Screening reactive metabolites bioactivated by multiple enzyme pathways using a multiplexed microfluidic system†
Dhanuka P.
Wasalathanthri
a,
Ronaldo C.
Faria
ab,
Spundana
Malla
a,
Amit A.
Joshi
a,
John B.
Schenkman
d and
James F.
Rusling
*acd
aDepartment of Chemistry, University of Connecticut, Storrs, Connecticut 06269, USA. E-mail: james.rusling@uconn.edu
bDepartamento de Química, Universidade Federal de São Carlos, São Carlos, SP, Brazil
cNational University of Ireland at Galway, Ireland
dDepartment of Cell Biology, University of Connecticut Health Center, Farmington, Connecticut 06032, USA
First published on 11th October 2012
Abstract
A multiplexed, microfluidic platform to detect reactive metabolites is described, and its performance is illustrated for compounds metabolized by oxidative and bioconjugation enzymes in multi-enzyme pathways to mimic natural human drug metabolism. The device features four 8-electrode screen printed carbon arrays coated with thin films of DNA, a ruthenium-polyvinylpyridine (RuPVP) catalyst, and multiple enzyme sources including human liver microsomes (HLM), cytochrome P450 (cyt P450) 1B1 supersomes, microsomal epoxide hydrolase (EH), human S9 liver fractions (Hs9) and N-acetyltransferase (NAT). Arrays are arranged in parallel to facilitate multiple compound screening, enabling up to 32 enzyme reactions and measurements in 20–30 min. In the first step of the assay, metabolic reactions are achieved under constant flow of oxygenated reactant solutions by electrode driven natural catalytic cycles of cyt P450s and cofactor-supported bioconjugation enzymes. Reactive metabolites formed in the enzyme reactions can react with DNA. Relative DNA damage is measured in the second assay step using square wave voltammetry (SWV) with RuPVP as catalyst. Studies were done on chemicals known to require metabolic activation to induce genotoxicity, and results reproduced known features of metabolite DNA-reactivity for the test compounds. Metabolism of benzo[a]pyrene (B[a]P) by cyt P450s and epoxide hydrolase showed an enhanced relative DNA damage rate for DNA compared to cyt P450s alone. DNA damage rates for arylamines by pathways featuring both oxidative and conjugative enzymes at pH 7.4 gave better correlation with rodent genotoxicity metric TD50. Results illustrate the broad utility of the reactive metabolite screening device.
1. Introduction
Reliable risk assessment at early stages of chemical and drug development is a major challenge faced by the chemical industry.1 We know most about these issues from experiences in the drug industry, in which development of a single new product costs about US $2 billion.2 In spite of a sophisticated array of in vivo, in vitro and in silico assessment tools,3–5 toxicity issues surface in nearly one third of drug candidates only after reaching human clinical trials, and a few even after marketing. Thus, prediction of genotoxicity at the earliest stages of drug and chemical development is an important goal that would increase development success rates and lower health care costs.Covalent DNA nucleobase adducts formed by reactive chemicals or their metabolites are important biomarkers of genotoxicity testing,6–8 a term used to denote in vitro and in vivo tests designed to detect compounds that induce genetic damage by various mechanisms. We have developed novel molecular-based genotoxicity screening technologies that measure the propensity of metabolites of parent chemicals to react with DNA, with the implicit assumption such reactive metabolites are likely to cause genotoxic effects. These screening approaches rely on ultrathin films that combine metabolic enzymes with DNA by alternate electrostatic layer-by-layer (LbL) fabrication.5,9–11 The rate of DNA damage caused by metabolites is detected electrochemically12,13 or electro-optically14,15 using specific ruthenium-based redox complexes or polymers. We recently reported a metabolic genotoxicity array based on an electro-optical detection method known as electrochemiluminescence (ECL) that was validated for a range of common genotoxic compounds and demonstrated reasonable correlations with standard in vivo and in vitro genotoxicity bioassays.16 These molecular genotoxicity screening arrays are complimented by high throughput methods utilizing magnetic biocolloid reactor particles that also feature films of DNA and metabolic enzymes and produce nucleobase adducts of metabolites for analysis by LC-MS/MS.17–20
We recently achieved bioelectronic actuation that enables electrolysis to drive the natural cyt P450 reductase/cyt P450 catalytic pathway.21,22 This approach was used to simplify the genotoxicity screening arrays by replacing NADPH regeneration to drive cyt P450 reactions. We reported a prototype microfluidic screening array based on this approach to detect reactive metabolites formed by cyt P450s only. This device employed films containing microsomes, DNA and [Ru(bpy)2(PVP)10](ClO4) {(PVP = poly(4-vinylpyridine)} (Ru-PVP) fabricated on 8-electrode screen printed carbon arrays in a simple microfluidic system.23 After running the cyt P450 reactions in this microfluidic device, catalytic square wave voltammetry (SWV) was used to detect DNA damage.9 SWV peaks are generated by catalytically oxidizing the guanines in DNA using Ru-PVP. Increased SWV peaks result as guanines in the damaged DNA become more accessible to catalytic sites in Ru-PVP due to partial unfolding of DNA double helices.9,13
These molecular approaches are important complements to traditional and emerging bioassays for genotoxic metabolite screening.5,11 However, they need to utilize a representative range of metabolic oxidative and bioconjugation enzymes to correlate better with human in vivo responses.11 The present paper describes the first multiplexed microfluidic platform combining oxidative and conjugative metabolic enzymes to enable multi-enzyme pathways for reactive metabolite generation, thus providing an enzyme panel that more accurately mimics human drug metabolism. A switching valve and multiplexed electronics was used so that up to 64 sensor measurements are possible in rapid sequence.
The multiplexed microfluidic array reported here features metabolic enzymes including cyt P450s and the bioconjugation enzymes N-acetyltransferase (NAT) and epoxide hydrolase (EH) to produce potential DNA-reactive metabolites. We also employed human liver S9 fraction (Hs9) as an enzyme source, since it contains both cyt P450s and other conjugation enzymes including NAT. Hs9 is used to compare with other assays utilizing cyt P450 and NAT. Arylamines and benzo[a]pyrene (B[a]P) were chosen as test pollutants because of their well known metabolite–DNA interactions. The fluidic platform features parallel-connected single-use 8-electrode sensor arrays with inlets connected to a sample valve via a switch used to select the specific 8-electrode device (Scheme 1). Each electrode is coated with a thin film of Ru-PVP, DNA, and an enzyme source. The enzyme reactions are run first under constant reactant flow, and followed by square wave voltammetry (SWV) detection of the resulting DNA damage.23
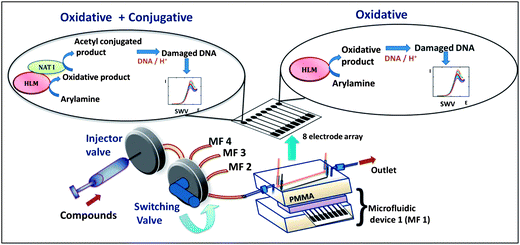 | ||
| Scheme 1 Multiplexed genotoxicity screening platform with reference to arylamine metabolism. Each microfluidic device features a 63 μL polydimethylsiloxane (PDMS) channel in a poly(methylmethacrylate) (PMMA) housing. Ag/AgCl reference and Pt counter wire electrodes are symmetrically located along the channel housing a replaceable 8-sensor array.23 In this study, four microfluidic systems were connected to a switching valve to enable sequential experiments. | ||
2. Experimental
2.1 Chemicals
Ruthenium metallopolymer [Ru(bpy)2(PVP)10](ClO4) (RuPVP (bpy = 2,2-bipyridyl; PVP = poly(4-vinylpyridine)) was synthesized and characterized by established protocols,24 screen printed carbon arrays were from Kanichi, UK. Sources and full description of other chemicals are reported in the ESI† section.2.2 Microfluidic system
The system features four microfluidic channels connected in parallel, each containing 8-electrode arrays with inlets connected to a sample valve via a switching valve (Rheodyne, 9060). The switching valve is used to direct solutions to a specific 8-electrode device. Full fabrication details are included in the ESI† file. A CHI 1040A multipotentiostat coupled with CHI 685 multiplexer (CH Instruments Inc.) was used to acquire electrochemical measurements in rapid sequence.2.3 Film construction
Enzyme/Ru-PVP/DNA films were constructed (Table 1) on the sensor elements from 1 μL droplets of stock solutions, depositing a layer at a time using optimized solution compositions reported previously.10,23,25 To achieve steady state adsorption, 1.0 μL of each adsorbate solution was incubated on electrodes for 20 min at 4 °C, except for enzymes and DNA where 30 min incubations were used.10,25 Adsorbate solutions were (a) poly(diallyldimethylammonium) chloride (PDDA), 2 mg mL−1 in 0.05 M NaCl; (b) poly(sodium 4-styrenesulfonate) (PSS), 3 mg mL−1 in 0.5 M NaCl; (c) Ru-PVP, 2.5 mg mL−1 50% v/v ethanol; (d) calf thymus DNA, 2 mg mL−1 in 10 mM TRIS + 0.5 M NaCl, pH 7.4; (e) human liver microsomes (HLM), 20 mg mL−1 in 250 mM sucrose; (f) N-acetyltransferase1 (NAT), 2.5 mg mL−1 in 20 mM Tris–HCl + 1.0 mM EDTA + 1.0 mM DTT of pH 7.5; (g) human microsomal epoxide hydrolase (EH), 10 mg mL−1 in 100 mM potassium phosphate buffer of pH 7.4; (h) pooled human liver S9 (Hs9) 20.0 mg mL−1 in 150 mM KCl, 50 mM Tris–HCl + 2.0 mM EDTA; (i) baculovirus-insect cell expressed cyt P450 1B1 supersomes (cyt P450 1B1), 4.5 mg mL−1 in 100 mM potassium phosphate buffer of pH 7.4. All the films have the general film architecture, PDDA/PSS/(RuPVP/DNA)2/RuPVP/oxidative enzyme source/conjugative enzyme source or PDDA/DNA (Table 1). Films will be referred to below by the name of the enzyme source as in Table 1.| Study | Film assembly | Abbreviation |
|---|---|---|
| B[a]P | PDDA/PSS/(RuPVP/DNA)2/RuPVP/HLM/PDDA/DNA | HLM |
| PDDA/PSS/(RuPVP/DNA)2/RuPVP/HLM/EH/DNA | HLM + EH | |
| PDDA/PSS/(RuPVP/DNA)2/RuPVP/cytP4501B1/PDDA/DNA | cyt P450 1B1 | |
| PDDA/PSS/(RuPVP/DNA)2/RuPVP/cytP4501B1/EH/DNA | cyt P450 1B1 + EH | |
| Arylamine | PDDA/PSS/(RuPVP/DNA)2/RuPVP/HLM/PDDA/DNA | HLM |
| PDDA/PSS/(RuPVP/DNA)2/RuPVP/HLM/NAT/DNA | HLM + NAT | |
| PDDA/PSS/(RuPVP/DNA)2/RuPVP/Hs9/PDDA/DNA | Hs9 |
2.4 Film characterization
The nominal thickness and mass densities of each film were estimated by quartz crystal microbalance (QCM, USI Japan).10,25 QCM was done on 9 MHz gold-coated quartz crystals coated with mercaptopropionic acid before film fabrication.13 Frequency shift was monitored after the deposition and drying of each layer. Bicinchoninic acid (BCA) assays26 using a commercial μBCA assay kit (model 23235, Thermoscientific Inc.) were employed to determine the total protein content of the films. CHI 660A electrochemical analyzer (CH Instruments Inc.) was used to obtain voltammograms with CHI 685 electrochemical multiplexer. Full details are in ESI† file.2.5 Metabolite generation and measurements
Safety note: B[a]P, 2-naphthylamine (2-NA), 2-aminofluorene(2-AF), 4-aminobiphenyl (4-ABP), N-(9H-fluoren-2-yl)acetamide (2-AAF), and their metabolites are known carcinogens. All manipulations were done under a closed hood while wearing gloves.The 8-electrode chips are single use, and are replaced with a new set of chips before each new run.
2.6 Reproducibility
SWV signals were acquired from arrays as described above using 0.2 mM 2-AF. Data were analyzed using GraphPad Prism software V5.01, (GraphPad Software, San Diego, CA).3. Results
3.1 Film construction and characterization
Films of RuPVP, DNA and enzymes fabricated a layer at a time were optimized to achieve the best SWV responses in the array. QCM weighing demonstrated stable and reproducible film formation, with an average nominal thickness of 38–56 nm (Table 2, Fig. S3, ESI† file). Protein assays provided the total amount of protein in each enzyme source (Table 2, Fig. S4, ESI† file).| Film | HLM + NAT | HLM | HLM + EH | Cyt P450 1B1 + EH | Cyt P450 1B1 | Hs9 |
|---|---|---|---|---|---|---|
| a From BCA total protein assay26 (ESI†), 56.8 (±0.1) μg of protein per mg of HLM, 32.1 (±0.1) μg of protein per mg of cyt P450 1B1, 35.2 (±0.4) μg of protein per mg of EH, and 26.8 (±0.2) μg of protein per mg of NAT, electrode area of an array electrode = 3.85 × 10−3 cm2. | ||||||
| Nominal thickness/nm | 56 | 52 | 51 | 38 | 37 | 40 |
| RuPVP/μg cm−2 | 9.5 ± 0.8 | 8.4 ± 0.2 | 6.6 ± 0.2 | 6.6 ± 0.2 | 7.5 ± 0.3 | 6.3 ± 0.4 |
| DNA/μg cm−2 | 2.3 ± 0.5 | 2.9 ± 0.4 | 4.1 ± 0.5 | 4.2 ± 0.5 | 3.6 ± 0.5 | 5.2 ± 0.2 |
| HLM/μg cm−2 | 6.0 ± 0.4 | 5.8 ± 0.7 | 5.8 ± 0.6 | — | — | — |
| NAT/μg cm−2 | 1.2 ± 0.3 | — | — | — | — | — |
| EH/μg cm−2 | — | — | 0.6 ± 0.1 | 0.7 ± 0.1 | — | — |
| cyt P450 1B1/μg cm−2 | — | — | — | 0.7 ± 0.6 | 0.8 ± 0.6 | — |
| Hs9/μg cm−2 | — | — | — | — | — | 1.5 ± 0.4 |
Cyclic voltammograms (CV) of HLM, cyt P450 1B1 and Hs9 films showed quasi-reversible peak pairs (Fig. S5(a)–(c), ESI†) with oxidation–reduction midpoint potentials of −0.50 V for HLM and Hs9, and −0.51 V for cyt P450 1B1 vs. saturated calomel electrode (SCE). Midpoint potentials of these enzyme films were characteristic of those of cyt P450 reductases in similar film assemblies.27–29
We chose benzo[a]pyrene (B[a]P) and arylamines to test the screening device because both have known multi-enzyme metabolic pathways that produce DNA-reactive metabolites. Bioactivation of B[a]P involves initial epoxidation by cyt P450s yielding B[a]P-7,8-epoxide (eqn (1)), followed by hydrolysis of B[a]P-7,8-epoxide by epoxide hydrolase to form B[a]P-7,8-dihydrodiol (eqn (2)).30 Subsequent cyt P450 oxidation of the dihydrodiol generates reactive B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE) (eqn (3)), which reacts with nucleobases in DNA.30,31
The primary pathway of arylamine metabolism involves initial exocyclic N-hydroxylation by cyt P450s to form N-arylhydoxylamine (eqn (4)).32,33N-Arylhydoxylamine hydrolyzes in acidic media to form highly reactive arylnitrenium ion (eqn (4)), or is transformed by N-acetyltransferase (NAT) or sulfotransferase (SULT) in the hepatic cytosol to O-substituted intermediates.32,34 When N-arylhydoxylamine undergoes O-acetylation by NAT, N-acetoxyarylamine is formed (eqn (5)).32 However, these O-substituted intermediates can also undergo spontaneous heterolysis of the N–O bond to form arylnitrenium ion.32,34 Highly electrophilic arylnitrenium ion forms DNA adducts at the C8 position of guanine affecting gene transcription and replication.35 NAT also induces detoxification of arylamine xenobiotics by direct N-acetylation (eqn (6)). N-Acetylarylamines do not react with nucleobases and are ultimately eliminated by excretion from the body.32,34
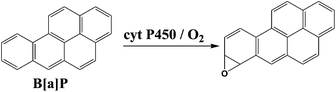 | (1) |
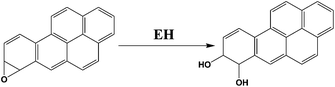 | (2) |
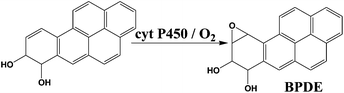 | (3) |
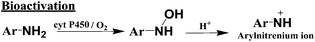 | (4) |
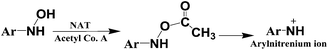 | (5) |
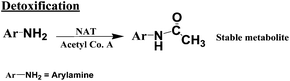 | (6) |
3.2 Reproducibility
We first determined the variance of SWV responses from four parallel microfluidic arrays connected in the multiplexed device (Scheme 1). The system chosen for this test employed human liver microsome (HLM) films as enzyme sources on all sensors and 0.2 mM 2-aminofluorene as test reactant in oxygenated 10 mM acetate buffer of pH = 5.8. 2-Aminofluorene is converted to N-hydroxy-2-aminofluorene by cyt P450 enzymes in HLM, and subsequently to reactive arylnitrenium ion (eqn (4)). We used the ratio of the SWV difference peak current after the enzyme reaction (Ip,f) to that of controls with no enzyme reaction (Ip,i)) as the measured quantity indicative of DNA damage.13,23Slopes obtained from each microfluidic chip were compared with the global regression slope (Fig. 1(a)) by t-tests. Slopes of the SWV peak ratios (Ip,f/Ip,i) (Fig. 1(a)) did not differ significantly at the 95% confidence interval (Fig. 1(b)). The microfluidic chambers are designed so that chips are easily replaced before each new experiment. The above data, in addition to assessing reproducibility of a multi-chip system for a single set of experiments, also reflects chip-to-chip reproducibility.
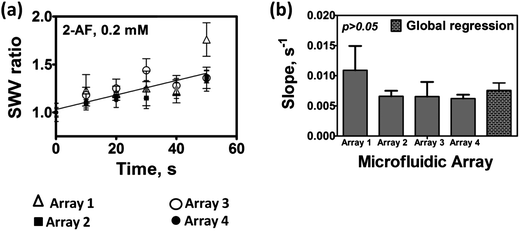 | ||
| Fig. 1 Reproducibility of microfluidic arrays used in parallel for 0.2 mM 2-AF in anaerobic 10 mM acetate buffer pH = 5.8: (a) SWV peak current ratios (Ip,f/Ip,i) obtained at each time interval are shown with a global regression line; error bars represent standard deviations for n = 4; and (b) slopes of SWV peak current ratios for each microfluidic array, as well as the global slope. | ||
3.3 Studies with B[a]P and arylamines
Increases in SWV peak current density with enzyme reaction time are directly related to the extent of DNA damage, mostly due to formation of covalent adducts of metabolites with nucleobases that disorder the DNA double helix.5,10,11 SWVs taken in the microfluidic system using B[a]P as reactant with different enzyme sources all gave increases in peak current with enzyme reaction time (Fig. 2). The variation of SWV peak ratio (Ip,f/Ip,i) with reaction time is depicted in Fig. 3 where the initial slope of SWV peak ratio vs. reaction time are proportional to relative rates of DNA damage.11,36 The largest rate of DNA damage was found for B[a]P when epoxide hydrolase was added to cyt P450 1B1 or HLM (Fig. 3 and 4), suggesting that multienzyme bioactivation occurred to produce the DNA-reactive diol epoxide (BPDE, eqn (3)). Parallel studies reacting B[a]P with metabolic enzyme/DNA biocolloids have detected the diol epoxide metabolite and its guanine and adenine adducts by LC-MS/MS.37![Background-subtracted averaged (n = 4) difference SWV current densities measured after each enzyme reaction for 25 μM B[a]P in anaerobic pH = 7.4 buffer on multiplexed array sensors featuring optimized films with different enzyme sources: (A) cyt P450 1B1, (B) cyt P450 1B1 + EH, (C) HLM, (D) HLM + EH (SWV ampl. 25 mV; freq. 15 Hz; step 4 mV).](/image/article/2013/AN/c2an35993f/c2an35993f-f2.gif) | ||
| Fig. 2 Background-subtracted averaged (n = 4) difference SWV current densities measured after each enzyme reaction for 25 μM B[a]P in anaerobic pH = 7.4 buffer on multiplexed array sensors featuring optimized films with different enzyme sources: (A) cyt P450 1B1, (B) cyt P450 1B1 + EH, (C) HLM, (D) HLM + EH (SWV ampl. 25 mV; freq. 15 Hz; step 4 mV). | ||
![Influence of enzyme reaction time on SWV peak current ratio (Ip,f/Ip,i) for microfluidic sensor arrays for 25 μM B[a]P at pH 7.4 using (A) cyt P450 1B1, (B) cyt P450 1B1 + EH, (C) HLM, (D) HLM + EH. Controls are without substrate or with substrate but without electrolysis, which gave equivalent results. Error bars represent standard deviations for n = 4.](/image/article/2013/AN/c2an35993f/c2an35993f-f3.gif) | ||
| Fig. 3 Influence of enzyme reaction time on SWV peak current ratio (Ip,f/Ip,i) for microfluidic sensor arrays for 25 μM B[a]P at pH 7.4 using (A) cyt P450 1B1, (B) cyt P450 1B1 + EH, (C) HLM, (D) HLM + EH. Controls are without substrate or with substrate but without electrolysis, which gave equivalent results. Error bars represent standard deviations for n = 4. | ||
![Influence of epoxide hydrolase on sensor array relative DNA damage rate ({μg protein−1} s−1 mM−1) for 25 μM B[a]P at pH 7.4, (a) cyt P450 1B1 + EH and (b) HLM + EH.](/image/article/2013/AN/c2an35993f/c2an35993f-f4.gif) | ||
| Fig. 4 Influence of epoxide hydrolase on sensor array relative DNA damage rate ({μg protein−1} s−1 mM−1) for 25 μM B[a]P at pH 7.4, (a) cyt P450 1B1 + EH and (b) HLM + EH. | ||
Relative DNA damage rate due to the formation of DNA-reactive metabolites was estimated as SWV peak ratio slope per μg of protein per mM of substrate (Fig. 4).23 Addition of epoxide hydrolase as an enzyme source increased the relative DNA damage rates as shown in Fig. 4. Approximately 5-fold higher turnover was observed for cyt P450 1B1 supersomes + EH compared to that of sensor arrays without EH (Fig. 4(a)). A 1.3-fold larger DNA damage rate was found for the HLM + EH (Fig. 4(b)), probably because of the known presence of microsomal EH in HLM.38 Significantly higher DNA damage rates for cyt P450 1B1 compared to HLM are attributed to the presence of higher concentrations of cyt P450 1B1 isoform in the supersomes.
SWV data for arylamines at both pH 7.4 (Fig. S6) and pH 5.8 (Fig. S7) are shown in the ESI† file. SWV ratio plots at both pHs are depicted in Fig. 5. These results show that arylamines demonstrated complex behavior with strong dependence on pH. A significant decrease in rate of relative DNA damage at pH 5.8 was observed in all cases. This is attributed to the attenuation of enzyme activity at this relatively low pH, since cyt P450s have maximum activity at pH 7.4, while NAT maxima are between pH 7.0–7.8.39 In contrast, a spectrum of relative DNA damage rates was observed with enzyme reactions at pH 7.4. In most cases, films containing both HLM and NAT show smaller relative DNA damage rates (column B, Fig. 5) at pH 7.4 due to the formation of N-acetylarylamines by direct N-acetylation by NAT.32,34N-Acetylarylamines are stable metabolites owing to the partial double bond character of the amide linkage, and are generally poor substrates for subsequent hydroxylation by cyt P450 enzymes.32 We observed slightly higher DNA damage rates at pH 5.8 than pH 7.4 with films containing HLM and NAT for 2-AF and 4-ABP possibly due to variation in arylamine enzyme specificity or idiosyncratic reactions.32,40
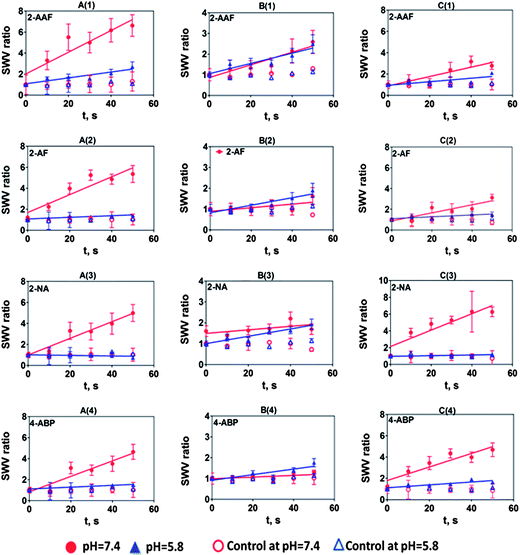 | ||
| Fig. 5 Influence of substrate incubation time on SWV peak current ratio (Ip,f/Ip,i) for multiplexed microfluidic genotoxicity sensor arrays of (A) HLM, (B) HLM + NAT, (C) Hs9, for the exposure of (1) 0.25 mM 2-AAF, (2) 0.2 mM 2-AF, (3) 0.2 mM 2-NA, (4) 0.05 mM 4-ABP/0.01 mM 4-ABP at pH 7.4 phosphate buffer and pH 5.8 acetate buffer. Controls are incubations without substrate or exposure to the substrate without electrolysis, which gave equivalent results. Error bars represent standard errors for n = 4. Negative control 0.2 mM THF did not show SWV peak increases under our assay conditions (Fig. S8, ESI†). | ||
Carcinogenic potency value (TD50) is the “dose-rate (mg kg−1 body weight per day) which, if administered chronically for the standard lifespan of the species, will halve the probability of remaining tumorless throughout that period”.41,42 Mouse liver TD50 values used here are the harmonic mean of each positive experiment from the Carcinogenic Potency Database (CPDB) by taking only liver as the target site.41,42 Relative DNA damage rates of the arylamines were compared with the reciprocal of the in vivo rodent liver TD50 (Fig. 6), which correlates directly with rodent liver carcinogenicity. The correlations of the reciprocal of rodent median lethal dose (LD50), the lethal dose (mg kg−1 body weight) of a chemical given all at once that causes the death of 50% of the rodents, values with relative DNA damage rates are included in the ESI file (Fig. S9, ESI†).43 Data from the array featuring oxidative and conjugative enzymes at pH 7.4 showed good correlation with 1/TD50 (Fig. 6(a)). The formation of DNA adducts was confirmed previously by LC-MS/MS using biocolloid reactors with enzyme–DNA films.19,20,44 In contrast, HLM and Hs9 correlate poorly with 1/TD50 values at pH 7.4 (Fig. 6(a)). Even though, Hs9 fractions contain both oxidative and conjugative enzymes,45 poor correlation was found at pH 7.4. The presence of relatively low concentrations of NAT in Hs9 fractions accounts for this behavior45 so that detoxification of arylamines to N-acetylated products is comparatively less pronounced than for HLM + NAT.
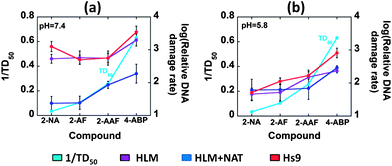 | ||
| Fig. 6 Plots illustrating correlations of the reciprocal of mouse liver TD50 values with log of relative DNA damage rates ({μg protein−1} s−1 mM−1) measured by sensor arrays containing HLM, HLM + NAT and Hs9 as enzyme sources, for 4 arylamines (a) at pH 7.4 and (b) at pH 5.8. | ||
4. Discussion
Results above validate the successful development of a multiplexed genotoxicity screening platform for detecting reactive metabolites that can arise from multiple-enzyme bioactivation pathways. Sensors were equipped with HLM and cyt P450 1B1 supersomes as oxidative enzyme sources and NAT and epoxide hydrolase as conjugative enzymes reproducing the spectrum of bioactivation events for the test compounds used. The microfluidic platform facilitated rapid multiplexed detection of reactive metabolites via their reactions with DNA by using four 8-electrode microfluidic devices in sequence (Fig. 2–6), and the system is adaptable to accommodate up to 8 such devices. The versatility of the device allows other enzymes to be added at will.The microfluidic system was used to access 32 experiments involving first running enzyme reactions and then measuring resulting DNA damage within 20–30 minutes. We previously showed that microfluidics enhance reproducibility and sensitivity by rapidly processing reaction chemistries with well-controlled hydrodynamics.23 Laminar flow of oxygenated substrate at low Reynolds numbers to the reactive sensor surfaces facilitates better sensitivity by maintaining oxygen and substrate concentrations constant throughout the enzyme reaction,46,47 as opposed to our earlier non-microfluidic ECL arrays in which reactants are spotted and concentrations are influenced by the reaction itself, access to atmospheric oxygen, and evaporation.14–16
Data in this paper clearly illustrate that the system described can mimic the known influence of bioconjugation enzymes that participate in multi-enzyme metabolism of test compounds. For example, increased DNA damage rate upon addition of epoxide hydrolase to cyt P450 enzyme sources for B[a]P reflect as expected an increased rate of DNA damage in the presence of the hydrolase (Fig. 3 and 4). Epoxide hydrolase significantly enhances turnover due to the formation of DNA-reactive BPDE metabolite (eqn (3)). A significantly lower DNA damage rate was found when array spots contained only cyt P450 1B1 supersomes because it forms B[a]P-7,8-epoxide (eqn (1)), which is not known to cause a large amount of DNA damage.30,31
Relative DNA damage rates obtained with arylamines varied with buffer acidity as expected.32,34 Good correlations may not result when comparing human enzymes (such as HLM and the S9 fraction) with rodent 1/TD50 since animal models may be poor indicators of human toxicity.1,6,16,40 Nevertheless, we feel that such comparisons are useful to place our results in the context of in vivo toxicity. Sensors containing HLM and NAT demonstrated better correlation of relative DNA damage rates with rodent tumorgenicity metric as 1/TD50 at the physiological pH 7.4 (Fig. 6) highlighting the significance of multienzyme bioactivation as in the human liver. At pH 5.8, this correlation is less strong, reflecting that liver TD50 values correspond to the typical liver pH of 7.4. Moreover, relative DNA damage rates obtained for HLM + NAT at pH 7.4 showed comparatively smaller values than those for only HLM or Hs9. (To obtain clearer comparison with HLM + NAT films, Hs9 factions were supplied only with cofactors of NAT, not the other conjugation enzymes present.) Behavior of HLM/NAT in our assays reflects the known competition for arylamines between bioactivation and detoxification at physiological pH.32,34 Thus, in HLM/NAT films, after oxidative metabolism of arylamines, acetyl conjugation follows leading to N-acetoxyarylamines (eqn (5)), which in turn undergo heterolytic cleavage to form arylnitrenium ions (eqn (5)). On the other hand, arylamines can undergo N-acetylation to form N-acetylarylamines (eqn (6)), which are not known to cause DNA damage.32,34 Imperfect correlations can be attributed to species differences, difference in measurement endpoints, and metabolism and detoxification differences in vivo and in vitro.
In contrast with pH 7.4, reasonable correlation with 1/TD50 in the presence or absence of NAT was found at pH 5.8 (Fig. 6(b)) for all the enzyme sources. This reflects that the DNA damage is caused by a common precursor, the arylnitrenium ion. Arylnitrenium ion can be formed from N-arylhydoxylamine (eqn (4)), which is a major product generated from oxidative metabolism of arylamines under acidic conditions.32 We used weakly acidic buffer pH 5.8, a value approximating that in the bladder, to facilitate hydrolysis of N-arylhydoxylamines which we previously observed at this pH in single sensor protocols and confirmed by LCMS/MS.44
In summary, the present study reports a versatile multiplexed microfluidic platform with electrochemical cyt P450 activation and DNA damage detection for metabolic genotoxicity screening featuring oxidative and conjugative metabolism, using a genotoxicity-related DNA damage endpoint. To our knowledge, this is the first report of an electrochemical microfluidic platform designed to detect reactive metabolites generated from multienzyme pathways designed to mimic human metabolism. The new instrumental set up facilitates 64 sensor measurements in rapid sequence. Results obtained with the system were consistent with the known DNA-reactivity of metabolites of arylamines and B[a]P, and with LC-MS/MS studies using similar films on biocolloid reactors. Electrode driven cyt P450 catalysis eliminates the requirement for expensive NADPH cofactors, and microfluidics provides constant reactant and cofactor feed during the enzyme conversion step. Rapid detection of reactive metabolites formed from up to four compounds within 20–30 minutes reflects very good screening throughput. Cooperation of multienzyme sources in the array provides realistic predicting capability and is necessary for consistent correlation with in vivo bioassays. This in vitro screening platform holds considerable promise as a molecular chemistry complement to genotoxicity bioassays. The niche of this approach is to test the possibility of metabolites reacting with DNA. A criterion for potential genotoxicity could be identified as a slope of SWV ratio vs. reaction time exceeding that of the control slope by 3 × (standard deviation of measurements). For compounds that meet this criterion, identities and formation rates of metabolite–DNA adducts can then be determined using DNA–enzyme biocolloid reactors and LC-MS/MS.5,10,20
Of course, full screening of unknown compounds with such devices will require a more complete set of bioconjugation enzymes, but they can be easily accommodated within the device described in this paper. Future progress would also benefit from miniaturization and simplification of the device and new protocols for faster, more convenient screening. These goals are currently being pursued in our laboratory.
Acknowledgements
This work was supported financially by US PHS grant no. ES03154 from the National Institute of Environmental Health Sciences (NIEHS), NIH, USA.References
- E. Stokstad, Science, 2009, 325, 294–295 CrossRef
.
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203–215 CAS
- J. A. Kramer, J. E. Sagartz and D. L. Morris, Nat. Rev. Drug Discovery, 2007, 6, 636–649 CrossRef CAS
- A. E. F. Nassar, A. M. Kamel and C. Clarimont, Drug Discovery Today, 2004, 9, 1055–1064 CrossRef CAS
- J. F. Rusling, E. G. Hvastkovs and J. B. Schenkman, Curr. Opin. Drug Discovery Dev., 2007, 10, 67–73 CAS
- B. K. Park, A. Boobis, S. Clarke, C. E. P. Goldring, D. Jones, J. G. Kenna, C. Lambert, H. G. Laverty, D. J. Naisbitt, S. Nelson, D. A. Nicoll-Griffith, R. S. Obach, P. Routledge, D. A. Smith, D. J. Tweedie, N. Vermeulen, D. P. Williams, I. D. Wilson and T. A. Baillie, Nat. Rev. Drug Discovery, 2011, 10, 292–307 CrossRef CAS
- F. P. Guengerich, Chem. Res. Toxicol., 2001, 14, 611–650 CrossRef CAS
- M. Tarun and J. F. Rusling, Anal. Chem., 2005, 77, 2056–2062 CrossRef CAS
- J. F. Rusling, Biosens. Bioelectron., 2004, 20, 1022–1028 CrossRef CAS
- J. F. Rusling, E. G. Hvastkovs, D. O. Hull and J. B. Schenkman, Chem. Commun., 2008, 141–154 RSC
-
J. F. Rusling, E. G. Hvastkovs and J. B. Schenkman, in Drug Metabolism Handbook, ed. A. Nassar, P. F. Hollenburg and J. Scatina, Wiley, NJ, 2009, pp. 307–340 Search PubMed
- L. Zhou and J. F. Rusling, Anal. Chem., 2001, 73, 4780–4786 CrossRef CAS
- L. Zhou, J. Yang, C. Estavillo, J. D. Stuart, J. B. Schenkman and J. F. Rusling, J. Am. Chem. Soc., 2003, 125, 1431–1436 CrossRef CAS
- E. G. Hvastkovs, M. So, S. Krishnan, B. Bajrami, M. Tarun, I. Jansson, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2007, 79, 1897–1906 CrossRef CAS
- S. Krishnan, E. G. Hvastkovs, B. Bajrami, I. Jansson, J. B. Schenkman and J. F. Rusling, Chem. Commun., 2007, 1713–1715 RSC
- S. Pan, L. Zhao, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2011, 83, 2754–2760 CrossRef CAS
- B. Bajrami, E. G. Hvastkovs, G. Jensen, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2008, 80, 922–932 CrossRef CAS
- B. Bajrami, L. Zhao, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2009, 81, 9921–9929 CrossRef CAS
- L. Zhao, S. Krishnan, Y. Zhang, J. B. Schenkman and J. F. Rusling, Chem. Res. Toxicol., 2009, 22, 341–347 CrossRef CAS
- L. Zhao, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2010, 82, 10172–10178 CrossRef CAS
- S. Krishnan, D. Wasalathanthri, L. Zhao, J. B. Schenkman and J. F. Rusling, J. Am. Chem. Soc., 2011, 133, 1459–1465 CrossRef CAS
- S. Krishnan, J. B. Schenkman and J. F. Rusling, J. Phys. Chem. B, 2011, 115, 8371–8380 CrossRef CAS
- D. P. Wasalathanthri, V. Mani, C. K. Tang and J. F. Rusling, Anal. Chem., 2011, 83, 9499–9506 CrossRef CAS
- L. Dennany, R. J. Forster and J. F. Rusling, J. Am. Chem. Soc., 2003, 125, 5213–5218 CrossRef CAS
-
Y. Lvov, in Handbook of Surfaces and Interfaces of Materials, ed. R. W. Nalwa, Academic Press, San Diego, CA, 2001, vol. 3, pp. 170–189 Search PubMed
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS
- N. Sultana, J. B. Schenkman and J. F. Rusling, J. Am. Chem. Soc., 2005, 127, 13460–13461 CrossRef CAS
- S. Krishnan and J. F. Rusling, Electrochem. Commun., 2007, 9, 2359–2363 CrossRef CAS
- N. Sultana, J. B. Schenkman and J. F. Rusling, Electroanalysis, 2007, 24, 2499–2506 CrossRef
- R. Benigni and C. Bossa, Chem. Rev., 2011, 111, 2507–2536 CrossRef CAS
- J. H. Kim, K. H. Stansbury, N. J. Walker, M. Trush, P. T. Strickland and T. R. Sutter, Carcinogenesis, 1998, 19, 1847–1853 CrossRef CAS
-
R. J. Turesky, in The Chemical Biology of DNA Damage, ed. N. E. Geacintov and S. Broyde, Wiley, NJ, 2010, pp. 157–184 Search PubMed
- M. A. Butler, M. Iwasaki, F. P. Guengerich and F. F. Kadlubar, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7696–7700 CrossRef CAS
- D. Kim and F. P. Guengerich, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 27–49 CrossRef CAS
- P. F. Guengerich, Drug Metab. Rev., 2002, 34, 607–623 CrossRef
- M. So, E. G. Hvastkovs, J. B. Schenkman and J. F. Rusling, Biosens. Bioelectron., 2007, 23, 492–498 CrossRef CAS
- S. Pan, D. Li and J. F. Rusling, unpublished work.
- A. J. Fretland and C. J. Omiecinski, Chem.-Biol. Interact., 2000, 129, 41–59 CrossRef CAS
-
(a) J. R. Cashman, Biochem. Biophys. Res. Commun., 2005, 338, 599–604 CrossRef CAS
- L. Liu, A. V. Vett, N. Zhang, K. J. Walters, C. R. Wagner and P. E. Hanna, Chem. Res. Toxicol., 2007, 20, 1300–1308 CrossRef CAS
- L. S. Gold, The Carcinogenic Potency Database, http://potency.berkeley.edu.
- R. Peto, M. C. Pike, L. Bernstein, L. S. Gold and B. N. Ames, Environ. Health Perspect., 1984, 58, 1–8 CAS
- United States National Library of Medicine, http://chem.sis.nlm.nih.gov/chemidplus.
- M. So, E. G. Hvaskovs, B. Bajrami, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2008, 80, 1192–1200 CrossRef CAS
- E. F. A. Brandon, C. D. Raap, I. Meijerman, J. H. Beijnen and J. H. M. Schellens, Toxicol. Appl. Pharmacol., 2003, 189, 233–246 CrossRef CAS
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS
- D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153–1182 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2an35993f |
| This journal is © The Royal Society of Chemistry 2013 |