Mass spectrometry methods for intrinsically disordered proteins
Rebecca Beveridgea, Quentin Chappuisa, Cait Macpheeb and Perdita Barran*a
aSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh EH9 3JJ, Scotland. E-mail: perdita.barran@ed.ac.uk
bSchool of Physics and Astronomy, University of Edinburgh, West Mains Road, Edinburgh EH9 3JJ, Scotland
First published on 25th September 2012
Abstract
In the last ten years mass spectrometry has emerged as a powerful biophysical technique capable of providing unique insights into the structure and dynamics of proteins. Part of this explosion in use involves investigations of the most recently ‘discovered’ subset of proteins: the so-called ‘Intrinsically Disordered’ or ‘Natively Unstructured’ proteins. A key advantage of the use of mass spectrometry to study intrinsically disordered proteins (IDPs) is its ability to test biophysical assertions made about why they differ from structured proteins. For example, from the charge state distribution presented by a protein following nano-electrospray (n-ESI) it is possible to infer the range of conformations present in solution and hence the extent of disorder; n-ESI is highly sensitive to the degree of folding at the moment of transfer from the liquid to the gas phase. The combination of mass spectrometry with ion mobility (IM-MS) provides rotationally averaged collision cross-sections of molecular ions which can be correlated with conformation; this too can be applied to IDPs. Another feature which can be monitored by IM-MS is the tendency of disordered proteins to form amyloid fibrils, the protein aggregates involved in the onset of neurodegenerative diseases such as Parkinson's and Alzheimer's. IM-MS provides a useful insight into events that occur during the early stages of aggregation including delineating the structure of the monomer, identifying oligomer distributions, and revealing mechanistic details of the aggregation process. Here we will review the use of MS and IM-MS to study IDPs using examples from our own and other laboratories.
Rebecca Beveridge is a post-graduate student in the School of Chemistry at Edinburgh University where she is undertaking a PhD in biological mass spectrometry sponsored by the BBSRC and LGC. Her current research uses ion mobility mass spectrometry to study the aggregation of intrinsically disordered proteins. She received her BSc in Biochemistry and Chemistry from the University of Leeds in 2011 and completed her final year project under the supervision of Professor Sheena Radford studying the folding pathways of small proteins. |
Quentin Chappuis is a student from Geneva, Switzerland who studies chemistry at l'Ecole Polytechnique Fédérale de Lausanne (EPFL) and is currently on an Erasmus exchange for the full year at the University of Edinburgh. As a part of his degree, he led a research project on protein biosynthesis and purification and brought his contribution to the present work. |
Cait MacPhee is a Professor of Biological Physics at the University of Edinburgh, and a Fellow of the Royal Society of Chemistry. She is expert in the formation and characterisation of amyloid-type fibrils, and her research interests lie in an understanding of the fundamental processes underlying fibril assembly, as well as the use of fibrillar architectures as components in novel materials. Cait was the first to provide experimental evidence that the formation of amyloid-type fibrils is a generic property of the polypeptide chain, rather than a property intrinsic to a small subset of disease-related proteins, by demonstrating that fibrils can be assembled from a mixture of two entirely unrelated polypeptide species. |
Perdita Barran is a Reader in Biophysical Chemistry at the University of Edinburgh. The Barran group have considerable experience in gas-phase ion chemistry, instrument development and the application of mass spectrometry to complex chemical and biological problems. Dr Barran was awarded an EPSRC Advanced Research Fellowship (March 2003) to study the structure and dynamics of model peptides and proteins in the gas phase. The Barran group have developed IM-MS instrumentation to investigate changes in protein conformation and aim to understand biological systems using mass spectrometry based techniques in conjunction with collaboration with biologists and biomedical research groups. In 2009 in recognition of her achievements Barran was awarded the inaugural Joseph Black award by the RSC Analytical Division. Recent work has focussed on the development of new methods to determine gas-phase structures, protein–protein and protein–ligand interactions, prefibrillar oligomeric species and intrinsically disordered proteins. |
Introduction
Mass spectrometry was originally applied to elucidate atomic isotopes and for most of the 20th century was principally used to obtain the molecular weight of small chemical compounds, as well as structural information via the use of dissociation. Due to developments in soft ionisation methods,1 it is now an increasingly popular technique with which to investigate conformations of intact biological molecules under a variety of conditions.2 Whole protein mass spectrometry relies on the transfer of either a single protein or a protein complex, which may often contain several components, into the gas phase from a solution.3 Several mass spectrometry based studies have addressed the process of protein folding and unfolding; both based on altering solution conditions and observing how this effects the distribution of charge states into the gas phase4,5 and also by trapping ions in the gas phase and observing the evolution of conformations over the time scale of mass spectrometry analysis (μs-s).6,7 This ability to discern conformational flexibility, by the measurement of discrete conformers in a mixture, is one of the key strengths of mass spectrometry over other biophysical techniques.8–10 Mass spectrometry can also be used to detect specific regions of disorder in proteins,11 measure the impact of metal ions on protein conformation12 and monitor conformational changes that occur upon the formation of protein complexes.13Mass spectrometry is now positioned as an informative biophysical tool, which can be used along with other techniques to gain structural and functional information about biological molecules. Under particular scrutiny at present is the newly discovered subset of proteins, the intrinsically disordered proteins (IDPs).14 IDPs are polypeptide chains which exist and function without a well-defined three-dimensional structure.15 They have increased flexibility, are more dynamic than folded globular proteins and can populate several heterogeneous conformations of similar energy.16,17 It follows that gathering information on the structures of IDP's, often intractable by crystallography, is proving a great challenge requiring the development of new techniques,18,19 or at least new ways to interpret data. This review considers the emerging role of mass spectrometry to interrogate IDPs.
The discovery of IDPs strongly contradicted the original protein paradigm which stated that the function of a protein is derived entirely from its folded 3-dimensional structure.20,21 Therefore, new definitions have since been developed. Dunker proposed the ‘protein trinity’18 which declares that proteins reside in one of the three states: the ordered state, the molten globule and the random coil. Dyson and Wright22 expand this to a quartet model which is similar to the protein trinity except for the discrimination between two states within the ordered state into ‘mostly folded with localised disorder’ and ‘linked folded domains’ in which independently folded globular domains are separated by flexible linker regions. Two points should be stressed: firstly, while some proteins can be found in each of these different states, not all can. For example, many IDPs do not adopt folded, ordered states. Secondly, these states are not absolute limits and proteins actually populate a continuum containing these different states.
Analysis of the primary structure of IDPs reveals features that enable us to predict disordered regions and also give information about their behaviour. Tertiary folds in structured proteins are maintained by a high proportion of hydrophobic groups that interact favourably with each other inside the fold, away from solvent; by contrast charged and polar residues will most likely be located on the surface of the protein fold where they can favourably interact with solvent.23,24 Too many of these hydrophilic groups will destabilise any hydrophobic core and cause the protein to be unstructured or disordered. For these reasons, amino acids considered as ‘ordering’ are Val, Leu, Ile, Met, Phe, Trp, Tyr and those considered as a source of disorder are Gln, Ser, Pro, Glu, Lys, and, on occasion, Gly and Ala.25–27 This categorisation enables a coarse prediction of disorder from primary sequence information alone. Common features identified across IDPs have been developed into predictors to determine if a given protein is likely to be structured or not, more than 50 of these predictors have so far been developed28 but they are by no means infallible,29 which suggests that disorder may not be encoded into the primary sequence, rather that it is a combination of intrinsic and extrinsic interactions.
IDPs in native conditions are not to be considered as ordered proteins in the denatured state; their hydrodynamic behaviour often differs. Most IDP's do not behave as random coils. Their lack of structure is not absolute and often they show a high degree of ‘compactness’.18 Transient elements of secondary structure (mainly α-helix) are observed and tend to reduce the hydrodynamic radius of IDPs.14 Other non-covalent interactions, such as hydrogen bonds and electrostatic interactions, also lead to dynamic tertiary structures.14 Of course, such interactions are highly dependent on the environment and the presence of specific extrinsic species will have an effect on the ‘compactness’ of a protein.25
An interesting and important characteristic of IDPs is their ability to bind to multiple partners, and this allows them to play a key role in many cellular signalling networks.30–33 This can result in three different behaviours:30
(1) An IDP binds to many different structured proteins.31
(2) Multiple IDPs bind to one structured protein.30
(3) Intrinsically disordered regions act as flexible linkers between ordered domains, allowing binding promiscuity.30,31,34
Often, but not always, IDPs are induced to fold upon interaction with specific binding partners.35–37 Upon binding to an ordered domain, the free energy required for the disorder to order transition is subtracted from the contact free energy, resulting in a highly specific interaction along with a low energy of association; such highly specific but weak interactions are perfect for signalling cascades as they ensure reversibility.18 As well as binding to other proteins, some IDPs are also known to bind to small molecules which can have an effect on the structure of the protein.
Furthermore, even under denaturing conditions, the behaviour of IDPs differs from ordered proteins. Indeed, a frequent feature of IDP's is their significantly low mobility via SDS-PAGE and gel filtration chromatography. For example, the apparent mass on SDS-PAGE of the repair protein xenopus XPA is 40–45 kDa, but its actual mass (measured by mass spectrometry and predicted from the amino acid sequence) is 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 922 Da.11 This example and the reason for this reduced mobility will be discussed below.
922 Da.11 This example and the reason for this reduced mobility will be discussed below.
IDPs are flexible and because they are unfolded – or partially unfolded – they have a high solvent accessible surface.14 These two features make them more sensitive to proteolysis than globular proteins, since proteases require a protein to be unfolded over ten residues or more for cleavage to occur.18 The amino acids in the inside of a globular protein are not accessible to proteases and they are mostly at external loops or at the termini that a protease can attack a globular protein. For IDPs, none or very few of the amino acids are buried in a protective core so there are no preferential cleavage sites beyond those that a given protease will select for.11
This review will cover the different ways in which MS can be applied to the study of IDPs, the methods that MS has been coupled to, and examples of IDPs which have been analysed by these techniques.
ESI and charge state distribution
Since the development of electrospray ionisation1 and matrix assisted laser desorption/ionisation (MALDI)38 mass spectrometry has become a widely used method for the analysis of intact proteins with impacts on biochemistry, structural biology and medicine. These ‘soft’ ionisation methods allow proteins to be transferred intact into the gas phase without excessive fragmentation, and in the case of ESI, multiprotein complexes can also be maintained.39 ESI is of particular importance in the study of IDPs because it is highly sensitive to the degree of disorder in solution.40 Proteins are observed in ESI MS in a range of charge states. In positive ionisation mode these charge states are due to protonated forms of the protein. Proteins with a lower degree of compactness have a wider charge state distribution since more