPs, especially stability, can be tuned by the choice of metal ion. Zn2+ was originally chosen as the metal linker for an MTX-containing NCP due to its biocompatibility. Zn-MTX (1) was synthesized in 84% yield by first preparing a microemulsion of 0.1 M CTAB (CTAB = cetyltrimethylammonium bromide) and 0.5 M 1-hexanol in isooctane with a water-to-surfactant ratio (W) of 15, containing [CH3NH3]2(MTX) and Zn(NO3)2. Surfactant-assisted synthesis was carried out by microwave heating of the microemulsion in a sealed vessel at 120 °C for 10 min, with stirring. The particles were isolated by centrifugation and washed with ethanol. Both scanning electron microscopy (SEM) and transmission electron microscopy (TEM) showed that particles of 1 are spherical with a diameter between 40 and 100 nm (Fig. 1a). Dynamic light scattering (DLS) in ethanol gives a number average size of 161 nm, slightly larger than the size observed by microscopy due to slight aggregation in this medium (Table 1). These particles were shown by thermogravimetric analysis (TGA) (Fig. 2a) to contain 79.1 wt% MTX based on the organic weight loss. Powder X-ray diffraction (PXRD) reveals the amorphous structure of these particles by the lack of diffraction peaks (Fig. S3†). Attempts were made to coat 1 with a biocompatible shell (either silica or a lipid bilayer), but the particles were not stable under coating conditions. Coating 1 with a layer of silica by hydrolysis and condensation of tetraethylorthosilicate (TEOS) was unsuccessful because the ammonium hydroxide catalyst causes severe erosion of the particles. Lipid-coating of 1 also failed due to particle agglomeration in aqueous media. Additionally, the zeta potential of the particles is close to neutral (−1.9 mV), precluding efficient electrostatic interactions with positively-charged liposomes. | ||
| Fig. 1 TEM images of 1 (a), 2 (b), 3a (c, e), and 3b (d, f). The samples shown in e and f have been stained with uranyl acetate to highlight the lipid bilayer coating on 3b. The scale bars represent 400 nm for a–d, and 100 nm for e–f. | ||
| Sample | No. avg size (nm) | PDI | Zeta potential (mV) | wt % MTX |
|---|---|---|---|---|
| a Obtained in 1 mM aq. KCl unless otherwise noted. b Determined from TGA data. c Obtained in ethanol. d Estimated based on lipid coating content. | ||||
| 1 | 161c | 0.20c | −1.9 | 79.1 |
| 2 | 88.4 | 0.18 | −27.2 | 78.2 |
| 3a | 77.7 | 0.04 | −10.0 | 71.6 |
| 3b | 64.6 | 0.24 | 32.6 | ∼69.2d |
| 3c | 76.8 | 0.15 | 25.3 | ∼69.0d |
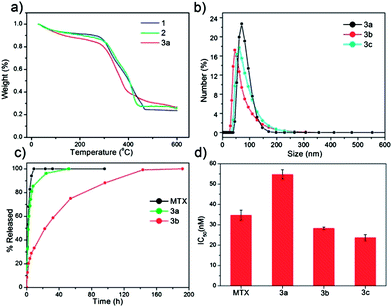 | ||
| Fig. 2 (a) TGA curves for 1, 2, and 3a. (b) Particle size distributions for 3a, 3b, and 3c obtained by DLS. (c) Release profiles in PBS for free MTX, 3a, and 3b obtained by measuring the release of MTX by UV-Vis spectroscopy. (d) IC50 values for free MTX, 3a, 3b, and 3c obtained from in vitro cytotoxicity assays with a range of MTX concentrations against Jurkat human ALL cells. Error bars represent one standard deviation. | ||
We hypothesized that more robust NCPs could be synthesized using Zr4+ in place of Zn2+ as the metal-connecting points, due to the strength of the Zr-carboxylate bond.55 Zr-MTX (2) was synthesized in 54% yield by microwave heating of a solution of MTX and ZrCl4 in N,N-dimethylformamide (DMF) at 60 °C for 5 min in a sealed vessel. The resulting spherical particles had a diameter of 70 to 180 nm by both SEM and TEM (Fig. 1b), and 88.4 nm by DLS (Fig. 2b). Particles of 2 had a high MTX loading of 78.2% by TGA. As-synthesized 2 had a zeta potential (1 mM aq. KCl) of −27.2 mV. This negative zeta potential, along with stability in water, allows for the coating of these particles with a cationic lipid bilayer to provide biocompatibility and stabilization in physiologically relevant media.56–582 was coated with different liposomal formulations, such as 4:48:48 (by mol) DOTAP/DOPE/cholesterol (DOTAP = dioleoyl trimethylammonium propane and DOPE = dioleoyl L-α-phosphatidylethanolamine), 1:1 (by mol) DOTAP/cholesterol, DOTAP, DSTAP (DSTAP = 1,2-stearoyl-3-trimethylammonium propane), and 1:1 (by mol) DSTAP/DOPE. Different liposomal formulations were made by drying a chloroform solution of lipids to create a film that was then hydrated with 1 mM aq. KCl. The resulting liposomes were extruded through polycarbonate membranes several times to obtain unilamellar vesicles with sizes ranging from 25–160 nm, depending on the formulation. Zeta potentials ranged from 50 to 60 mV. Dispersions of liposomes and 2 were mixed together, and empty liposomes were removed by centrifugation at low speed. Coating occurs by rearrangement of the cationic liposomes onto the anionic surface of the particles, resulting in the encapsulation of 2 in a lipid bilayer. Lipid coating of 2 was verified by an increase in the zeta potential from −27.2 mV to 25–50 mV. However, lipid coating failed to provide any significant stabilization over the as-synthesized particles in phosphate buffered saline (PBS) or simulated body fluid (SBF). The half-life for both coated and bare particles was approximately 2.5 h in PBS and 0.5 h in SBF at 37 °C and pH = 7.4 (Fig. S6, S7†). We speculate that lipid coating failed to stabilize 2 owing to the strong driving force in forming Zr3(PO4)4 in the presence of phosphate ions. For comparison, Zr3(PO4)4 has a Ksp value of 10−134 while Zn3(PO4)2 has a Ksp value of 10−34.
Considering our inability to stabilize 1 and 2, we rationalized that a formulation containing a metal less labile than zinc, but with a phosphate Ksp much greater than that of Zr3(PO4)4, would prove easier to stabilize by lipid coating. Trivalent lanthanide ions have much greater Ksp values (10−23 for Gd(PO4) and should provide a good balance between particle stability and lipid stabilization toward lanthanide phosphate formation. Gd-MTX NCP, 3a, was synthesized in 85% yield by microwave heating of a solution of MTX and Gd(NO3)3 in DMF at 80 °C for 5 min in a sealed vessel. The as-synthesized particles of 3a were spheres of 70 to 200 nm by SEM and TEM (Fig. 1c) and 77.7 nm by DLS (Fig. 2b). By TGA, these particles contain 71.6 wt% MTX (Fig. 2a). With a zeta potential (1 mM aq. KCl) of −10.0 mV, 3a can be coated with a cationic liposome in order to further stabilize these particles.
Stabilization of Gd-MTX NCP with a lipid bilayer
The half-life of 3a is only 2 h in 8 mM PBS at 37 °C, and needs to be prolonged for many biological applications (Fig. 2c). Stabilization with a lipid bilayer was pursued, due to the biocompatibility and known biological stability of this coating.56–58 Thus, 3a was coated with a 1:1 (by mol) DSTAP/DOPE liposomal formulation. These liposomes were made as previously discussed and had an average size of 94.3 nm and a zeta potential of 59.5 mV by DLS. The liposomes were mixed with a particle dispersion of 3a in 1 mM aq. KCl at a 10:1 particle:lipid weight ratio to obtain 3a encapsulated in a lipid bilayer (3b). Complete coverage is indicated by an increase in the zeta potential to 32.6 mV, and a size similar to the as-synthesized particles. Complete coverage of individual particles with a lipid bilayer was confirmed by TEM (Fig. 1f). 3b, when stained with 5% aq. uranyl acetate, exhibited dark rings around each particle due to strong binding of uranyl ions with the phosphates of DOPE in the lipid bilayer.59 The lipid bilayer stabilized the particles against dissolution, as evidenced by a release profile in 8 mM PBS (37 °C, pH = 7.4). MTX release was monitored by UV-Vis spectroscopy as the particles were dialyzed. The half-life of 3b was 23 h, compared to t1/2 = 2 h for 3a, with 100% release at 192 h (Fig. 2c). It is worth noting that lipid-coated NCPs are individual, lipid-stabilized, solid particles and are different from conventional liposomes.60
In order to improve delivery of nanoparticles in vitro, particles can be modified with affinity molecules to target receptors which are overexpressed on the surface of tumor cells. For example, sigma receptors are overexpressed on a number of different human cancer cell lines, including lung, colorectal, breast, and others.61 These sigma receptors can be targeted by small benzamides, such as anisamide (AA).62–64 For this work, we were able to conjugate AA to the primary amine of DOPE and integrate approximately 10 mol% of this lipid into the liposomal formulation before lipid coating of 3a, resulting in AA-functionalized particles (3c). The particle size of 3c is similar to that of the as-synthesized particles, and the zeta potentials are positive, indicating coverage by the cationic liposome.
Synthesis and characterization of Gd-Folate NCP control vehicles
As a control, a non-cytotoxic analog of 3a was synthesized by substituting folic acid, a naturally-occurring nutrient, for MTX to give a Gd-folate NCP (4a). Folic acid has a structure similar to MTX, with two carboxylic acid groups which can coordinate to metal ions. These particles were synthesized in DMF with microwave heating in a similar fashion to 3a. The resulting particles were 200–300 nm by SEM, and 262 nm by DLS. These particles have a zeta potential of −11.8 mV (1 mM aq. KCl), and can be coated with 1:1 (by mol) DSTAP/DOPE liposomes (4b) and functionalized with AA (4c) by the methods described for 3a. Control vehicles 4a–c do not contain any anticancer agent and were utilized in cytotoxicity and confocal microscopy experiments as analogs of 3a–c.
Enhanced in vitro efficacy of lipid-coated Gd-MTX NCPs
3a, 3b, and 3c all showed enhanced efficacy compared to free MTX in vitro against Jurkat human ALL cells. Jurkat cells are known to overexpress sigma receptors,65 and can therefore be subjected to AA-targeting. Cells were incubated with MTX or NCPs for 72 h, and then viability was determined by the trypan blue exclusion assay. 3a was slightly less cytotoxic than free MTX (IC50 = 34.7 ± 2.5 nM), with an IC50 of 54.7 ± 2.3 nM. Improved efficacy of 3a over free MTX is not expected due to the rapid degradation of these particles in biological media. The lipid coating on 3b significantly improved efficacy with an IC50 value of 28.3 ± 0.6 nM; this enhancement is contributed to the affinity of cationic lipid bilayers for cell membranes. AA targeting proved successful, with 3c having a slightly lower, statistically significant, IC50 (23.7 ± 1.5 nM) than the lipid-coated formulation (Fig. 2d). Free Gd3+ salt and 4a–c were also tested (Fig. S16†), and did not exhibit any cytotoxic effect in the same concentration range, proving that MTX, not Gd3+, is responsible for the cytotoxicity of 3a–c. 1 and 2 were also tested against human cancer cell lines (Fig. S13, S14†), and no enhancement in efficacy over free MTX was observed, presumably due to their rapid degradation in biological media to release free MTX.Optical imaging of Gd-MTX NCPs with Jurkat human ALL cells
3a and 4a were doped with approximately 10 mol% of a carboxylic acid derivative of the phosphor Ru(bpy)32+ so that this platform can be used for optical imaging. Additionally, this doping strategy allows for further verification of successful lipid coating when the lipid is functionalized with FITC (fluoroscein isothiocyanate) (Fig. S9†). The doped particles were synthesized in DMF by the same method as 3a and 4a but with the addition of 10 mol% of the Ru(bpy)32+ derivative. The doped particles had similar sizes, ligand loadings, and zeta potentials to their undoped counterparts, allowing phosphorescent versions of 3b, 3c, 4b, and 4c to be made from doped 3a and 4a. Laser scanning confocal fluorescence microscopy was performed on Jurkat cells incubated with NCPs to assess the imaging and targeting efficiency of the NCPs. The cells were also stained with Annexin V FITC conjugate to assay for early apoptosis. As displayed in Fig. 3, co-localized fluorescence signal is observed in the cells from the Ru(bpy)32+-doped particles (red) and the apoptosis stain (green) for 3b. Significantly stronger red and green fluorescence signals are seen for 3c, due to increased cell uptake of the targeted particles, and hence increased cytotoxicity. No red or green fluorescence was observed for 3a, as these particles disintegrate quickly, and do not have a mechanism to permeate the cell membrane during the 1 h incubation time for the experiment. Fluorescence from Ru(bpy)32+ was observed for the cells incubated with 4b and 4c, similar to 3b and 3c. However, as expected, no apoptosis (green fluorescence) was observed for 4a–c (Fig. S18†).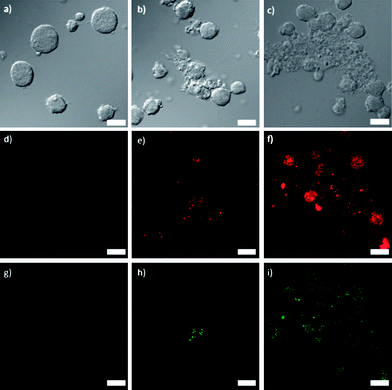 | ||
| Fig. 3 (a–c) Differential interference contrast (DIC) images and (d–i) fluorescence images of Jurkat cells incubated with 3a (d, g), 3b (e, h), and 3c (f, i). Red fluorescence (d–f) is from the Ru(bpy)32+-doped particles and green fluorescence (g–i) is from the Annexin V FITC conjugate early apoptosis stain. Scale bars represent 25 μm. | ||
Conclusions
We have successfully formulated novel NCPs containing up to 79.1 wt% methotrexate. Gd-MTX NCPs were stabilized by a DSTAP/DOPE lipid bilayer. A DOPE-AA conjugate was incorporated into the lipid bilayer and used to target sigma receptors overexpressed on leukemia cancer cells. The NCPs were doped with 10 mol% of a carboxylated Ru(bpy)32+ derivative, which serves as an optical imaging agent, as evidenced by confocal microscopy of Jurkat human acute lymphoblastic leukemia cells. Finally, lipid-coated and targeted NCPs were shown to have superior efficacy compared to the as-synthesized particles or free drug in in vitro cytotoxicity assays with Jurkat cells. We believe that this NCP formulation strategy is general and can be applied to deliver many other organic anticancer drugs.Acknowledgements
We acknowledge financial support from NIH (U01-CA1511455) and NSF (DMR-0906662). We thank Ms. Stephanie Kramer for experimental help.Notes and references
- R. Robson, J. Chem. Soc., Dalton Trans., 2000, 3735–3744 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- B. Kesanli and W. B. Lin, Coord. Chem. Rev., 2003, 246, 305–326 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS.
- F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS.
- O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS.
- C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS.
- K. Uemura, S. Kitagawa, K. Fukui and K. Saito, J. Am. Chem. Soc., 2004, 126, 3817–3828 CrossRef CAS.
- K. Uemura, S. Kitagawa, M. Kondo, K. Fukui, R. Kitaura, H.-C. Chang and T. Mizutani, Chem.–Eur. J., 2002, 8, 3586–3600 CrossRef CAS.
- B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui and G. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS.
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS.
- A. Lan, K. Li, H. Wu, D. H. Olsson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem. Int. Ed., 2009, 48, 2334–2338 CAS.
- Z. Xie, L. Ma, K. E. deKrafft, A. Jin and W. Lin, J. Am. Chem. Soc., 2009, 132, 922–923 CrossRef.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS.
- M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef CAS.
- H. Furukawa, N. Ko, Y.-B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS.
- L. Ma, D. J. Mihalcik and W. Lin, J. Am. Chem. Soc., 2009, 131, 4610–4612 CrossRef CAS.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS.
- J. Della Rocca and W. Lin, Eur. J. Inorg. Chem., 2010, 3725–3734 CrossRef CAS.
- R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS.
- W. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650–658 CrossRef CAS.
- A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC.
- R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata, C. Adachi, Y. Katayama, M. Hashizume and N. Kimizuka, J. Am. Chem. Soc., 2009, 131, 2151–2158 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024–9025 CrossRef CAS.
- M. D. Rowe, C. C. Chang, D. H. Thamm, S. L. Kraft, J. F. Harmon, A. P. Vogt, B. S. Sumerlin and S. G. Boyes, Langmuir, 2009, 25, 9487–9499 CrossRef CAS.
- M. D. Rowe, D. H. Thamm, S. L. Kraft and S. G. Boyes, Biomacromolecules, 2009, 13, 983–993 CrossRef.
- K. M. L. Taylor, A. Jin and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 7722–7725 CrossRef CAS.
- K. M. L. Taylor, W. J. Rieter and W. Lin, J. Am. Chem. Soc., 2008, 130, 14358–14359 CrossRef CAS.
- K. E. deKrafft, Z. Xie, G. Cao, S. Tran, L. Ma, O. Z. Zhou and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9901–9904 CrossRef CAS.
- D. Liu, R. C. Huxford and W. Lin, Angew. Chem., Int. Ed., 2011, 50, 3696–3700 CrossRef CAS.
- M. Roming, H. Lunsdorf, K. E. J. Dittmar and C. Feldmann, Angew. Chem. Int. Ed., 2010, 49, 632–637 CAS.
- W. J. Rieter, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2007, 129, 9852–9853 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Ferey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6 CrossRef CAS.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS.
- K. M. L. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS.
- I. Imaz, M. Rubio-Martinez, L. Garcia-Fernandez, F. Garcia, D. Ruiz-Molina, J. Hernando, V. Puntes and D. Maspoch, Chem. Commun., 2010, 46, 4737–4739 RSC.
- S. R. Miller, D. Heurtaux, T. Baati, P. Horcajada, J. M. Greneche and C. Serre, Chem. Commun., 2010, 46, 4526–4528 RSC.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–4 CrossRef CAS.
- J. Rabone, Y. F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–7 CrossRef CAS.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–7 CrossRef CAS.
- M. Krajinovic and A. Moghrabi, Pharmacogenomics, 2004, 5, 819–834 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- G. P. Stathopoulos, Anti-Cancer Drugs, 2010, 21, 732–736 CrossRef CAS.
- Y.-H. Chen, C.-Y. Tsai, P.-Y. Huang, M.-Y. Chang, P.-C. Cheng, C.-H. Chou, D.-H. Chen, C.-R. Wang, A.-L. Shiau and C.-L. Wu, Mol. Pharmaceutics, 2007, 4, 713–722 CrossRef CAS.
- R. S. Dhanikula, A. Argaw, J.-F. Bouchard and P. Hildgen, Mol. Pharmaceutics, 2008, 5, 105–116 CrossRef CAS.
- N. Kohler, C. Sun, A. Fichtenholtz, J. Gunn, C. Fang and M. Zhang, Small, 2006, 2, 785–792 CrossRef CAS.
- N. Kohler, C. Sun, J. Wang and M. Zhang, Langmuir, 2005, 21, 8858–8864 CrossRef CAS.
- K. Wosikowski, E. Biedermann, B. Rattel, N. Breiter, P. Jank, R. Loser, G. Jansen and G. J. Peters, Clin. Cancer Res., 2003, 9, 1917–1926 CAS.
- X. Yang, Q. Zhang, Y. Wang, H. Chen, H. Zhang, F. Gao and L. Liu, Colloids Surf., B, 2008, 61, 125–131 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. L. Guillou, C., S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. W. Chackerian, W., D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS.
- J. Li, Y.-C. Chen, Y.-C. Tseng, S. Mozumdar and L. Huang, J. Controlled Release, 2010, 142, 416–421 CrossRef CAS.
- J. Liu, A. Stace-Naughton, X. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 1354–1355 CrossRef CAS.
- A.-L. Troutier, T. Delair, C. Pichot and C. Ladavire, Langmuir, 2005, 21, 1305–1313 CrossRef CAS.
- H. Chen, R. C. MacDonald, S. Li, N. L. Krett, S. T. Rosen and T. V. O'Halloran, J. Am. Chem. Soc., 2006, 128, 13348–13349 CrossRef CAS.
- B. J. Vilner, C. S. John and W. D. Bowen, Cancer Res., 1995, 55, 408–413 CAS.
- E. Aydar, C. P. Palmer and M. B. A. Djamgoz, Cancer Res., 2004, 64, 5029–5035 CrossRef CAS.
- R. Banerjee, P. Tyagi, S. Li and L. Huang, Int. J. Cancer, 2004, 112, 693–700 CrossRef CAS.
- C. S. John, B. J. Vilner, B. C. Geyer, T. Moody and W. D. Bowen, Cancer Res., 1999, 59, 4578–4583 CAS.
- M. E. Ganapathy, P. D. Prasad, W. Huang, P. Seth, F. H. Leibach and V. Ganapathy, J. Pharmacol. Exp. Ther., 1999, 289, 251–260 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: general experimental; SEM images of 1, 2, 3a, 4a, Ru(bpy)32+-doped 3a, and Ru(bpy)32+-doped 4a; TEM images of liposomes, 1, 2, 3a, and 3b; PXRD of 1; DLS of liposomes, 1, 2, and 3a; UV-Vis spectra of MTX, 3a, and Ru(bpy)32+-doped 3a; release profiles of 2 and coated 2 in PBS and SBF; preparation of SBF; confocal images of Ru(bpy)32+-doped 3b coated in 10 mol% DOPE-FITC liposomes; synthesis of DOPE-FITC; in vitro cytotoxicity procedure and curves for 1 and 2; in vitro cytotoxicity curves for 3a–c; in vitro viability assays for 4a–c and Gd3+; and confocal images of Jurkat cells incubated with 4a–c. See DOI: 10.1039/c1sc00499a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |