Structural and kinetic study of self-assembling macrocyclic dimer natural product aminoglycoside66-40C and unnatural variants†
Stephen
Hanessian
*,
Juan Pablo
Maianti
,
Vu Linh
Ly
and
Benoît
Deschênes-Simard
Department of Chemistry, Université de Montréal, C. P. 6128, Succ. Centre-Ville, Montréal, QC, Canada H3C 3J7. E-mail: stephen.hanessian@umontreal.ca; Fax: +1-514-343-5728; Tel: +1-514-343-6738
First published on 23rd September 2011
Abstract
Aminoglycoside 66-40C, co-produced with antibiotic sisomicin by Micromonospora inyoensis is unique among natural products, featuring a 16-membered macrocyclic bis-imine (X-ray) stable in aqueous solution and produced by quantitative self-assembly from self-selecting condensation of 6′-aldehyde monomers, in complex mixtures and in the presence of competing amine and aldehyde substrates.
Introduction
Aminoglycoside antibiotics comprise a group of diverse polyaminated pseudosaccharides varying in the structure, connectivity and stereochemistry of component sugar residues that are glycosidically linked to a 2-deoxystreptamine core unit.1 They are unified by their broad-spectrum antibacterial actions against Gram-positive and Gram-negative strains, including important pathogenic bacteria.1Aminoglycoside antibiotics target the prokaryotic ribosome within an RNA binding site at the level of the decoding region of the 16S subunit,2,3 resulting in codon misreading and inhibiting translocation during protein biosynthesis.4 Members of the 4,5- and 4,6-disubstituted 2-deoxystreptamine classes of aminoglycosides such as neomycin, gentamicin and tobramycin, have been in clinical use for many decades as topical, intravenous or nebulizer formulations, respectively.1 In the course of the characterization and scale-up efforts of the fermentation medium of Micromonospora inyoensis at the Schering–Plough corporation from which the antibiotic sisomicin (1) was identified,5 a natural congener defying the commonly known structural characteristics of aminoglycosides was also isolated and named aminoglycoside66-40C.6 The speculated structure comprised an unprecedented C2-symmetrical 16-membered bis-azadiene macrocycle (3), which to this day, represents the first example of a naturally-derived dimeric bis-imine isolated from culture media.6The structure was proposed in the 1970s from evidence that included controlled degradation identifying rings B and C common to sisomicin (1), osmometric determination of molecular weight, and chemical transformation to the macrocyclic bis-amine counterpart by reductive amination.6,7 The intriguing biosynthetic origin of aminoglycoside66-40C remained unchallenged for over four decades, and likewise, its potential biological roles are subject to speculation. In our preliminary studies on the synthesis of aminoglycoside66-40C,8 we hypothesised that it may arise as the product of a non-enzymatic self-condensation of a putative monomeric 6′-aldehydo sisomicin (2), itself potentially produced via transamination or oxidation of the 6′-allylic amino group in sisomicin.8 The plausible biosynthetic origin and speculative biological roles of aminoglycoside66-40C are illustrated in Fig. 1.
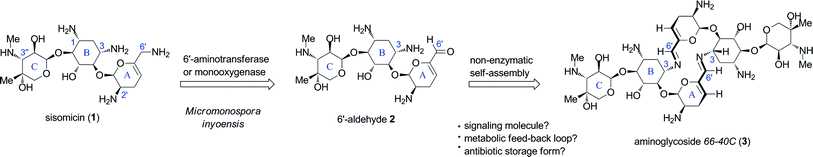 | ||
| Fig. 1 Proposed biosynthetic origin and putative biological functions of aminoglycoside66-40C (3). | ||
We have previously reported the validation of our hypothesis through the concise biomimetic synthesis of aminoglycoside66-40C (3), and demonstrated its quantitative self-condensation from aldehyde 2 in aqueous solution.8 Furthermore, using heteronuclear correlation spectroscopic analysis of the macrocyclic core we confirmed the imine location on N-3 of the 2-deoxystreptamine ring B as originally presumed.8 Moreover, nuclear Overhauser effects (nOe) observed across neighbors of the 6′-imine proton led us to propose a refined s-trans conformation for the macrocycle bis-azadiene bridges (Fig. 1).8
Herein we report an in-depth exploration of the dimerization event leading to aminoglycoside66-40C (3) relying on a diversified synthetic, kinetic and structural approach. The synthesis of related, non-natural macrocyclic bis-imine analogs was also embarked, allowing the detailed study of their individual self-condensing properties, and the dynamic behaviour of these bis-imino macrocycles as evidenced by NMR spectroscopy and LCMS. We also report the X-ray crystal structures of the macrocyclic pseudotetrasaccharide core of aminoglycoside66-40C and a ring-expanded analog.
Results
Aminoglycoside 66-40C (3) was synthesised using our concise biomimetic route from sisomicin sulphate (1) in 7 steps and 41% overall yield (Scheme 1). Application of optimised copper-catalysed azide transfer to sisomicin sulphate (1) provided tetraazido sisomicin (4) as a practical starting material.8,9,10 In this instance, we modified the original route by switching to Fmoc protection at N3′′ (5) in order to simplify the endgame global deprotection of all nitrogen functionalities. The requisite chemoselective oxidation of the 6′-allylic azide 5 to the aldehyde level was performed in excellent yield using our previously reported selenium dioxide conditions (Scheme 1).9 The resulting aldehyde 6 was masked as its acetal 7, following which we effected concomitant deblocking of azides and the 3′′N-Fmoc group with trimethylphosphine and piperidine in one-pot. This procedure provided key intermediate sisomicin 6′-dimethoxy acetal 8 with improved purity on a practical scale compared to the original procedure.8 The monomeric 6′-aldehyde (2) precursor of aminoglycoside66-40C (3) was formed instantaneously by treatment of acetal 8 with dilute acid solutions of H2SO4 (or TFA), and was stable as a monomer under these conditions. We found that neutralization to pH 7.5–8 with a solution of Ba(OH)2 cooled at 0 °C, and removal of the BaSO4 precipitate by filtration was the method of choice for synthetic purposes (Scheme 1).8Freeze-drying followed by further desalting using bench-top chromatographic purification provided aminoglycoside66-40C (3) in quantitative yield and >90% purity by HPLC and NMR analyses.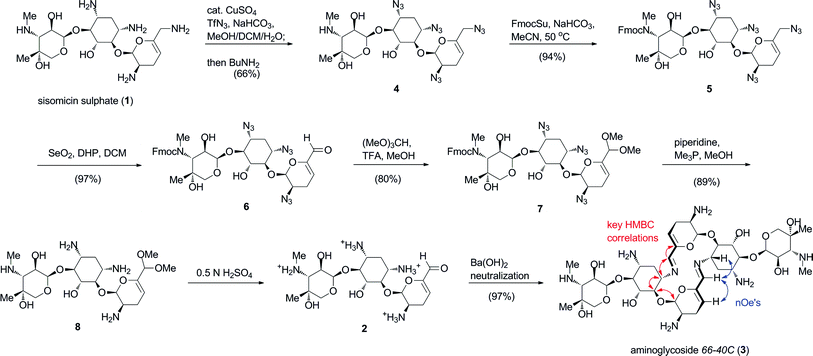 | ||
| Scheme 1 Synthesis and biomimetic self-assembly of aminoglycoside66-40C (3) by neutralization of an aqueous solution of aldehyde 2. Characteristic HMBC and nOe correlations that revealed the macrocyclic structure are shown in red and blue arrows, respectively. | ||
To assess the generality of the self-condensation process leading to macrocyclic bis-azadienes, we extended our studies to the 4,5-disubstituted aminoglycoside paromomycin (9) (Scheme 2). Per-O-acetylation of the readily available 4′,6′-O-benzylidene per-N-Cbz paromomycin 10,11 followed by treatment with 80% AcOH gave the partially-protected diol intermediate 11. Subsequent manipulation of the diol group led to 4′-mesylate 12. Parikh–Doering oxidation of the 6′-alcohol with concomitant β-elimination produced the α,β-unsaturated aldehyde 13. Finally, formation of the 6′-dimethylacetal, and deprotection of the amino groups under Birch conditions provided the paromomycin 6′-dimethylacetal derivative 14. Following the established protocol for self-condensation led to the bis-azadiene macrocycle 15, which was isolated after chromatography in 62% yield. HMQC and nOe correlation patterns for 15 were identical with those observed with aminoglycoside66-40C (Scheme 2). Thus, the extended aminated pseudosaccharide structure in the paromomycin 6′-aldehyde produced from 14 had no consequence on the self-condensation event to produce dimer 15.
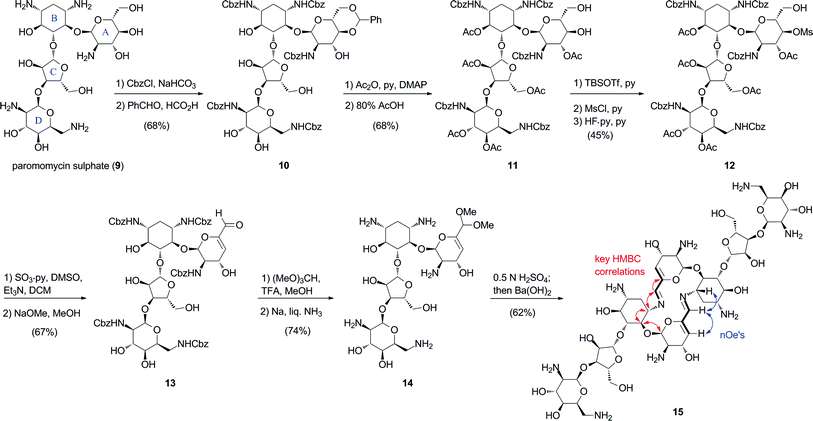 | ||
| Scheme 2 Synthesis and self-assembly of paromomycin bis-imino dimer (15). Red and blue arrows indicate key HMBC and nOe correlations. | ||
Likewise, we expected that the 6′-aldehyde corresponding to a truncated pseudosaccharide formed from controlled degradation of per-azido sisomicin (4) would also spontaneously undergo self-condensation to produce the pseudotetrasaccharide macrocyclic core of aminoglycoside66-40C (Scheme 3).8 Cleavage of the vicinal aminodiol group in ring C of 4 with periodate led to tetrazido sisamine 16.8 Thereafter, selenium dioxide allylic oxidation to aldehyde 17,9 6′-acetalization and reduction of the azido groups under the established Staudinger conditions afforded 19. Applying the optimised self-condensation protocol, the 6′-aldehyde 20 was transformed to the sisamine bis-imino dimer 21 in excellent yield.8
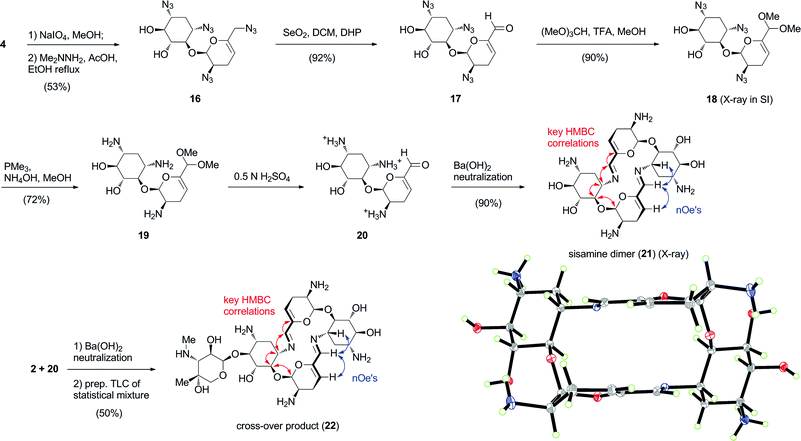 | ||
| Scheme 3 Synthesis and self-assembly of sisamine dimer 21 (ORTEP at 30% probability level). Heterodimeric self-assembly of aldehydes 2 and 20 to produce cross-over product 22 in a statistical mixture with dimers 3 and 21. Red and blue arrows indicate key HMBC and nOe correlations, respectively. | ||
With two additional bis-azadiene macrocycles in hand, we concluded that the formation of 16-membered rings as originally found in aminoglycoside66-40C (3), was the expected norm in this series of 4,5- or 4,6-disubstituted aminoglycosides. We next deemed it of interest to study the intrinsic molecular and topological properties of these macrocycles and their preference for self-condensation to afford homo-dimeric macrocycles, by attempting cross-over experiments to give hetero-dimers. To this end, we mixed equimolar amounts of aldehydes 2 and 20, and analysed the process of self-condensation using LCMS quantitation.8 A statistical mixture in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 ratio containing dimers 3, 21 and their cross-over dimeric product 22 was observed, indicative of preservation of the supramolecular properties of aminoglycoside66-40C (3) in the core dimer 21, and its respective 6′-aldehyde monomeric precursor.8 Preparative TLC allowed the isolation of 22 and its full characterization.
:1:2 ratio containing dimers 3, 21 and their cross-over dimeric product 22 was observed, indicative of preservation of the supramolecular properties of aminoglycoside66-40C (3) in the core dimer 21, and its respective 6′-aldehyde monomeric precursor.8 Preparative TLC allowed the isolation of 22 and its full characterization.
We undertook comprehensive crystallization trials with sisamine dimer 21 in order to obtain structural information regarding the bis-azadiene macrocyclic unit as well as the disposition of the pseudodisaccharide moieties in the solid phase. Adopting a water/ethanol diffusion procedure in the presence of substoichiometric sources of sulphate, such as H2SO4 or (NH4)2SO4, we reliably obtained X-ray quality crystals of the heavily-hydrated N1,N2′-di-sulphate salt (Scheme 3).† The crystals obtained showed a phase transition between 170 K (orthorhombic crystal system R1 = 0.0335 and wR2 = 0.1016) and 200 K (monoclinic crystal system R1 = 0.0437 and wR2 = 0.1022).†
The next challenge was to attempt an expansion of the now familiar 16-membered bis-imine aminoglycoside dimers to the 20-membered homolog (Scheme 4). Treatment of 17 with triethyl phosphonoacetate under standard Horner–Wadsworth–Emmons conditions furnished the E-olefinic ester as a single isomer (Scheme 4). Reduction of the ester with DIBAL-H, followed by oxidation with MnO2 afforded the extended 5′-dienylaldehyde 23. Following the previously described protocol led to the dimethyl acetal 24, which was hydrolysed to key dienal 25. Under the standard dimerization conditions, the expected 20-membered C2-symmetrical bis-azatriene macrocycle 26 was formed in near quantitative yield upon neutralization of a solution of 25. The extended macrocycle product 26 was also crystallized by the water/ethanol diffusion protocol with ammonium sulphate as an additive (Scheme 4). The X-ray structure of 26 was also a heavily-hydrated N1,N2′-di-sulphate salt, which also co-crystallized with ethanol molecules and ammonium ions.‡ Final R indices were R1 = 0.0766 and wR2 = 0.1888.† The all-s-trans bis-imine bridges in the crystal structures of dimers 21 and 26 share a similar antiparallel planar zig-zag arrangement. In dimer 26 ring B accommodates with slight rotation along the C4–O4 bond to a conformation that points the extended bridge with 21° angle difference based on the C1–C5′ axes. However, disturbance of the disaccharide scaffold conformation is minimal, displaying a root-mean-square-distance difference of 0.26 Å over the 20 common atoms. Thus, we infer that the conformational preference for the N1-bridged macrocycle remained relatively unperturbed by its expansion. Moreover, tetraazido 6′-acetal intermediate 18,† which was readily crystallized, permitted further comparison with a fully protected monomeric pseudodisaccharide. Its conformation in the solid state was remarkably similar to both 21 and 26 (RMS 0.23 Å and 0.37 Å, respectively, over the 19 common atoms).
 | ||
| Scheme 4 Synthesis and self-assembly of 20-membered macrocyclic analog 26 (ORTEP at 30% probability level). Red and blue arrows indicate key HMBC and nOe correlations, respectively. | ||
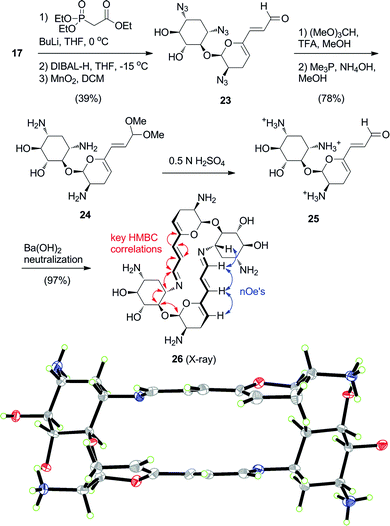
With a clear picture of the structure of the preferred thermodynamic macrocyclic products in solution and the solid state, we sought to examine the kinetic aspect of the self-assembly process. Using the clear individual 1H-NMR signals belonging to 6′-aldehyde (9.10 ppm) and 6′-imine groups (7.40 ppm) we followed in real-time the evolution of the mixture during self-assembly of aminoglycoside66-40C in buffered D2O (Fig. 2). Freeze-drying solutions of acetal 8 in the presence of excess TFA led to isolation of aldehyde 2 as a solid salt, which was indefinitely stable. Upon mixing of the TFA salt of aldehyde 2 in an appropriately buffered solution (100 mM buffer) the signal for the 6′-aldehyde 2 (9 mM) began to decay while, correspondingly, the imine signal of dimer 66-40C (3) increased. Automatic acquisition of 1H-NMR spectra was performed every 10 min for several hours while the reaction mixture remained inside the magnet at 21 °C.8 The serial spectra obtained were integrated over the aldehyde and imine signals, whose relative values were analysed with non-linear curve fitting (Fig. 2). Under these conditions, a slight concentration-dependence was observed (Fig. S1†), therefore the calculated half-lives were obtained using data points up to 75% conversion.†
 | ||
| Fig. 2 Plots of signal integrals from sequential 1H-NMR spectra (400 MHz, D2O) for aldehyde 2 (9.05 ppm, 9 mM concentration), aminoglycoside66-40C3 (7.40 ppm) and the transient imine species (7.75 ppm) at A) pH 6.5, 100 mM MES buffer and B) pH 8.0, 100 mM HEPES buffer. C) Dependence of self-assembly half-life on pH. D) Calculated rate of dimerization (red line, mM min−1) and maximal percentage of transient imine observed (blue bars). | ||
We first examined the pH-dependence of the process, including the pH region of potential biological significance (Fig. 2). The calculated half-life of self assembly to 66-40C, and likewise aldehyde 2 consumption, at pH 6.0 was 34 min (Fig. 2A), and at pH 8.0 it decreased to 10 min (Fig. 2B). Conversion of half-lives to rate constants produced a straight line extrapolating to null conversion below pH ∼4.5 (Fig. 2C and 2D), consistent with the requirement of a minimum amount of N-3 in nucleophilic form.
Concomitantly with the rate enhancement obtained with increasing pH, we noted the accumulation of transient unproductive imine species, whose signal was evident at 7.7 ppm (Fig. 2B). The relative proportion of such transient species rose rapidly at the onset of the experiments (up to ∼30% of the mixture at pH 10, Fig. 2D), and thereafter this proportion decreased slowly, in essence undergoing “repair” and converging to aminoglycoside66-40C (3) as the sole product. Based on pKa considerations, a likely transient species is presumed to be an open pseudodimer held through an N1-imine. Taken together, our 1H-NMR observations indicate that aminoglycoside66-40C (3) is invariably the major kinetic and thermodynamic product throughout the pH spectrum we examined.
In order to establish if the self-assembly process leading to the macrocyclic bis-azadienes is irreversible or in a dynamic equilibrium akin to a dynamic combinatorial library (DCL),12 we used dimer 21 as a labelling entity to trap aldehyde 2 in fleeting existence as a hybrid product 22 (Scheme 3). In the event of cross-over, LCMS would discern the mixture of the three dimers based on their mass and polarity. Firstly, we noted that incubation of an equimolar mixture of aminoglycoside66-40C (3) and sisamine dimer 21 in free base or neutral salt states for 72 h produced negligible cross-over product 22 upon LCMS analysis (Table 1, entries 1–4). Furthermore, treatment of an equimolar mixture of 3 and 21 with excess acetic acid (pH 4) for 72 h led to hydrolysis of a significant proportion of the two parent dimers. Yet, upon neutralization and LCMS analysis, we noted that the cross-over dimer 22 distribution did not reach the statistical 1:1:2 ratio (Table 1, entry 5 vs. entry 6), initially observed upon mixing equimolar amounts of aldehydes 2 and 20. Our pH and cross-over experiments indicate that aminoglycoside66-40C (3) and its analog 21 are kinetically stable, do not exist in a dynamic equilibrium with monomer aldehydes, and that the hydrolysis and self-condensation reactions are irreversible and occur within distinct pH ranges. Meanwhile other imines potentially formed by the polyaminated structures, particularly at high pH, are apparently labile and undergo “repair” under all tested conditions.
| Entry | Treatment/description | Relative ion counts (%) | ||
|---|---|---|---|---|
| 3 | 21 | 22 | ||
| 1 | control, 3 alone | 100 | — | — |
| 2 | control, 21 alone | — | 100 | — |
| 3 | free-bases, 72 h | 41 | 55 | 3 |
| 4 | acetate salts, 72 h | 48 | 52 | <1 |
| 5 | AcOH pH 4, 72 h; freeze-drying | 37 | 31 | 32 |
| 6 | 0.5 M H2SO4, 5 min; sat. Ba(OH)2 | 23 | 22 | 54 |
| 7 | control, 22 alone | — | — | 100 |
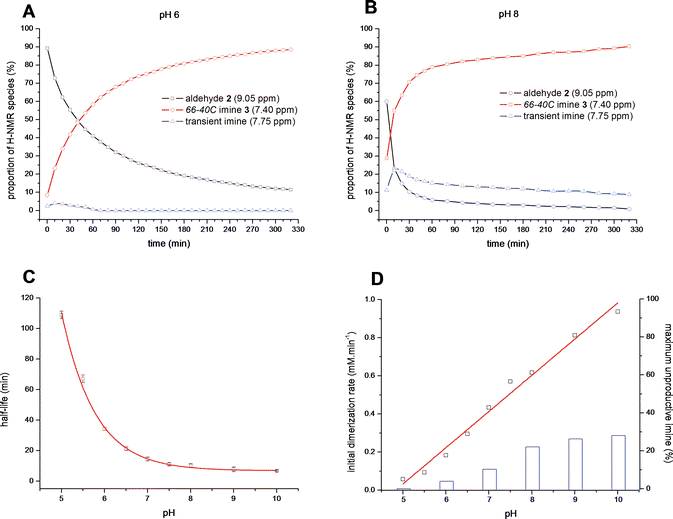
We next examined the possibility of self-selection of aldehyde 2 in the presence of sisomicin (1), the major component of the M. inyoensisfermentation broth, which also bears N1- and N3-amino groups of reduced pKa.1 Notably, the overall process of macrocycle self-assembly at pH 6.5 was relatively unperturbed in the presence of 1, 2 or even 4 equivalents of sisomicin-TFA salt (Fig. 3), despite the clear increase in the amount of transient imine species observed at 7.70 ppm at the onset of such experiments. The swift decline of these signals apparently resulting from competition by the amines in sisomicin (1) indicates that these by-products were transient and subject to “repair”, effectively operating within the time-frame of self-assembly at pH 6.5 (Fig. 3).
 | ||
| Fig. 3 Plot of signal integrals from sequential 1H NMR spectra (400 MHz, D2O) for aldehyde 2 (9.05 ppm), aminoglycoside66-40C3 (7.40 ppm) and transient imine species (7.75 ppm) at pH 6.5, 100 mM MES buffer in the presence of sisomicin-TFA salt (0, 1, 2 and 4 equiv.). | ||
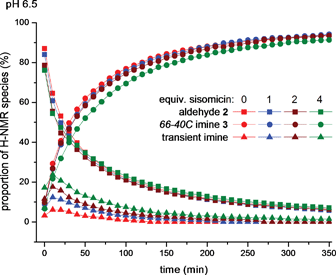
Finally, we performed a cross-over experiment to challenge the self-selection of the monomeric precursor aldehyde 2 in the presence of homologated aldehyde 25 (Fig. 4). When each aldehyde was combined as their stable TFA salt (9 mM 2, 7 mM 25), then exposed to standard buffered conditions at pH 6.5, we observed the expected decay of the signals corresponding to the aldehyde functions with unaffected half-lives compared to control experiments. On the other hand, the newly formed imine signals included two more peaks than those belonging to dimer 3 (s, 7.40 ppm) and 26 (d, 7.70 ppm), accompanied by a minor singlet at 7.35 ppm and minor doublet at 7.85 ppm (Fig. 4). These minor imine signals may belong to cross-over metastable products at pH 6.5, but which decreased gradually over 72 h, leaving a solution in which the known dimers 3 and 26 were dominant products. Although the apparent “repair” in this mixture was relatively slower compared to the previous experiments (e.g.Fig. 2A, 2B and 3), the immutable thermodynamic end-products from the complex mixture were the two C2-symmetric macrocyclic dimers 3 and 26 exclusively (Fig. 4).
 | ||
| Fig. 4 Plot of signal integrals from sequential 1H NMR spectra (400 MHz, D2O) of a mixture of aldehyde 2 (s, 9.05 ppm) and aldehyde 25 (d, 9.50 ppm) in 9 mM and 7 mM respective final concentrations, mixed in pH 6.5, 100 mM MES buffer. Rising signals identified were aminoglycoside66-40C3 (7.40 ppm), 20-membered macrocycle 26 and two transient imine species (d, 7.85 ppm and s, 7.70 ppm). | ||
Discussion
Spontaneous self-assembly is a general phenomenon for several natural products, which have been validated through total synthesis of precursors followed by the biomimetic formation of dimeric or pseudodimeric entities.13 Notable examples include trichodimerol,14,15bisorbicillinol,15 torreyanic acid,16panepophenanthrin,17 rugulosin18 and stephacidin B.19In general, imine formation is often thermodynamically unfavorable or kinetically unstable in water,20 requiring dehydration, or predisposing substitution to favor condensation (e.g. aromatic aldehydes, N-acyl hydrazones, N-oximes)12 or templating through metal chelation (e.g.salen ligands).21,22 In contrast, the unique bis-imino supramolecular structure of aminoglycoside66-40C and its analogs can achieve quantitative self-condensation with kinetic and thermodynamic stability in water under a particularly broad pH spectrum, including the range of biological significance.
The vast number of potential imines that can be formed during the self-assembly and self-selection experiments performed with aminoglycoside66-40C (3) are reminiscent of dynamic combinatorial libraries (DCL).12 The relevance of imines in the DCL field stems from their rapid exchange between reactive monomers so that the outcome of the mixture can be selected, modulated or templated by metal chelation21 or host–guest interactions.12,23 For example, macrocyclic crown-ether compounds held by bis-imines increase in stability when chelated to calcium in solution,21 and in serendipitous cases it has been documented that monomers containing a reactive aldehyde and hydrazone group would invariably provide a particularly stable dimer or trimer entity as a single product.24 Such stabilising and self-selecting behaviors are analogous to those observed for the dimerization of aldehyde 2 leading to aminoglycoside66-40C (3), albeit in the biological environment of a culture medium, as well as our recreated conditions in the laboratory.5,6
The symmetrical nature of the macrocyclic bis-imines appears to be an intrinsic property as a result of a mutual selection process between competent 6′-aldehydes leading to unstrained 16- and 20-membered rings as in 3, 15, and 26. Furthermore, while the hybrid cross-over dimer 22 is readily formed, an analogous stable hybrid could not be isolated from cross-over of aldehydes 2 and 25, underscoring the geometric preference for “matched” monomers toward C2-symmetrical bis-imino macrocycles, which were the ultimate products observed. Aminoglycoside66-40C and its analogs have features which have been theoretically ascribed as ideal for the design of any self-assembling macrocycle under thermodynamic control.25 Firstly, the building block aldehyde 2 is rigid, and lacking rotatable bonds, which maximizes the effective molarity for cyclizationvia a second imine formation. Secondly, there is a structural predisposition to assemble a unique privileged structure without strain, provided that the two halves of the dimer are in the lowest energy conformation for the AB-ring pseudodisaccharide.8,26 Data from the X-ray structures of 21 and 26 concur with these observations, when compared with 18 and with RNA-bound aminoglycosides.3,26 This underscores the intrinsic supramolecular predisposition provided by the AB-ring pseudodisaccharide and the matching lengths of the s-trans bis-azadiene bridges, without disturbance of the lowest energy conformation dictated by the anomeric effect of the C1′–O4 glycosidic bond.
Conclusions
Our detailed synthetic, structural and kinetic experimental observations have delineated the remarkable events leading to the formation of the natural product aminoglycoside66-40C (3). Unique among aminoglycosides and other secondary metabolites, this natural product is an unprecedented example of a self-assembling macrocycle held by stable bis-imine functionalities. We have validated and demonstrated the generality of the supramolecular self-assembly and propensity of self-selection displayed by aldehyde monomers such as 2, 14, 20 and 25, derived from diverse aminoglycoside antibiotic scaffolds. Comparative study of the X-ray structures of dimers 21 and 26 reveal that the AB-ring system of such aminoglycosides is conformationally predisposed toward self-selection of bis-imine dimers at N3, an opportunely accessible nucleophile at neutral pH owing to its lower pKa induced by the peripheral charges of the polyaminated structure. The general reversibility of imine condensations in water endows the precursor 6′-aldehyde 2 with a “repair” pathway, permitting quantitative condensation to aminoglycoside66-40C (3), both as the major thermodynamic and kinetic product in a broad pH range and under the presence of excess competitor amine and aldehyde substrates. Without doubt, such repair properties would be essential for efficient self-assembly of aminoglycoside66-40C (3) in the fermentation broth of its producer M. inyoensis while in the presence of a multitude of other amine-containing metabolites, including antibiotic sisomicin (1).Acknowledgements
This study has been possible owing to the outstanding technical support at the MS and NMR core facilities in our institution, led by Dr Alexandra Furtos and Dr Minh Tan Phan Viet, respectively. We thank Dr Raissa Trend at Achaogen Inc. for the HPLC trace of aminoglycoside 66-40C. We thank Fonds de Recherche en Santé du Québec (FRSQ) for fellowship to JPM, and continued support from the Natural Sciences and Engineering Research Council of Canada (NSERC).Notes and references
- Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery, ed. D. P. Arya, Wiley-Interscience, Hoboken, N.J., 2007 Search PubMed; C. Walsh, Antibiotics: Actions, Origins, Resistance, ASM Press, Washington, D.C., 2003 Search PubMed.
- D. E. Brodersen, W. M. Clemons, Jr., A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Cell, 2000, 103, 1143–1154 CrossRef CAS; A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Nature, 2000, 407, 340–348 CrossRef CAS; J. M. Ogle and V. Ramakrishnan, Annu. Rev. Biochem., 2005, 74, 129–177 CrossRef CAS.
- B. Francois, J. Szychowski, S. S. Adhikari, K. Pachamuthu, E. E. Swayze, R. H. Griffey, M. T. Migawa, E. Westhof and S. Hanessian, Angew. Chem., Int. Ed., 2004, 43, 6735–6738 CrossRef CAS; S. Hanessian, J. Szychowski, S. S. Adhikari, G. Vasquez, P. Kandasamy, E. E. Swayze, M. T. Migawa, R. Ranken, B. Francois, J. Wirmer-Bartoschek, J. Kondo and E. Westhof, J. Med. Chem., 2007, 50, 2352–2369 CrossRef CAS; J. Kondo, K. Pachamuthu, B. Francois, J. Szychowski, S. Hanessian and E. Westhof, ChemMedChem, 2007, 2, 1631–1638 CrossRef CAS.
- D. N. Wilson, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 393–433 CrossRef CAS; K. B. Gromadski and M. V. Rodnina, Mol. Cell, 2004, 13, 191–200 CrossRef CAS; M. Johansson, M. Lovmar and M. Ehrenberg, Curr. Opin. Microbiol., 2008, 11, 141–147 CrossRef CAS; M. B. Feldman, D. S. Terry, R. B. Altman and S. C. Blanchard, Nat. Chem. Biol., 2010, 6, 244 CrossRef CAS.
- M. J. Weinstein, J. A. Marquez, R. T. Testa, G. H. Wagman, E. M. Oden and J. A. Waitz, J. Antibiot. (Tokyo), 1970, 23, 551–554 CAS; D. H. Davies, D. Greeves, A. K. Mallams, J. B. Morton and R. W. Tkach, J. Chem. Soc., Perkin Trans. 1, 1975, 814–818 RSC; M. Kugelman, R. S. Jaret, S. Mittelman and W. Gau, J. Antibiot. (Tokyo), 1978, 31, 643–645 CAS.
- D. H. Davies, A. K. Mallams, J. McGlotten, J. B. Morton and R. W. Tkach, J. Chem. Soc., Perkin Trans. 1, 1977, 1407–1411 RSC.
- D. H. Davies, A. K. Mallams, M. Counelis, D. Loebenberg, E. L. Moss, Jr. and J. A. Waitz, J. Med. Chem., 1978, 21, 189–193 CrossRef CAS.
- S. Hanessian and J. P. Maianti, Chem. Commun., 2010, 46, 2013–2015 RSC.
- S. Hanessian, J. Szychowski and J. P. Maianti, Org. Lett., 2009, 11, 429–432 CrossRef CAS.
- P. T. Nyffeler, C. H. Liang, K. M. Koeller and C. H. Wong, J. Am. Chem. Soc., 2002, 124, 10773–10778 CrossRef CAS; P. B. Alper, S.-C. Hung and C.-H. Wong, Tetrahedron Lett., 1996, 37, 6029–6032 CrossRef CAS; A. Vasella, C. Witzig, J.-L. Chiara and M. Martin-Lomas, Helv. Chim. Acta, 1991, 74, 2073–2077 CAS; J. Zaloom and D. C. Roberts, J. Org. Chem., 1981, 46, 5173–5176 CrossRef CAS; C. J. Cavender and V. J. Shiner, J. Org. Chem., 1972, 37, 3567–3569 CrossRef CAS; W. Fischer and J. P. Anselme, J. Am. Chem. Soc., 1967, 89, 5284–5285 CrossRef CAS.
- S. Hanessian, T. Ogawa and T. Takamoto, Can. J. Chem., 1978, 56, 1500–1508 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- E. Gravel and E. Poupon, Eur. J. Org. Chem., 2008, 27–42 CrossRef.
- D. Barnes-Seeman and E. J. Corey, Org. Lett., 1999, 1, 1503–1504 CrossRef CAS.
- K. C. Nicolaou, K. B. Simonsen, G. Vassilikogiannakis, P. S. Baran, V. P. Vidali, E. N. Pitsinos and E. A. Couladouros, Angew. Chem., Int. Ed., 1999, 38, 3555–3559 CrossRef CAS; K. C. Nicolaou, G. Vassilikogiannakis, K. B. Simonsen, P. S. Baran, Y.-L. Zhong, V. P. Vidali, E. N. Pitsinos and E. A. Couladouros, J. Am. Chem. Soc., 2000, 122, 3071–3079 CrossRef CAS.
- C. Li, R. P. Johnson and J. A. Porco, J. Am. Chem. Soc., 2003, 125, 5095–5106 CrossRef CAS.
- J. E. Moses, L. Commeiras, J. E. Baldwin and R. M. Adlington, Org. Lett., 2003, 5, 2987–2988 CrossRef CAS.
- K. C. Nicolaou, C. D. Papageorgiou, J. L. Piper and R. K. Chadha, Angew. Chem., Int. Ed., 2005, 44, 5846–5851 CrossRef CAS; K. C. Nicolaou, Y. H. Lim, J. L. Piper and C. D. Papageorgiou, J. Am. Chem. Soc., 2007, 129, 4001–4013 CrossRef CAS.
- P. S. Baran, C. A. Guerrero, B. D. Hafensteiner and N. B. Ambhaikar, Angew. Chem., Int. Ed., 2005, 44, 3892–3895 CrossRef CAS; S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342–5344 CrossRef CAS; P. S. Baran, B. D. Hafensteiner, N. B. Ambhaikar, C. A. Guerrero and J. D. Gallagher, J. Am. Chem. Soc., 2006, 128, 8678–8693 CrossRef CAS; G. D. Artman, A. W. Grubbs and R. M. Williams, J. Am. Chem. Soc., 2007, 129, 6336–6342 CrossRef.
- C. Godoy-Alcantar, A. K. Yatsimirsky and J. M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef CAS; V. Saggiomo and U. Luning, Tetrahedron Lett., 2009, 50, 4663–4665 CrossRef CAS.
- V. Saggiomo and U. Lüning, Eur. J. Org. Chem., 2008, 4329–4333 CrossRef CAS.
- E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS; T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS; J. F. Larrow and E. N. Jacobsen, Top. Organomet. Chem., 2004, 6, 123–152 CAS.
- I. Huc and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS; M. Hochgurtel, H. Kroth, D. Piecha, M. W. Hofmann, C. Nicolau, S. Krause, O. Schaaf, G. Sonnenmoser and A. V. Eliseev, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3382–3387 CrossRef; M. Hochgurtel, R. Biesinger, H. Kroth, D. Piecha, M. W. Hofmann, S. Krause, O. Schaaf, C. Nicolau and A. V. Eliseev, J. Med. Chem., 2003, 46, 356–358 CrossRef CAS; A. Herrmann, Org. Biomol. Chem., 2009, 7, 3195–3204 RSC.
- M. G. Simpson, M. Pittelkow, S. P. Watson and J. K. Sanders, Org. Biomol. Chem., 2010, 8, 1173–1180 RSC; M. G. Simpson, M. Pittelkow, S. P. Watson and J. K. Sanders, Org. Biomol. Chem., 2010, 8, 1181–1187 RSC.
- G. Ercolani, L. Mandolini, P. Mencarelli and S. Roelens, J. Am. Chem. Soc., 1993, 115, 3901–3908 CrossRef CAS; G. Ercolani, J. Phys. Chem. B, 1998, 102, 5699–5703 CrossRef CAS.
- D. H. Fong and A. M. Berghuis, EMBO J., 2002, 21, 2323–2331 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra of novel compounds, crystallographic reports, CIF files, PDB files, raw data and calculation reports from kinetic experiments and extra figures. CCDC reference numbers 838347–838350. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00538c |
| ‡ For the final model of the crystal structure of compound 26, highly disordered sectors required squeezing out, including a molecule of ethanol, one ammonium ion and several water molecules. For more details see crystallographic report files. |
| This journal is © The Royal Society of Chemistry 2012 |