Asymmetric dearomatization of pyrrolesviaIr-catalyzed allylic substitution reaction: enantioselective synthesis of spiro-2H-pyrroles†‡
Chun-Xiang
Zhuo
,
Wen-Bo
Liu
,
Qing-Feng
Wu
and
Shu-Li
You
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai, 200032, China. E-mail: slyou@sioc.ac.cn; Fax: (+86) 21-5492-5087
First published on 12th September 2011
Abstract
Asymmetric dearomatization of pyrroles has been accomplished by using Ir-catalyzed intramolecular asymmetric allylic alkylation reactions. Reactions of allylic carbonate tethered pyrroles in the presence of [Ir(cod)Cl]2 and a BINOL-derived phosphoramidite ligand lead to efficient generation of spiro-2H-pyrrole derivatives with up to 96% ee.
The pyrrole moiety serves as the key structural core of numerous biologically active natural products and pharmaceutical agents.1,2 However, enantioselective transformations of pyrroles have not been fully explored.3Pyrroles have similar structural and electronic properties as do indoles, which readily participate in selective Friedel–Crafts alkylation reactions as a consequence of the fact that the 3-position of indoles is much more reactive with electrophiles.4 In contrast, relatively few Friedel–Crafts alkylation reactions of pyrroles are known, perhaps a result of a regioselectivity issue caused by the fact that the 2- and 3-positions of pyrroles display similar reactivity with electrophiles.5 In addition, alkylated pyrroles are generally more reactive than their parent pyrroles, a property that makes highly selective formation of mono-alkylated products a challenging issue.6
Enantioselective preparation of substituted spiro-2H-pyrrole derived substructures, which are interesting synthetic intermediates and scaffolds for drug discovery, remains a significant goal in synthetic organic chemistry (Fig. 1).7 Important problems confront the synthesis of substituted spiro-2H-pyrroles, including regioselectivity and dearomatization, both of which are known to be unfavourable processes. Although seemingly straightforward, to our knowledge no catalytic enantioselective synthetic methods exist for generating these important synthetic intermediates from pyrroles(eqn (a)).
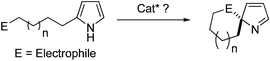 | (a) |
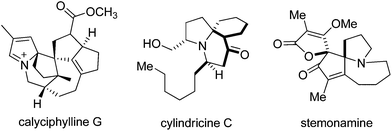 | ||
| Fig. 1 Selected naturally occurring compounds bearing spiro-2H-pyrrole and spiro-pyrrolidine units. | ||
As part of a continuing research program focusing on asymmetric dearomatization reactions,8,9 we have explored catalytic asymmetric dearomatization reactions of pyrroles. Recently, Reddy and Davies have elegantly demonstrated that N-protected pyrroles undergo [4 + 3] cycloaddition reactions with vinyldiazoacetates in the presence of a chiral Rh-complex to produce tropanes in good yields and with excellent ee.10 In an exploration aimed at uncovering methods to carry out efficient synthesis of enantioenriched spiro-2H-pyrroles bearing a chiral quaternary carbon center, we recently probed intramolecular allylic dearomatization reactions of pyrroles. In spite of the reactivity issues mentioned above, these reactions proceed in high yields with excellent levels of chemo-, regio-, and enantioselectivities. Below, we describe the results of this effort which has resulted in the development of a method for the enantioselective synthesis of spiro-2H-pyrroles that utilizes an Ir-catalyzed asymmetric allylic dearomatization reaction of pyrroles.
Studies of this problem were initiated by an exploration of the reaction of the allylic carbonate tethered pyrrole 2a with an Ir-catalytic system comprised of [Ir(cod)Cl]2 and the phosphoramidite ligand 1a (Table 1).11 In the presence of 2 mol% of [Ir(cod)Cl]2, 4 mol% of 1a, and 1.0 equiv. of Cs2CO3, reaction of 2a in THF for 15 h gave the spiro-2H-pyrrole 3a in 69% yield, 99/1 dr and 87% ee (entry 1, Table 1). In order to optimize the reaction conditions, various bases such as K3PO4, DBU, DABCO and Li2CO3 were screened (entries 2–5, Table 1). This exploration led to the finding that Cs2CO3 is the optimal base for the process. The effects of different chiral ligands were examined next. Ligands 1b and 1c were observed to form catalysts that catalyze the reaction of 2a with good ee, but with only moderate yields and dr. In addition, Ir catalysts containing ligands 1d and 1e were not effective in promoting the allylic dearomatization process. Interestingly, screening of ligands 1f, 1g, and 1h that were developed in our earlier work12 demonstrated that the catalyst derived from 1f gave satisfactory results in terms of yield, dr and ee (entry 10, Table 1). Also, screening of various solvents (entries 13–16, Table 1) led to the identification of THF as being ideal. Finally, the effects of substrate concentration, base loading and temperature were probed (entries 17–19, Table 1). In combination with observations made in other optimization studies, the results showed that the best conditions for the reaction of 2a are THF (0.1 M) with 2 mol% of [Ir(cod)Cl]2, 4 mol% 1f, and 1.0 equiv. of Cs2CO3 at 50 °C. This process produces spiro-2H-pyrrole 3a in 80% yield, 99/1 dr and 93% ee (entry 10, Table 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Base | t/h | Yield (%)b | drc | ee (%)d |
| a Reaction conditions: 0.1 mmol of 2a, 0.1 mmol of base in solvent (1.0 mL) at 50 °C or under reflux. b Isolated yield of the major diastereoisomer. c Determined by 1H NMR of the crude reaction mixture. d Determined by HPLC analysis. e Reaction was performed in 2.0 mL solvent. f Reaction was performed with 2.0 equiv. of base. g Reaction was performed at room temperature. | |||||||
| 1 | 1a | THF | Cs2CO3 | 15 | 69 | 99/1 | 87 |
| 2 | 1a | THF | K3PO4 | 34 | 60 | 99/1 | 86 |
| 3 | 1a | THF | DBU | 34 | 46 | 99/1 | 88 |
| 4 | 1a | THF | DABCO | 34 | 16 | 85/15 | 86 |
| 5 | 1a | THF | Li2CO3 | 34 | 62 | 99/1 | 85 |
| 6 | 1b | THF | Cs2CO3 | 38 | 64 | 89/11 | 83 |
| 7 | 1c | THF | Cs2CO3 | 38 | 53 | 88/12 | 84 |
| 8 | 1d | THF | Cs2CO3 | 38 | Trace | — | — |
| 9 | 1e | THF | Cs2CO3 | 38 | Trace | — | — |
| 10 | 1f | THF | Cs2CO3 | 21 | 80 | 99/1 | 93 |
| 11 | 1g | THF | Cs2CO3 | 21 | 69 | 91/9 | 84 |
| 12 | 1h | THF | Cs2CO3 | 39 | Trace | — | — |
| 13 | 1f | DCM | Cs2CO3 | 21 | 82 | 83/17 | 88 |
| 14 | 1f | Toluene | Cs2CO3 | 21 | 72 | 93/7 | 92 |
| 15 | 1f | Dioxane | Cs2CO3 | 21 | 74 | 95/5 | 89 |
| 16 | 1f | Et2O | Cs2CO3 | 21 | 80 | 91/9 | 93 |
| 17e | 1f | THF | Cs2CO3 | 21 | 71 | 94/6 | 88 |
| 18f | 1f | THF | Cs2CO3 | 21 | 69 | 99/1 | 90 |
| 19g | 1f | THF | Cs2CO3 | 39 | 67 | 96/4 | 88 |

Employing the optimized conditions, reactions of various 2-pyrrolyl allylic carbonates were explored to examine the generality of the process (Table 2). The stereochemistry of the products of these processes was assigned based on the results of X-ray crystallographic analysis of enantiopure 3g.‡ Reactions of allylic carbonates containing various protecting groups (Bn, allyl, 4-Br-C6H4CH2) on the amine moiety in the tether all gave the corresponding spiro-2H-pyrrole products in good yields with excellent dr and ee (79–85% yield, 95/5–99/1 dr, 86–93% ee, entries 1–3, Table 2). Notably, substrate 2d, containing an N-methyl tertiary amine group, did not react smoothly (47% yield, 91/9 dr, 81% ee) under the optimized conditions. However, by switching the ligand in the catalyst to 1a, this reaction generated 3d in good yield with excellent dr and ee (77% yield, >99/1 dr, 94% ee, entry 4, Table 2). Substrates bearing either electron-donating (4-Me, 4-MeO, 3,4-(MeO)2) (entries 5–8, Table 2) or electron-withdrawing groups (4-Cl, 4-F) (entries 9–10, Table 2) on the 5-phenyl moiety (R2) on the pyrrole core all reacted to form the corresponding products in good yields, dr and ee (82–90% yield, 92/8–97/3 dr, 84–95% ee). Importantly, reaction took place smoothly on the substrate 2k, in which an alkyl group is present at the pyrrole 5-position (forming 3k, 83% yield, 96% ee, 90/10 dr, entry 11, Table 2). However, the pyrrole 2l, bearing no substituent at C-5, reacted inefficiently under the optimized conditions outlined above to give a low yield and ee of 3l (21% yield, 57% ee). In contrast, by using the catalyst containing ligand 1a, the yield of this process was improved and excellent dr and ee were obtained (61% yield, 97/3 dr, 96% ee, entry 12, Table 2). Unfortunately, when the carbon tethered substrate was used, only a trace amount of the corresponding product could be observed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2, R1, R2 | 3 | Yield (%)a | drb | ee (%)c |
| a Isolated yields of the major diastereoisomer. b Determined by 1H NMR of the crude reaction mixture. c Determined by HPLC analysis. d Reaction was performed with 1a as ligand. | |||||
| 1 | 2a, Bn, Ph | 3a | 80 | 99/1 | 93 |
| 2 | 2b, allyl, Ph | 3b | 85 | 95/5 | 86 |
| 3 | 2c, 4-Br-C6H4CH2, Ph | 3c | 79 | 96/4 | 90 |
| 4d | 2d, Me, Ph | 3d | 77 | >99/1 | 94 |
| 5 | 2e, Bn, 4-Me-C6H4 | 3e | 83 | 93/7 | 84 |
| 6 | 2f, Bn, 4-MeO-C6H4 | 3f | 88 | 97/3 | 95 |
| 7 | 2g, 4-Br-C6H4CH2, 4-MeO-C6H4 | 3g | 82 | 97/3 | 95 |
| 8 | 2h, Bn, 3,4-(MeO)2-C6H3 | 3h | 82 | 94/6 | 91 |
| 9 | 2i, Bn, 4-Cl-C6H4 | 3i | 88 | 92/8 | 86 |
| 10 | 2j, Bn, 4-F-C6H4 | 3j | 90 | 92/8 | 89 |
| 11 | 2k, Bn, Et | 3k | 83 | 90/10 | 96 |
| 12d | 2l, Bn, H | 3l | 61 | 97/3 | 96 |

To demonstrate the utility of the newly developed methodology, several transformations of the spiro-2H-pyrrole products were carried out. As shown in Scheme 1, treatment of 3l with sodium borohydride afforded the spiro-2,5-dihydropyrrole 4 in 90% yield (Scheme 1, eqn (1)). The spirolactam 5 was obtained via a two-step route, involving sequential treatment with NaClO2 and sodium borohydride (Scheme 1, eqn (2)).13,14 When compound 3l was subjected to Pd/C-catalyzed hydrogenation conditions using 600 psi of hydrogen, spiropyrroline 7 was formed in 75% yield. Interestingly, when this process was carried out under 1 atm of hydrogen, spiro-3,4-dihydropyrrole 6 was produced in 65% yield (Scheme 1, eqn (3) and (4)). The results suggest that selective hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C over the CN bond is possible, a phenomenon that should enhance the synthetic utility of the spiro-2H-pyrrole products. Notably, the CN bond of product 3g, bearing an aryl substituted imine, could not be reduced using sodium borohydride but it was transformed to amine 8 when sodium cyanoborohydride in acetic acid was employed (Scheme 1, eqn (5)).13,15 In all processes, no notable loss of enantiomeric purity took place.
C over the CN bond is possible, a phenomenon that should enhance the synthetic utility of the spiro-2H-pyrrole products. Notably, the CN bond of product 3g, bearing an aryl substituted imine, could not be reduced using sodium borohydride but it was transformed to amine 8 when sodium cyanoborohydride in acetic acid was employed (Scheme 1, eqn (5)).13,15 In all processes, no notable loss of enantiomeric purity took place.
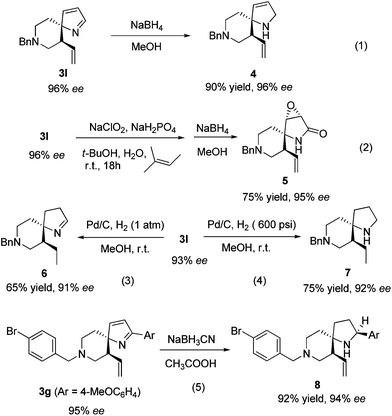 | ||
| Scheme 1 Transformation of the products. | ||
In summary, the investigation described above has led to the development of the first Ir-catalyzed intramolecular asymmetric allylic dearomatization reaction of pyrroles. The spiro-2H-pyrrole derivatives were generated in these reactions in good yields with up to >99/1 dr and 96% ee. Extensions of the scope and applications of the spiro-2H-pyrrole forming processes are currently under study in our laboratory.
Acknowledgements
We thank the National Basic Research Program of China (973 Program 2009CB825300), the NSFC (20821002, 20923005, 21025209), and the Chinese Academy of Sciences for generous financial support of this research.Notes and references
- For reviews: (a) H. Hoffmann and T. Lindel, Synthesis, 2003, 1753 CAS; (b) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582 CrossRef; (c) G. Balme, Angew. Chem., Int. Ed., 2004, 43, 6238 CrossRef CAS; (d) H. Fan, J. N. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS.
- Selected examples: (a) J. A. Johnson, N. Li and D. Sames, J. Am. Chem. Soc., 2002, 124, 6900 CrossRef CAS; (b) P. S. Baran, J. M. Richter and D. W. Lin, Angew. Chem., Int. Ed., 2005, 44, 609 CrossRef CAS; (c) A. Fürstner, K. Radkowski and H. Peters, Angew. Chem., Int. Ed., 2005, 44, 2777 CrossRef.
- Selected examples: (a) W. Zhuang, N. Gathergood, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2001, 66, 1009 CrossRef CAS; (b) N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370 CrossRef CAS; (c) D. A. Evans and K. R. Fandrick, Org. Lett., 2006, 8, 2249 CrossRef CAS; (d) B. M. Trost and G. Dong, J. Am. Chem. Soc., 2006, 128, 6054 CrossRef CAS; (e) D. A. Evans, K. R. Fandrick, H. J. Song, K. A. Scheidt and R. S. Xu, J. Am. Chem. Soc., 2007, 129, 10029 CrossRef CAS; (f) B. M. Trost and G. Dong, Org. Lett., 2007, 9, 2357 CrossRef CAS; (g) B. M. Trost and C. Müller, J. Am. Chem. Soc., 2008, 130, 2438 CrossRef CAS; (h) H. Liu, S.-F. Lu, J.-X. Xu and D.-M. Du, Chem.–Asian J., 2008, 3, 1111 CrossRef CAS; (i) N. Yokoyama and T. Arai, Chem. Commun., 2009, 3285 RSC; (j) Y.-F. Sheng, G.-Q. Li, Q. Kang and S.-L. You, Chem.–Eur. J., 2009, 15, 3351 CrossRef CAS; (k) Y.-F. Sheng, Q. Gu, A.-J. Zhang and S.-L. You, J. Org. Chem., 2009, 74, 6899 CrossRef CAS; (l) B. M. Trost, M. Osipov and G. Dong, J. Am. Chem. Soc., 2010, 132, 15800 CrossRef CAS; (m) Z. Cao, Y. Liu, Z. Liu, X. Feng, M. Zhuang and H. Du, Org. Lett., 2011, 13, 2164 CrossRef CAS; (n) during the preparation of this manuscript, Zhou and co-workers reported a Pd-catalyzed enantioselective partial hydrogenation of pyrroles. For details, see: D.-S. Wang, Z.-S. Ye, Q.-A. Chen, Y.-G. Zhou, C.-B. Yu, H.-J. Fan and Y. Duan, J. Am. Chem. Soc., 2011, 133, 8866 CrossRef CAS.
- For reviews: (a) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS; (b) M. Bandini, A. Melloni, S. Tommasi and A. Umani-Ronchi, Synlett, 2005, 1199 CrossRef CAS; (c) T. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS; (d) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (e) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS; (f) S.-L. You and M. Zeng, Synlett, 2010, 1289 CrossRef.
- (a) I. Fleming, in Frontier Orbitals and Organic Chemical Reactions, Wiley, New York, 1976, p. 58 Search PubMed; (b) K. Fukui, T. Yonezawa, C. Nagata and H. Shingu, J. Chem. Phys., 1954, 22, 1433 CrossRef CAS; for an elegant example on regioselective control: (c) I. T. Raheem, P. S. Thiara and E. N. Jacobsen, Org. Lett., 2008, 10, 1577 CrossRef CAS.
- (a) J. A. Joule, K. Mills and G. F. Smith, in Heterocyclic Chemistry, 3rd edn, Chapman & Hall, London, 1995, pp. 231–238 Search PubMed; (b) K. Schofield, in Heteroaromatic Nitrogen Compounds: Pyrroles and Pyridines, Plenum Press, New York, 1967 Search PubMed; (c) D. A. Williams, in Foye's Principles of Medicinal Chemistry, ed. T. L. Lemke, D. A. Williams, V. F. Roche and S. W. Zito, Lippincott Williams & Wilkins, Philadelphia, PA, 2008, vol. 6, p. 253 Search PubMed; (d) R. B. Silverman, in The Organic Chemistry of Drug Design and Drug Action, 2nd edn, Elsevier, Oxford, 2004, vol. 2, p. 7 Search PubMed; for a recent example: (e) Y. R. Jorapur, C. H. Lee and D. Y. Chi, Org. Lett., 2005, 7, 1231 CrossRef CAS.
- (a) R. Aumann, Z. Yu, R. Fröhlich and F. Zippel, Eur. J. Inorg. Chem., 1998, 1623 CrossRef CAS; (b) S. Saito, K. Takaaki and J. Kobayashi, Tetrahedron Lett., 2007, 48, 5693 CrossRef CAS; (c) F. C. Frebault and N. S. Simpkins, Tetrahedron, 2010, 66, 6585 CrossRef CAS; (d) M. A. Brodney, I. V. Efremov, C. J. Helal and B. T. O'Neill, PCT Int. Appl. WO 2010058333A1, 2010 Search PubMed; (e) Y.-M. Zhao, P. Gu, Y.-Q. Tu, C.-A. Fan and Q. Zhang, Org. Lett., 2008, 10, 1763 CrossRef CAS; (f) T. J. Donohoe, P. M. Brian, G. C. Hargaden and T. J. C. O'Riordan, Tetrahedron, 2010, 66, 6411 CrossRef CAS.
- (a) Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056 CrossRef CAS; (b) Q.-F. Wu, H. He, W.-B. Liu and S.-L. You, J. Am. Chem. Soc., 2010, 132, 11418 CrossRef CAS; (c) Q.-F. Wu, W.-B. Liu, C.-X. Zhuo, Z.-Q. Rong, K.-Y. Ye and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 4455 CrossRef CAS; (d) Q. Gu and S.-L. You, Chem. Sci., 2011, 2, 1519 RSC.
- Selected examples from other groups: (a) M. Takamura, K. Funabashi, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 6327 CrossRef CAS; (b) K. Funabashi, H. Ratni, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 10784 CrossRef CAS; (c) E. Ichikawa, M. Suzuki, K. Yabu, M. Albert, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 11808 CrossRef CAS; (d) J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS; (e) C. Y. Legault and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 8966 CrossRef CAS; (f) B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314 CrossRef CAS; (g) S. Quideau, G. Lyvinec, M. Marguerit, K. Bathany, A. Ozanne-Beaudenon, T. Buffeteau, D. Cavagnat and A. Chénedé, Angew. Chem., Int. Ed., 2009, 48, 4605 CrossRef CAS; (h) J. García-Fortanet, F. Kessler and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 6676 CrossRef; (i) S. Jones, B. Simmons and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 13606 CrossRef CAS; (j) M. Uyanik, T. Yasui and K. Ishihara, Tetrahedron, 2010, 66, 5841 CrossRef CAS; (k) T. Nemoto, Y. Ishige, M. Yoshida, Y. Kohno, M. Kanematsu and Y. Hamada, Org. Lett., 2010, 12, 5020 CrossRef CAS; (l) Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 440 CrossRef CAS; (m) S. Rousseaux, J. García-Fortanet, M. A. Del Aguila Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282 CrossRef CAS; (n) J. Qi, A. B. Beeler, Q. Zhang and J. A. Porco Jr, J. Am. Chem. Soc., 2010, 132, 13642 CrossRef CAS; (o) R. Leon, A. Jawalekar, T. Redert and M. J. Gaunt, Chem. Sci., 2011, 2, 1487 RSC; (p) A. Rudolph, P. H. Bos, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 5834 CrossRef CAS; (q) O. Lozano, G. Blessley, T. M. del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS; for a recent review, see: (r) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS.
- (a) R. P. Reddy and H. M. L. Davies, J. Am. Chem. Soc., 2007, 129, 10312 CrossRef CAS; for an early work using a chiral auxiliary see: (b) H. M. L. Davies, J. J. Matasi, L. M. Hodges, N. J. S. Huby, C. Thornley, N. Kong and J. H. Houser, J. Org. Chem., 1997, 62, 1095 CrossRef CAS.
- For reviews: (a) R. Takeuchi and S. Kezuka, Synthesis, 2006, 3349 CrossRef CAS; (b) G. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675 RSC; (c) G. Helmchen, in Iridium Complexes in Organic Synthesis, ed. L. A. Oro and C. Claver, Wiley-VCH, Weinheim, 2009, pp. 211–250 Search PubMed; (d) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS; for early examples: (e) R. Takeuchi and M. Kashio, Angew. Chem., Int. Ed. Engl., 1997, 36, 263 CrossRef CAS; (f) J. P. Janssen and G. Helmchen, Tetrahedron Lett., 1997, 38, 8025 CrossRef CAS.
- W.-B. Liu, H. He, L.-X. Dai and S.-L. You, Synthesis, 2009, 2076 CAS.
- The relative configurations of products 5 and 8 were determined by NOE spectra respectively. For details, see the ESI†.
- (a) C. J. Hayes, A. E. Sherlock, M. P. Green, C. Wilson, A. J. Blake, M. D. Selby and J. C. Prodger, J. Org. Chem., 2008, 73, 2041 CrossRef CAS; (b) M. A. Mohamed, K. Yamada and K. Tomioka, Tetrahedron Lett., 2009, 50, 3436 CrossRef CAS.
- U. von Fischer and F. Schneider, Helv. Chim. Acta, 1983, 66, 971 CrossRef.
Footnotes |
| † Dedicated to Professor Christian Bruneau on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and analysis data for new compounds, CIF file of 3g. CCDC reference number 824629. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00517k |
| This journal is © The Royal Society of Chemistry 2012 |