ionisable sites are exposed to the solvent,41 and hence ESI can be used to distinguish between different conformations of the same protein42,43 (see Fig. 1a). A noteworthy point is the importance of removal of the His-tag if one is employed for purification of a recombinant protein as this can create a second, higher charge state distribution which mimics that of a disordered protein. In addition we have often observed erroneous binding effects between His tagged proteins and their interaction targets in our laboratory and recommend against the use of His-tagged proteins for MS investigations. Other factors that must be considered when designing ESI MS experiments to examine proteins are the effects of source and solvent conditions on the observed species and the source of protein material, which can at times be extremely variable – even from commercial sources – in our laboratory we take care to desalt protein samples and to measure the concentration using BCA assays or other spectroscopic methods. The storage of proteins is also important, since all proteins are susceptible to decomposition if subjected to repeated freeze–thaw cycles.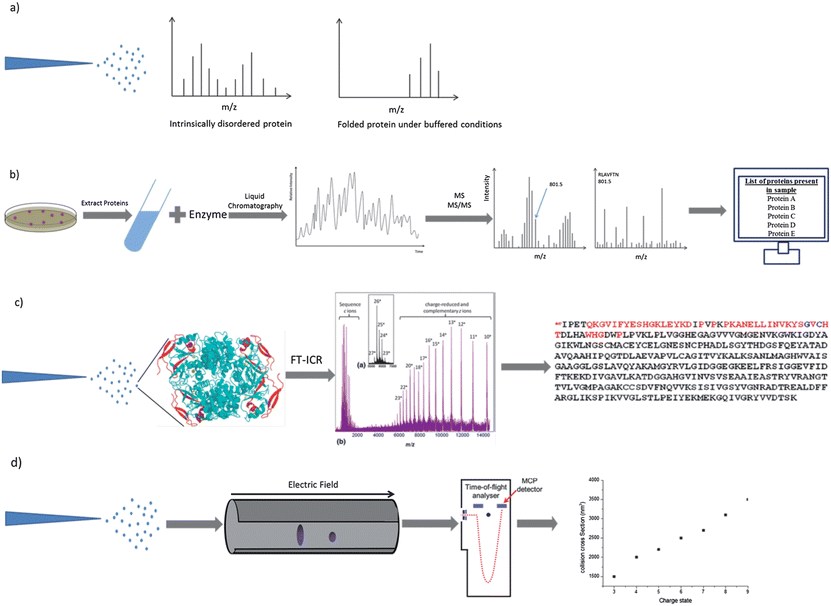 | ||
| Fig. 1 Schematic representations of procedures which involve the use of mass spectrometry. Electrospray ionisation charge state distribution analysis (a), MS-based proteomics (b), electron capture dissociation top-down sequencing (c) and ion mobility mass spectrometry (d). Components of image adapted from M. L. Gross et al., J. Am. Soc. Mass Spectrom., 2010, 21, 1966–1968. | ||
Frimpong et al.42 used ESI-MS to probe the structure of monomeric α-synuclein, a 140 amino acid neuronal protein implicated in the onset of Parkinson's disease due to its propensity to form intracellular fibrillar aggregates.44 Although classified as intrinsically disordered, α-synuclein has been found to populate four distinct conformations which coexist in solution, all which have different extents of disorder.45 The deconvolution of charge state distributions obtained from α-synuclein following ESI from solutions with pH in the ranges 2.5–8 yields 4 basis functions, each of which was selected as a Gaussian curve (Fig. 2).45 The most compact state spans charge states 5 ≤ z ≤ 10 (corresponding to 5–10 extra protons), the compact intermediate spans charge states 7 ≤ z ≤ 13, the more disordered intermediate spans charge states 10 ≤ z ≤ 17 and the most unfolded conformer spans charge states 14 ≤ z ≤ 24. The spectra are similar when the pH decreases from 8 through to 4, however at pH 2.5 the presence of the most disordered state (U) becomes negligible and the presence of the compact state (C) and the α-helix containing intermediate is significantly increased. This shows that conditions which would be usually considered highly denaturing for globular proteins induce compaction of α-synuclein into a solution form(s) with lower surface accessibility. A proposed theory for this is the protonation of negative amino acids which would otherwise repel each other due to electrostatic forces.40 Whether this hypothesis of collapse at low (or high) pH will extend to other IDPs remains to be investigated, but certainly the work of Kaltashov and co-workers shows how ESI-MS can offer detailed insight into the solution stability of disordered proteins.
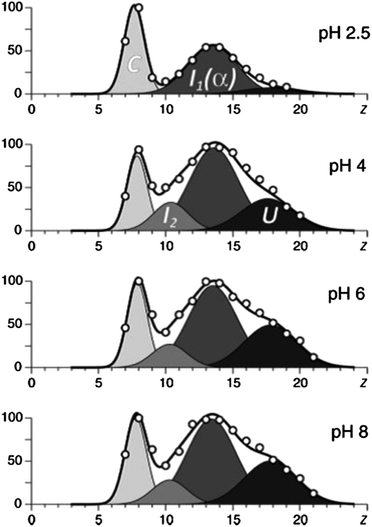 | ||
| Fig. 2 The results of deconvolution of charge state distributions of α-synuclein ions in ESI MS acquired in the pH range of 2.5–8. The four basis functions are assigned to the following putative states of the protein: U, unstructured; I1, helix-rich intermediate; I2, β-sheet-rich intermediate; and C, highly compact. Image taken from Frimpong et al.42 | ||
CD spectroscopy has previously shown that the α-helical content of α-synuclein can be increased by lowering the pH. By contrast, the presence of ethanol increases the amount of β-sheet.46 Frimpong et al.42 suggest that the two most compact conformations, which are enhanced at low pH, may be characterized by α-helix structures and the two more unfolded conformations by β-sheets. The study of these semi-folded states is of particular importance because they are thought to be the intermediates in the aggregation which is responsible for amyloid diseases (Fig. 3).
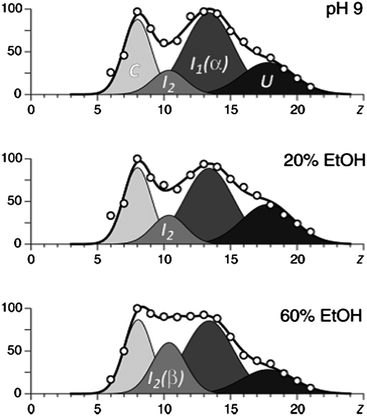 | ||
| Fig. 3 The results of deconvolution of charge state distributions of α-synuclein ions in ESI MS in different concentrations of ethanol, which has been proven to increase the amount of β-sheet. The four basis functions are assigned to the following putative states of the protein: U, unstructured; I1, helix-rich intermediate; I2, β-sheet-rich intermediate; and C, highly compact. Image taken from Frimpong et al.42 | ||
Work by Natalello et al.40 presents ESI-MS analysis of the same protein from identical solvent conditions (aqueous and 10 mM ammonium acetate) but in negative ionisation mode and employing nano-ESI rather than ESI. Surprisingly, the spectrum at pH 7.4 shows only three states. This discrepancy shows the limits (or subtleties) when using ESI-MS for conformational study: retention of protein conformation from solution to gas phase during ESI depends on many parameters which are yet not fully understood and can vary between instruments, and certainly as a function of solution, source and the polarity of the ionisation mode chosen (Fig. 4).
 | ||
| Fig. 4 α-Synuclein – a case study protein for the use of mass spectrometry to examine IDPs. | ||
Limited proteolysis
Resistance to proteolysis correlates strongly with structural stability58,59 and limited proteolysis in solution coupled with mass spectrometry10,60,61 has been shown to identify regions of reduced stability, for example domain borders58 and linker regions.59 Fragments resulting from partial proteolysis can be analysed by ESI-MS to allow identification of specific proteolytic peptides by comparison with a database of predicted cleavable regions. Because intrinsically disordered regions are more accessible to the protease and are therefore cleaved more frequently, it is possible to deduce from the fragments areas of structural stability and regions of disorder from the analysis of fragmentation patterns.11Using time resolved proteolysis coupled with ESI-FTICR, Iakoucheva et al.11 identified disordered regions of Xeroderma pigmentosum group A (XPA). XPA is a protein involved in nucleotide excision repair; it is able to recognize damaged DNA albeit with the help of other ligands and to trigger the repair process through mechanisms which are yet unclear.62 Trypsin cleavage, and lack of it, revealed that there are certain preferred trypsin cleavage sites and certain sites that are never cut. Disordered regions were revealed by partial proteolysis to be both termini of XPA, and a core fragment was found to be structurally stable. This core domain possesses 18 possible cleavage sites and no fragments cleaved at those sites were detected, which suggests that this domain is ordered while the two termini are disordered (Fig. 5). This result is in close agreement with the PONDR disorder prediction, a neural network predictor originally developed from literature searches of intrinsically ordered and disordered regions in proteins.63 The structured domain approximately corresponds to the minimal binding domain with DNA. We use the word ‘approximately’ because the limited proteolysis experiment was done on xenopus XPA and the minimal binding domain is only known for human XPA,11 but the comparison is relevant as the sequences of the two share 67% amino acids identity and 82% similarity.
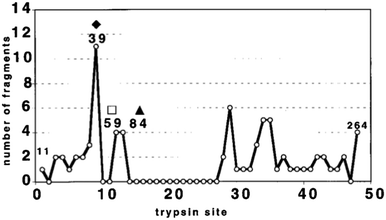 | ||
| Fig. 5 Summary of cleavage site frequency. Each of the 48 cleavage sites is indicated on the x axis with the number of unique peptide fragments resulting from limited proteolysis on the y axis.11 | ||
The same group of authors studied the aberrant mobility of XPA on SDS-PAGE and gel filtration chromatography.64 XPA has an expected molecular weight of 30922 Da while its apparent molecular weight is 40–45 kDa on SDS-PAGE and 92 kDa via gel filtration. Mass spectrometry measurements show that no post-translational modifications are responsible for those phenomena, and provide an exact mass of 30922.02 Da which corresponds well with the sequence mass of 30922.45 Da. Fragments of the protein also show low mobility but their respective deviations are less important. The structured domain is the one which is most mobile, with a deviation on SDS-PAGE of 15% while other fragments have a deviation of 30% and the full length protein has a deviation of 40%. This is consistent with the assertion that disorder results in lower mobility than expected for globular proteins. The authors of this paper concluded that this low mobility can be attributed to highly extended conformation(s) of all forms of xXPA.64
Mass spectrometry based proteomics
The study of proteomics involves the determination of gene and cellular function directly at the protein level,65 and at the forefront of techniques available in this field of research as mass spectrometry.65 Methods have been developed to examine specific subsets of proteins, for example, those containing post-translational modifications or those that change in response to specific stimuli, developmental events or during disease. The latter are of great importance to biomedical science because they may help to elucidate the mechanism of processes and could be extended to act as potential biomarkers.There are five stages in a typical MS-based proteomics experiment: fractionation, digestion, chromatography, MS and finally MS/MS (see Fig. 1b). In the first step the proteins of the cell or tissue in question are purified, either by affinity selection or biochemical fractionation, and then further separated, often by SDS-PAGE, or liquid chromatography to define a smaller set of proteins for characterisation. These proteins must then be enzymatically digested as intact protein masses provide insufficient information for certain identification by MS. These peptides are then separated by liquid chromatography, the eluent directly sprayed into the mass spectrometer by electrospray ionisation and a spectrum recorded. A prioritized list of peptides for fragmentation can then be generated by the computer, determined by intensity, charge state and/or other sample specific information. Peptide ions are isolated, fragmented by a high-energy collision with gas and a MS/MS spectrum recorded. The spectra of fragmented peptides can then be compared against protein sequence databases for identification, and the proteins in question can also be identified.
Washburn et al.66 used two-dimensional liquid chromatography (LC) coupled to tandem mass spectrometry (MS/MS) to characterise the proteome of yeast Saccharomyces cerevisiae. The method, which had initially been developed by Link et al.,67 involves filling a pulled microcapillary column with two independent chromatography phases and loading a complex peptide mixture which is eluted from the column directly into the mass spectrometer. The peptides and respective proteins were resolved which resulted in the identification of 1484 proteins from the S. cerevisiae proteome which included those with extremes in pI, molecular weight, abundance and hydrophobicity.
A review by Csizmok et al.68 outlines several proteomic approaches for the identification of structural disorder in a complex mixture of proteins. Cortese et al.69 exploit the resistance of IDPs to acid denaturation to enrich cell extracts with unfolded proteins. Although the reduction in the total amount of soluble Escherichia coli proteins was almost 100000-fold after treatment by 9% PCA, 158 spots were observed on silver-stained 2-D SDS-PAGE gels. It was therefore suggested that resistance to acid denaturation by IDPs, as well as to other denaturation methods such as high temperatures, can be exploited to separate unstructured and structured proteins to study IDPs on a proteomic scale.69
Galea et al. show that the heat treatment of NIH3T3 mouse fibroblast cell extracts at 98 °C also selects IDPs for proteomic analysis.70 It is likely that resistance to thermal aggregation is a result of the low mean hydrophobicity and high net charge characteristic of these proteins. The IDP enriched cell extracts were then separated by 2-D SDS-PAGE, excised from the gel and then digested with trypsin into smaller peptides for analysis by mass spectrometry. These peptides were then identified by comparison against sequence databases and classified according to their known subcellular location (cytoplasm, 38.1%; nucleus, 20.6%; mitochondria, 4.0%; cytoskeleton, 16.7%; extracellular matrix, 4.0%; ER, 7.1%) and further to their reported biological function. It was found that this heat treatment resulted in the enrichment of proteins involved in regulation and maintenance of cell structure and a corresponding depletion of metabolic proteins. Enriched to a lesser extent were proteins involved in cell signalling and protein folding as well as heat shock proteins. This demonstrates that exploiting the biophysical characteristic of IDPs (here their thermal stability) can be used in a MS based workflow.
Hydrogen–deuterium exchange
Hydrogen–deuterium exchange (HDX) experiments are frequently used to detect regions of disorder in proteins.71 The technique exploits the increase in the rate of exchange of amide protons (N–H) with solvent protons in areas of reduced stability, which is due to a lack of protection from strong intramolecular H-bonds which occur in structured regions. Thus, the observation of the hydrogen-exchange process can provide valuable information on structural stability at many sites along the polypeptide chain.72 HDX studies can be carried out in both directions. The labelling of a protonated protein with deuterium (‘exchange in’) is more widely used than the labelling of a deuterated protein with protons (‘exchange out’) as it eliminates the need for the initial step of complete deuteration.71 A distinct benefit of HDX is its ability to report on the entire length of the polypeptide chain because every amino acid (with the exception of proline) has an amide proton. This gives it an advantage over techniques such as fluorescence emission spectroscopy, which only probe the structural environments of a select few chromophores.Liquid chromatography followed by mass spectrometry of proteolytic fragments74 or a top-down fragmentation MS approach75 can be used to measure the extent of HDX in different regions of the polypeptide chain and hence provide information on disordered regions. The former approach has been used by Zhang and Smith74 to elucidate disordered regions of horse heart cytochrome c, and the latter approach has been used by Pan et al.75 to distinguish between helices and loops in horse myoglobin.
Keppel et al.73 used HDX in combination with pepsin digestion and mass spectrometry to investigate the disorder-to-order transition of IDPs that occurs upon the formation of a protein complex. The intrinsically disordered protein ACTR (activator of thyroid and retinoid receptors, NCOA3_HUMAN, residues 1018–1088) is known to bind to the molten globular protein CBP (the nuclear coactivator binding domain of the CREB binding protein, CBP_MOUSE, residues 2059–2117). The extent of HDX was investigated for each protein alone and in the complex. It was found that deuteration of the proteins in the complex was much slower than that of the individual proteins in isolation, indicating that the formation of the protein–protein complex confers structure to both of the participating polypeptide chains (Fig. 6).
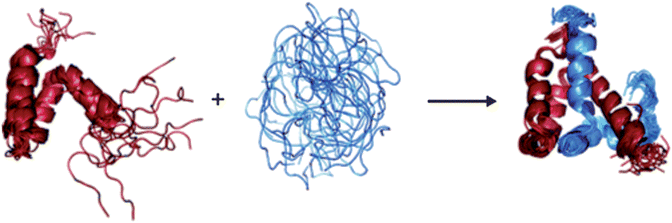 | ||
| Fig. 6 The disorder to order transition of ACTR and CBP upon complex formation, as determined by HDX.73 | ||
Ion mobility mass spectrometry
Ion mobility is a measure of how quickly a gas phase ion moves through a buffer gas under the influence of an electric field. A typical Drift tube Ion Mobility Mass Spectrometry (DT IM-MS) experiment involves the injection of a pulse of ions into a chamber filled with a known gas at a known pressure across which is applied a weak static electric field (5–50 V cm−1). Upon injection into the chamber the ions experience an electrostatic force pulling them through the cell. This force is countered by low energy collisions between the ions and the buffer gas. Two of the factors which influence how quickly the ions travel are the number of collisions which occur with the protein and the buffer gas which is determined by the size of the protein and the charge present on it which determines how quickly the ion is pulled through the drift cell.76,77 By measuring the drift time of an ion through the cell, the rotationally averaged collision cross-section can be measured which provides valuable information on the conformation of a protein (Fig. 1d).78Ion mobility is frequently coupled with mass spectrometry which allows separation of protein conformers based on their mass-to-charge ratios as well as their interactions with the buffer gas. This has proven to be a powerful analytical tool which has been used in several instances to interrogate IDPs.79–82
Maurizio et al.79 have used IM-MS to report on the High Mobility Group A (HMGA) proteins that are involved in an abundance of biological processes from transcription regulation to chromatin remodelling.83 Through different mechanisms the HMGA proteins are also known to be involved in both benign and malignant neoplasias.84 Based on the fact that the loss of the highly acidic C-terminal tail increases cell growth,85 ion mobility measurements of wildtype and C-terminal truncated HGMA2 were recorded. Despite the increase in mass of WT with respect to the C-terminal truncated HGMA2, the conformation of the former was more compact, indicating that the presence of the C-terminal tail is here responsible for further compacting HGMA2 and hence reducing oncogenic activity.
IM-MS has also been used to study the structure of the tumour suppressor protein p53. The p53 protein is known to bind to DNA, and this complex is stabilised by a single zinc ion which plays a regulatory role in the folding and DNA binding ability of p53.86–88 Removal of this zinc ion disrupts the structure of the DNA-binding domain, resulting in rapid cysteine oxidation and disulphide-linked aggregation.87 Faull et al.12 used IM-MS to investigate the conformations of p53 with and without the presence of zinc.
In the presence of zinc, the mass spectrum has a charge state distribution from 7 ≤ z ≤ 17 with highest intensity species assigned to the [M + 9H]9+ and [M + 10H]10+. Their dominance can be associated with compact conformations being prevalent in solution as there are few solvent-accessible sites available for protonation. Two dimeric species are seen, and low intensity monomeric species between charge states 11 ≤ z ≤ 16 can be attributed to unfolded states in solution. Ion mobility data revealed that for seven of the ten charge states, at least two conformations are present, further reflecting the conformation flexibility of this system. Low charge states adopt compact conformations and as the charge increases, unfolding occurs.
When zinc has been removed, the charge state distribution alters, with strong signal from 7 ≤ z ≤ 12, and a dominant peak for [M + 10H]10+, indicating that p53 has not denatured extensively in the absence of zinc. For protein conformations that carry a low number of positive charges (8 ≤ z ≤ 11) ion mobility shows that the cross-sections are smaller than those for the equivalent charge for zinc present by 12.3% for the large [M + 10H]10+ conformer. The collision cross-section is small at low charge, with a large increase between [M + 11H]11+ and [M + 13H]13+ which corresponds to an unfolding transition. For [M + 12H]12+ the arrival time distribution was very wide, indicative of a large number of conformations at the unfolding transition. The intensity of this [M + 12H]12+ species in the mass spectrum is low, indicative of an ion that is not as stable as those either side of it.
The protein α-synuclein has also been examined using IM-MS, illustrating how different mass spectrometry based techniques can provide complementary and at times conflicting information. Bernstein et al.55 used negative-ion IM-MS to decipher how the size of conformations of α-synuclein differed with charge state at initial solutions of neutral and acidic pH. Negative-ion mode was used because in pH 7 solution α-synuclein has an overall charge of −9. As shown by Frimpong et al.42 the mass spectrum of the protein sprayed from a pH 2.5 solution shows a narrow charge state distribution at low charge states corresponding to a tightly folded protein, whereas when sprayed from pH 7 there is a wide charge state distribution with a maximum intensity peak at a higher charge. Arrival time distributions (ATDs) were reported for the [M − 7H]7− [M − 8H]8− and [M − 9H]9− species obtained from pH 2.5 solutions at several injection energies. The ATD of [M − 7H]7− at an injection voltage of 20 V has a narrow distribution characteristic of a single conformer, and a short arrival time characteristic of a compact structure. As the injection energy increases to 40 V the ATD becomes broader, indicative of some less compact isoforms, and by 100 V a narrow distribution at longer time is observed, representative of the annealing of the compact structures to a more extended conformation via substantial internal excitation which is more stable in the gas phase. This effect of thermally induced structural reorganisation has also been reported for structured proteins,89 but it may be that disordered proteins will respond differently to increased injection energy. This would be analogous to the observation in solution assays of sharp unfolding transitions for ordered proteins versus gradual unfolding for disordered proteins.
The [M − 8H]8− ion acts much like the [M − 7H]7−, however the [M − 9H]9− charge state has only extended structures regardless of the injection energy, indicating that an extended structure is being sprayed from solution. Fig. 7 shows the collision cross-sections (CCSs) for species where −6 ≤ z ≤ −11 compared to those calculated by molecular modelling. The structures of charge states −6 ≤ z ≤ −8 are very compact, whereas for ions where z > −9 the structures are elongated. Between −8 and −9 a conformational rearrangement occurs which increases the CCS by over 50%. As the charge increases above −9 the CCSs also continue to increase, indicating that as more charges are added the structure continues to elongate. It can be seen that the CCSs of charge states −6, −7 and −8 are in good agreement with the theoretical cross-section of the compact globular structure confirming that these charge states are collapsed. The experimental CCSs of charge states −9 and higher lie between the globular and all-helical theoretical structures, in agreement with the hypothesis of substantially unfolded structures.
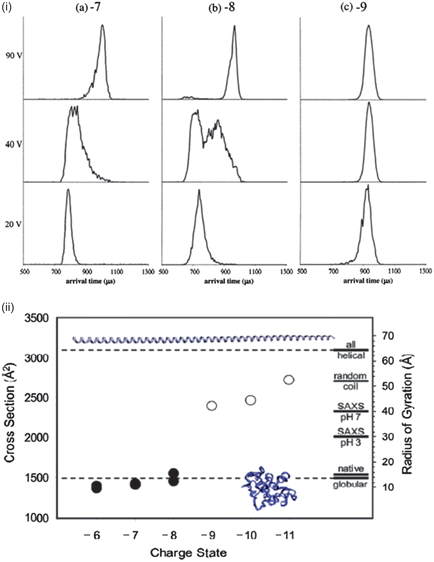 | ||
| Fig. 7 (i) Arrival time distributions for α-synuclein −7, −8 and −9 charge states at injection energies 20 V, 40 V and 90 V. (ii) CCS vs. charge for the dominant peaks in the ATD measurements. Theoretical CCSs are represented for globular and helical structures by dotted lines. Images taken from ref. 55. | ||
Vlad et al. also studied α-synuclein with IM-MS and identified a highly aggregating fragment formed from a cleavage between Val71 and Thr72. The aggregation of this carboxyl-terminal peptide has been shown to occur faster than full length α-synuclein by ThT fluorescence assays, as well as produce more autoproteolytic fragments as aggregation proceeds. HDX-MS was performed on both the full length structure and the fragment (α-Syn72-140). Full length α-Syn showed rapid exchange for 115 of the 134 backbone hydrogens, with 19 residues remaining resistant to exchange for more than 14 days. 19 amino acids were also resistant to HDX in the α-Syn(72-140) fragment.
Conclusion
This review has covered many of the mass spectrometry-based techniques used in researching intrinsically disordered proteins. New developments in this research area are continuing to emerge and are likely to be of great importance given the role of IDPs in cell signalling and regulation, errors in which can result in the onset of cancer.90,91 Mass spectrometry is a useful tool in the elucidation of IDP structure, providing detail on the conformational spread of a given protein at the single conformer level rather than averaged data for all structures adopted by the polypeptide. Undoubtedly, mass spectrometry will be developed further and will continue to yield results in the elucidation of protein structure–function relationships.Acknowledgements
The authors acknowledge the Schools of Chemistry and Physics at the University of Edinburgh for continuing support of their interdisciplinary work, and allowing them the space to develop new lines of research.Notes and references
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- A. Dobo and I. A. Kaltashov, Anal. Chem., 2001, 73, 4763–4773 CrossRef CAS.
- E. van Duijn, P. J. Bakkes, R. M. A. Heeren, R. H. H. van den Heuvell, H. van Heerikhuizen, S. M. van der Vies and A. J. R. Heck, Nat. Methods, 2005, 2, 371–376 CrossRef CAS.
- L. Konermann and D. J. Douglas, Rapid Commun. Mass Spectrom., 1998, 12, 435–442 CrossRef CAS.
- S. J. Eyles, J. P. Speir, G. H. Kruppa, L. M. Gierasch and I. A. Kaltashov, J. Am. Chem. Soc., 2000, 122, 495–500 CrossRef CAS.
- E. R. Badman, S. Myung and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2005, 16, 1493–1497 CrossRef CAS.
- M. A. Freitas, C. L. Hendrickson, M. R. Emmett and A. G. Marshall, Int. J. Mass Spectrom., 1999, 187, 565–575 CrossRef.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- B. L. Schwartz, J. E. Bruce, G. A. Anderson, S. A. Hofstadler, A. L. Rockwood, R. D. Smith, A. Chilkoti and P. S. Stayton, J. Am. Soc. Mass Spectrom., 1995, 6, 459–465 CrossRef CAS.
- S. L. Cohen, A. R. Ferredamare, S. K. Burley and B. T. Chait, Protein Sci., 1995, 4, 1088–1099 CrossRef CAS.
- L. M. Iakoucheva, A. L. Kimzey, C. D. Masselon, J. E. Bruce, E. C. Garner, C. J. Brown, A. K. Dunker, R. D. Smith and E. J. Ackerman, Protein Sci., 2001, 10, 560–571 CrossRef CAS.
- P. A. Faull, H. V. Florance, C. Q. Schmidt, N. Tomczyk, P. N. Barlow, T. R. Hupp, P. V. Nikolova and P. E. Barran, Int. J. Mass Spectrom., 2010, 298, 99–110 CrossRef CAS.
- E. van Duijn, P. J. Bakkes, R. M. A. Heeren, R. H. H. van den Heuvel, H. van Heerikhuizen, S. M. van der Vies and A. J. R. Heck, Nat.ure Methods, 2005, 2, 371–376 CrossRef CAS.
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1231–1264 CrossRef CAS.
- P. Tompa, Curr. Opin. Struct. Biol., 2011, 21, 419–425 CrossRef CAS.
- J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, J. Mol. Biol., 2004, 337, 635–645 CrossRef CAS.
- G. W. Daughdrill, L. J. Hanely and F. W. Dahlquist, Biochemistry, 1998, 37, 1076–1082 CrossRef CAS.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. R. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. H. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, M. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS.
- A. K. Dunker, J. Y. Yang, C. J. Oldfield, Z. Obradovic, J. W. Meng, P. Romero and V. N. Uversky, IEEE 7(th) BIBE Invited Plenary Keynote: Intrinsically Disordered Proteins: Predictions and Applications, IEEE, New York, 2007 Search PubMed.
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321–331 CrossRef CAS.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins: Struct., Funct., Genet., 2000, 41, 415–427 CrossRef CAS.
- S. Muller-Spath, A. Soranno, V. Hirschfeld, H. Hofmann, S. Ruegger, L. Reymond, D. Nettels and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14609–14614 CrossRef CAS.
- S. Mueller-Spaeth, A. Soranno, V. Hirschfeld, H. Hofmann, S. Rueegger, L. Reymond, D. Nettels and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14609–14614 CrossRef.
- R. M. Williams, Z. Obradovi, V. Mathura, W. Braun, E. C. Garner, J. Young, S. Takayama, C. J. Brown and A. K. Dunker, Pac. Symp. Biocomput., 2001, 89–100 CAS.
- P. Radivojac, L. M. Iakoucheva, C. J. Oldfield, Z. Obradovic, V. N. Uversky and A. K. Dunker, Biophys. J., 2007, 92, 1439–1456 CrossRef CAS.
- B. He, K. Wang, Y. Liu, B. Xue, V. N. Uversky and A. K. Dunker, Cell Res., 2009, 19, 929–949 CrossRef CAS.
- B. He, K. Wang, Y. Liu, B. Xue, V. N. Uversky and A. K. Dunker, Cell Res., 2009, 19, 929–949 CrossRef CAS.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129–5148 CrossRef CAS.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, J. Mol. Recognit., 2005, 18, 343–384 CrossRef CAS.
- G. P. Singh and D. Dash, Proteins, 2007, 68, 602–605 CrossRef CAS.
- H. B. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, V. N. Uversky and Z. Obradovic, J. Proteome Res., 2007, 6, 1882–1898 CrossRef CAS.
- V. N. Uversky, Int. J. Biochem. Cell Biol., 2011, 43, 1090–1103 CrossRef CAS.
- K. E. Olson, P. Narayanaswami, P. D. Vise, D. F. Lowry, M. S. Wold and G. W. Daughdrill, J. Biomol. Struct. Dyn., 2005, 23, 113–124 CAS.
- R. S. Spolar and M. T. Record, Science, 1994, 263, 777–784 CAS.
- B. W. Pontius, Trends Biochem. Sci., 1993, 18, 181–186 CrossRef CAS.
- M. Karas and F. Hillenkamp, Anal. Chem., 1988, 60, 2299–2301 CrossRef CAS.
- M. Sharon and C. V. Robinson, in Annual Review of Biochemistry, Annual Reviews, Palo Alto, 2007, vol. 76, pp. 167–193 Search PubMed.
- A. Natalello, F. Benetti, S. M. Doglia, G. Legname and R. Grandori, Proteins, 2011, 79, 611–621 CrossRef CAS.
- L. Konermann, J. Phys. Chem. B, 2007, 111, 6534–6543 CrossRef CAS.
- A. K. Frimpong, R. R. Abzatimov, V. N. Uversky and I. A. Kaltashov, Proteins, 2010, 78, 714–722 CAS.
- L. Konermann, B. A. Collings and D. J. Douglas, Biochemistry, 1997, 36, 5554–5559 CrossRef CAS.
- M. Baba, S. Nakajo, P. H. Tu, T. Tomita, K. Nakaya, V. M. Y. Lee, J. Q. Trojanowski and T. Iwatsubo, Am. J. Pathol., 1998, 152, 879–884 CAS.
- A. K. Frimpong, R. R. Abzatimov, V. N. Uversky and I. A. Kaltashov, Proteins, 2010, 78, 714–722 CAS.
- V. N. Uversky, J. Biomol. Struct. Dyn., 2003, 21, 211–234 CAS.
- X. Chen, H. A. R. de Silva, M. J. Pettenati, P. N. Rao, P. S. George-Hyslop, A. D. Roses, Y. Xia, K. Horsburgh, K. Uéda and T. Saitoh, Genomics, 1995, 26, 425–427 CrossRef CAS.
- L. Maroteaux, J. T. Campanelli and R. H. Scheller, J. Neurosci., 1988, 8, 2804–2815 CAS.
- M. H. Polymeropoulos, C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. DiIorio, L. I. Golbe and R. L. Nussbaum, Science, 1997, 276, 2045–2047 CrossRef CAS.
- M. G. Spillantini, M. L. Schmidt, V. M. Y. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 10737–10744 CrossRef CAS.
- L. C. Serpell, J. Berriman, R. Jakes, M. Goedert and R. A. Crowther, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4897–4902 CrossRef CAS.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS.
- S. L. Bernstein, D. F. Liu, T. Wyttenbach, M. T. Bowers, J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Soc. Mass Spectrom., 2004, 15, 1435–1443 CrossRef CAS.
- L. A. Munishkina, C. Phelan, V. N. Uversky and A. L. Fink, Biochemistry, 2003, 42, 2720–2730 CrossRef CAS.
- C. Vlad, K. Lindner, C. Karreman, S. Schildknecht, M. Leist, N. Tomczyk, J. Rontree, J. Langridge, K. Danzer, T. Ciossek, A. Petre, M. L. Gross, B. Hengerer and M. Przybylski, ChemBioChem, 2011, 12, 2740–2744 CrossRef CAS.
- S. J. Hubbard, F. Eisenmenger and J. M. Thornton, Protein Sci., 1994, 3, 757–768 CrossRef CAS.
- S. J. Hubbard, R. J. Beynon and J. M. Thornton, Protein Eng., 1998, 11, 349–359 CrossRef CAS.
- D. Massotte, M. Yamamoto, S. Scianimanico, O. Sorokine, A. Vandorsselaer, Y. Nakatani, G. Ourisson and F. Pattus, Biochemistry, 1993, 32, 13787–13794 CrossRef CAS.
- A. Fontana, G. Fassina, C. Vita, D. Dalzoppo, M. Zamai and M. Zambonin, Biochemistry, 1986, 25, 1847–1851 CrossRef CAS.
- O. V. Tsodikov, D. Ivanov, B. Orelli, L. Staresincic, I. Shoshani, R. Oberman, O. D. Scharer, G. Wagner and T. Ellenberger, EMBO J., 2007, 26, 4768–4776 CrossRef CAS.
- P. Romero, Z. Obradovic, C. Kissinger, J. E. Villafranca and A. K. Dunker, IEEE Identifying Disordered Regions in Proteins from Amino Acid Sequence, 1997 Search PubMed.
- L. M. Iakoucheva, A. L. Kimzey, C. D. Masselon, R. D. Smith, A. K. Dunker and E. J. Ackerman, Protein Sci., 2001, 10, 1353–1362 CrossRef CAS.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS.
- M. P. Washburn, D. Wolters and J. R. Yates, Nat. Biotechnol., 2001, 19, 242–247 CrossRef CAS.
- A. J. Link, J. Eng, D. M. Schieltz, E. Carmack, G. J. Mize, D. R. Morris, B. M. Garvik and J. R. Yates, Nat. Biotechnol., 1999, 17, 676–682 CrossRef CAS.
- V. Csizmok, Z. Dosztanyi, I. Simon and P. Tompa, Curr. Protein Pept. Sci., 2007, 8, 173–179 CrossRef CAS.
- M. S. Cortese, J. P. Baird, V. N. Uversky and A. K. Dunker, J. Proteome Res., 2005, 4, 1610–1618 CrossRef CAS.
- C. A. Galea, V. R. Pagala, J. C. Obenauer, C. G. Park, C. A. Slaughter and R. W. Kriwacki, J. Proteome Res., 2006, 5, 2839–2848 CrossRef CAS.
- L. Konermann, X. Tong and Y. Pan, J. Mass Spectrom., 2008, 43, 1021–1036 CrossRef CAS.
- S. W. Englander and N. R. Kallenbach, Q. Rev. Biophys., 1983, 16, 521–655 CrossRef CAS.
- T. R. Keppel, B. A. Howard and D. D. Weis, Biochemistry, 2011, 50, 8722–8732 CrossRef CAS.
- Z. Q. Zhang and D. L. Smith, Protein Sci., 1993, 2, 522–531 CrossRef CAS.
- J. Pan, J. Han, C. H. Borchers and L. Konermann, J. Am. Chem. Soc., 2009, 131, 12801–12808 CrossRef CAS.
- B. J. McCullough, J. Kalapothakis, H. Eastwood, P. Kemper, D. MacMillan, K. Taylor, J. Dorin and P. E. Barran, Anal. Chem., 2008, 80, 6336–6344 CrossRef CAS.
- S. R. Harvey, C. E. MacPhee and P. E. Barran, Methods, 2011, 54, 454–461 CrossRef CAS.
- B. C. Bohrer, S. I. Merenbloom, S. L. Koeniger, A. E. Hilderbrand and D. E. Clemmer, Annu. Rev. Anal. Chem., 2008, 1, 293–327 CrossRef CAS.
- E. Maurizio, L. Cravello, L. Brady, B. Spolaore, L. Arnoldo, V. Giancotti, G. Manfioletti and R. Sgarra, J. Proteome Res., 2011, 10, 3283–3291 CrossRef CAS.
- S. Brocca, L. Testa, F. Sobott, M. Samalikova, A. Natalello, E. Papaleo, M. Lotti, L. De Gioia, S. M. Doglia, L. Alberghina and R. Grandori, Biophys. J., 2011, 100, 2243–2252 CrossRef CAS.
- F. Canon, R. Ballivian, F. Chirot, R. Antoine, P. Sarni-Manchado, J. Lemoine and P. Dugourd, J. Am. Chem. Soc., 2011, 133, 7847–7852 CrossRef CAS.
- P. A. Faull, K. E. Korkeila, J. M. Kalapothakis, A. Gray, B. J. McCullough and P. E. Barran, Int. J. Mass Spectrom., 2009, 283, 140–148 CrossRef CAS.
- R. Sgarra, S. Zammitti, A. Lo Sardo, E. Maurizio, L. Arnoldo, S. Pegoraro, V. Giancotti and G. Manfioletti, Biochim. Biophys. Acta, Gene Regul. Mech., 2010, 1799, 37–47 CAS.
- A. Fusco and M. Fedele, Nat. Rev. Cancer, 2007, 7, 899–910 CrossRef CAS.
- G. M. Pierantoni, S. Battista, F. Pentimalli, M. Fedele, R. Visone, A. Federico, M. Santoro, G. Viglietto and A. Fusco, Carcinogenesis, 2003, 24, 1861–1869 CrossRef CAS.
- P. Hainaut and K. Mann, Antioxid. Redox Signaling, 2001, 3, 611–623 CrossRef CAS.
- C. Meplan, M. J. Richard and P. Hainaut, Oncogene, 2000, 19, 5227–5236 CrossRef CAS.
- C. Meplan, M. J. Richard and P. Hainaut, Biochem. Pharmacol., 2000, 59, 25–33 CrossRef CAS.
- Y. Mao, J. Woenckhaus, J. Kolafa, M. A. Ratner and M. F. Jarrold, J. Am. Chem. Soc., 1999, 121, 2712–2721 CrossRef CAS.
- L. M. Iakoucheva, C. J. Brown, J. D. Lawson, Z. Obradović and A. K. Dunker, J. Mol. Biol., 2002, 323, 573–584 CrossRef CAS.
- H. B. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, J. Proteome Res., 2007, 6, 1917–1932 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |