Biofuel cell-based self-powered biogenerators for online continuous monitoring of neurochemicals in rat brain
Hanjun
Cheng
a,
Ping
Yu
a,
Xulin
Lu
a,
Yuqing
Lin
a,
Takeo
Ohsaka
b and
Lanqun
Mao
*a
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, the Chinese Academy of Sciences (CAS), Beijing 100190, China. E-mail: lqmao@iccas.ac.cn; Fax: +86-10-62559373
bDepartment of Electronic Chemistry, Interdisciplinary Graduate School of Science and Engineering, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan
First published on 23rd October 2012
Abstract
This study demonstrates a new electrochemical method for continuous neurochemical sensing with a biofuel cell-based self-powered biogenerator as the detector for the analysis of microdialysate continuously sampled from rat brain, with glucose as an example analyte. To assemble a glucose/O2 biofuel cell that can be used as a self-powered biogenerator for glucose sensing, glucose dehydrogenase (GDH) was used as the bioanodic catalyst for the oxidation of glucose with methylene green (MG) adsorbed onto single-walled carbon nanotubes (SWNTs) as the electrocatalyst for the oxidation of dihydronicotinamide adenine dinucleotide (NADH). Laccase crosslinked onto SWNTs was used as the biocathodic catalyst for the O2 reduction. To enable the bioanode and biocathode to work efficiently in their individually favorable solutions and to eliminate the interference between the glucose bioanode and O2 biocathode, the biofuel cell-based biogenerator was built in a co-laminar microfluidic chip so that the bioanodic and biocathodic streams could be independently optimized to provide conditions favorable for each of the bioelectrodes. By using a home-made portable voltmeter to output the voltage generated on an external resistor, the biogenerator was used for glucose sensing based on a galvanic cell mechanism. In vitro experiments demonstrate that, under the optimized conditions, the voltage generated on an external resistor shows a linear relationship with the logarithmic glucose concentration within a concentration range of 0.2 mM to 1.0 mM. Moreover, the biogenerator exhibits a high stability and a good selectivity for glucose sensing. The validity of the biofuel cell-based self-powered biogenerator for continuous neurochemical sensing was illustrated by online continuous monitoring of striatum glucose in rat brain through the combination of in vivo microdialysis. This study offers a new and technically simple platform for continuously monitoring physiologically important species in cerebral systems.
Introduction
Extensive attention has long been given to the development of new analytical methods for the study of brain chemistry because brain chemistry basically allows the brain to function efficiently with various neurochemicals and its variations may consequently explain how those neurochemicals affect brain functions at the molecular level.1–5 Earlier efforts from our and other groups have demonstrated that the smart design and rational construction of electrode/electrolyte interfaces greatly enable electrochemical methods, which are particularly attractive for probing brain chemistry either based on the electrochemical properties of the neurochemical themselves or on the full exploration of biomacromolecules (e.g., enzymes), to specifically convert the neurochemicals into electrochemically detectable species.6–12 However, the ever-increasing interest in understanding brain chemistry still presents a pressing need for the development of theoretically simple and less technically demanding analytical methods for neurochemical sensing which can be easily adapted for the monitoring of other physiologically important species.13–16In this study, we demonstrate a new electrochemical method for neurochemical sensing with self-powered biogenerators as the biosensors by efficiently combining in vivo microdialysis with biofuel cell technology. As first demonstrated by Wang et al., self-powered generators represent one kind of energy scavenging devices that harvest energy in personal and daily environments not only for powering personal electronics but also for biomedical and healthcare applications.17,18 As one kind of self-powered biogenerators that harvest bio-energy from biochemical reactions to produce electricity, enzymatic biofuel cells utilize enzymes as the catalysts and have advantages in mild operation conditions (i.e. ambient temperature and neutral pH) and potential applications as in vivo power sources.19–26 Nowadays, biofuel cells have been of great concern both in fundamental bioelectrochemical studies27–33 and in practical applications.34–38 While the intrinsic properties of enzymatic biofuel cells have been demonstrated to offer a new approach to analytical applications, as elegantly reported recently,39−44 it remains a challenge to apply such self-powered biogenerators for in vivo neurochemical sensing through a galvanic cell mechanism, even though the self-powered feature of the biogenerators will simplify the analytical systems and potentially facilitate wireless neurochemical monitoring in freely moving animals. Firstly, the biocathodic catalysts, i.e., laccase used in this work, normally work only in weakly acidic media and lose almost all of their catalytic activity in neutral media.45–47 This property, unfortunately, invalidates the laccase-based biogenerators for neurochemical sensing under physiological conditions. Secondly, the O2 level in the brain is relatively low (ca. 50 μM)15,48 and often undergoes a large fluctuation during some physiological and pathological processes such as cerebral ischemia/reperfusion.49–52 These features actually make it quite difficult to constitute an analytical scheme for neurochemical sensing since the signal output from the as-prepared biogenerators will be dominated by that of the biocathode for the O2 reduction, rather than by that of the bioanode for the neurochemical oxidation. Thirdly, the co-existence of electroactive neurochemicals, ascorbic acid in particular, with the neurochemicals of interest renders difficulties in achieving selectivity for neurochemical sensing with biogenerators.
To circumvent these problems, this study utilizes a microfluidic technique to validate the biofuel cell-based self-powered biogenerators for in vivo neurochemical sensing, with glucose as an example. The utilization of a microfluidic technique enables the bioanode and biocathode to work efficiently in their individually favorable media and suppresses the interference from the microdialysates at the O2 biocathode as well as increasing the power output of the O2 biocathode so as to validate the microfluidic self-powered biogenerators for in vivo neurochemical sensing. As far as we know, this is the first demonstration of the use of biofuel cell-based biogenerators for neurochemical sensing, and is envisaged to be applicable to the monitoring of brain chemistry in quite a simple fashion.
Experimental
Reagents and solutions
Laccase (from Trametes versicolor) was purchased from Sigma and was purified as reported previously.53 Glucose dehydrogenase (GDH, from Pseudomonas sp.), bovine serum albumin (BSA), D-(+)-glucose and ascorbic acid (AA) were all purchased from Sigma and were used as received. The oxidized form of nicotinamide adenine dinucleotide (NAD+) and methylene green (MG) were purchased from Roche. Single-walled carbon nanotubes (SWNTs, with an average diameter less than 2 nm and a length of about 50 μm) were obtained from Shenzhen Nanoport Co. Ltd (Shenzhen, China) and were purified by refluxing the as-received samples in 2.6 M HNO3 for 5 h, followed by centrifugation, resuspension, filtration and drying to evaporate the solvent. Artificial cerebrospinal fluid (aCSF) was prepared by mixing NaCl (126 mM), KCl (2.4 mM), KH2PO4 (0.5 mM), MgCl2 (0.85 mM), NaHCO3 (27.5 mM), Na2SO4 (0.5 mM), and CaCl2 (1.1 mM) in water. Phosphate buffer (0.16 M) was prepared with Na2HPO4 and NaH2PO4 and the buffer pH was adjusted with NaOH. All aqueous solutions were prepared using 18.2 MΩ cm−1 Milli-Q water (Millipore).Microchip design and fabrication
A 60 mm × 60 mm poly(methyl methacrylate) (PMMA) glass was used as the master chip with desired patterns fabricated by mechanical microfabrication. A polydimethylsiloxane (PDMS) master with positive surface patterns was produced by molding against the as-prepared PMMA glass master, as described previously.54 Briefly, the PDMS precursor mixture prepared at a weight ratio of base to curing agent of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was first poured carefully onto the PMMA glass master, then vacuumed for 0.5 h to avoid the formation of bubbles, and finally cured at 100 °C for 1 h. After gently peeled off the master, the cured PDMS master was cleaned by an O2 plasma and then silanized with 1H,1H,2H,2H-perfluorodecyltriethoxysilane (Alfa Aesar, UK) for 12 h. To generate microfluidic channels, we replica-molded PDMS chips from the prepared PDMS master. The microchannels used in this study were of the dimensions indicated in Scheme 1 (i.e., width, 1 mm; depth, 0.5 mm; length, 20 mm). The inlets and outlets of the microfluidic channels (ca. 0.8 mm in diameter) were made with a sharpened needle.
:1 was first poured carefully onto the PMMA glass master, then vacuumed for 0.5 h to avoid the formation of bubbles, and finally cured at 100 °C for 1 h. After gently peeled off the master, the cured PDMS master was cleaned by an O2 plasma and then silanized with 1H,1H,2H,2H-perfluorodecyltriethoxysilane (Alfa Aesar, UK) for 12 h. To generate microfluidic channels, we replica-molded PDMS chips from the prepared PDMS master. The microchannels used in this study were of the dimensions indicated in Scheme 1 (i.e., width, 1 mm; depth, 0.5 mm; length, 20 mm). The inlets and outlets of the microfluidic channels (ca. 0.8 mm in diameter) were made with a sharpened needle.
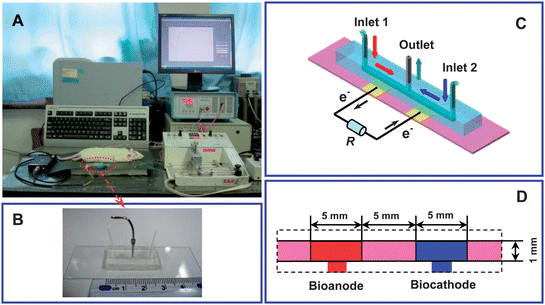 | ||
| Scheme 1 (A) Schematic illustration of the sensing system based on the efficient integration of a biofuel cell-based biogenerator with in vivo microdialysis for online continuous monitoring of glucose in rat brain. (B) Photograph, and (C and D) architecture of the microfluidic biofuel cell-based biogenerator. | ||
Preparation of the microfluidic biofuel cell-based biogenerator
Fluorine-doped tin oxide (FTO) glass plates were used as the substrate for the preparation of the biofuel cell-based biogenerator. Prior to the preparation, the FTO glass plates were cleaned by bath sonication with alcohol, acetone and Milli-Q water, each for 10 min. To prepare a GDH-based bioanode, 5 μL of SWNT dispersion in N,N-dimethylformamide (DMF) (1 mg mL−1) was dip-coated onto the FTO glass plate (1 mm × 5 mm). After the plate was dried at ambient temperature, 5 μL of the aqueous solution of MG (1 mg mL−1) was dip-coated onto the SWNT-modified FTO glass plate and the plate (MG/SWNT-modified plate) was carefully rinsed with Milli-Q water and air-dried. To confine GDH onto the MG/SWNT-modified plate, 4 μL of GDH (10 mg mL−1) was mixed with 2 μL of BSA (1 wt%) and 2 μL of glutaraldehyde (1 wt%) and the mixture was then totally dip-coated onto the MG/SWNT-modified plate. The plate was air-dried and used as the bioanode in the biogenerator for the oxidation of glucose. To prepare the laccase-based biocathode in the biogenerator, 4 μL of the purified laccase (50 mg mL−1), 4 μL of BSA (1%, wt%) and 2 μL of glutaraldehyde (1%, wt%) were mixed together and the mixture was totally dip-coated onto the SWNT-modified FTO glass plate (1 mm × 5 mm). The obtained plate was finally air-dried and used as the biocathode for the reduction of O2. Finally, a PDMS stamp (30 mm × 10 mm × 5 mm, Scheme 1) with an I-shaped microchannel was covered onto the two plates and the as-prepared microchip was then sealed with two steel clamps.Apparatus and electrochemical measurements
Three syringe pumps were used to perfuse the bioanodic and biocathodic streams into the microchannel. Electrochemical experiments employed to separately study the properties of the bioanode and the biocathodes integrated on the microchip were performed on a computer-controlled Autolab (Eco chemie). For such a purpose, a micro-sized Ag/AgCl electrode prepared in our earlier studies55 was carefully inserted into the microchannel and used as the reference electrode. A stainless steel needle outlet was used as the counter electrode. The bioanodic and biocathodic streams were separately perfused into the microchannel through inlets 1 and 2, respectively. For investigation of the performance of the biofuel cell-based biogenerator, the bioanodic and biocathodic streams were simultaneously perfused into the microchannel respectively from inlets 1 and 2, and flowed out from the outlet, as shown in Scheme 1C.In order to apply the assembled biogenerator for continuous sensing of glucose in the brain of living rats, in vivo microdialysis was efficiently coupled with the biogenerator to form an online detecting system (Scheme 1A). In this case, brain microdialysate and solutions were delivered from gas-impermeable syringes and pumped through tetrafluoroethylene hexafluoropropene (FEP) tubing by three microinjection pumps (i.e., pumps 1, 2 and 3). Brain microdialysates were sampled from the rat brain with pump 1 with pure aCSF as the perfusion solution at 1.5 μL min−1. To supply the NAD+ cofactor to the enzymatic reactions of GDH, aCSF containing 2.5 mM NAD+ was externally perfused with pump 2 at 1.5 μL min−1 and online mixed with the microdialysates in a T-joint. The mixture was perfused into the microchannel through inlet 1 and referred to as the bioanodic stream (Scheme 1C). Phosphate buffer (0.16 M, pH 6.0) was perfused with pump 3 into the microchannel through inlet 2 at 3 μL min−1 and serves as the biocathodic stream. The bioanodic and biocathodic streams formed in a waste stream that flowed out of the microchannel through the outlet. A home-made portable voltmeter was used to continuously measure the voltage generated on an external 600 kΩ resistor. The voltage was used as the signal readout for the cerebral glucose sensing.
In vivo microdialysis
Animal surgery and in vivo microdialysis were carried out with procedures demonstrated in our previous reports.56,57 Briefly, adult male Sprague-Dawley rats (250–300 g) purchased from the Health Science Center, Peking University were housed using a 12:12 h light–dark schedule with food and water ad libitum. A microdialysis guide cannula (BAS/MD-2250, BAS) was implanted into the striatum (0 mm caudal to bregma, 3.0 mm medial-lateral, and 2 mm ventral from the surface of the skull) using standard stereotaxic procedures. The guide cannula was kept in place with three skull screws and dental cement. Stainless steel dummy blockers were inserted into the guide cannula and fixed until the insertion of the microdialysis probe. Throughout the surgery, the body temperature of the animals was maintained at 37 °C with a heating pad. After the rats were allowed to recover for at least 24 h, a microdialysis probe (BAS; dialysis length, 4 mm; diameter, 0.24 mm) was first implanted into the rat striatum and then perfused with aCSF solution at 1.5 μL min−1. After continuously perfusing the probe for at least 90 min for equilibration, the microdialysate was continuously sampled and online measured with the biogenerator with voltage generated on the external resistor as the signal readout.
Results and discussion
Performance of microfluidic glucose/O2 biofuel cell-based biogenerators
Fig. 1 shows typical polarization curves of the GDH-based bioanode for glucose oxidation and the laccase-based biocathode for O2 reduction. As depicted in Fig. 1A, at the GDH/MG/SWNT-based bioanode, the oxidation of glucose commenced at ca. −0.14 V. This potential was almost identical to that obtained in our early studies with GDH as the biorecognition element and with MG as the electrocatalysts for the oxidation of NADH,25,58–60 demonstrating that the oxidation of glucose at the bioanode undergoes the GDH-based bioelectrocatalytic process shown in Fig. 1A (inset). The current density reached its maximum (350 μA cm−2) at about +0.10 V. The current density was comparable to or higher than those reported previously,25,58–60 presumably due to the enhanced mass transport in the microchannel employed in this study.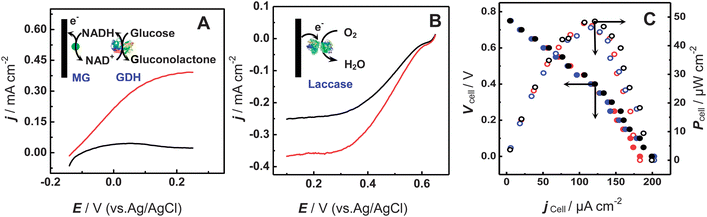 | ||
| Fig. 1 (A) Polarization curves of the bioanode for glucose oxidation in 0.16 M phosphate buffer (pH 7.0) containing 10 mM NAD+ in the absence (black curve) and presence (red curve) of 30 mM glucose. Flow rate, 1 μL min−1; potential scan rate, 1 mV s−1. (B) Polarization curves of the biocathode for the O2 reduction in 0.16 M phosphate buffer (pH 6.0) under ambient air (black curve) or saturated with O2 (red curve). Flow rate, 1 μL min−1; potential scan rate, 1 mV s−1. (C) Polarization curves (filled symbol) and the relationship between power output and current densities (open symbol) for the glucose/O2 biofuel cell-based biogenerator. The bioanodic stream was 0.16 M phosphate buffer (pH 7.0) containing 1 mM NAD+ and 2 mM glucose, while the biocathodic stream was 0.16 M phosphate buffer (pH 6.0). The flow rate for the bioanodic stream was kept as 3 μL min−1 while that for the biocathodic stream was set as 3 μL min−1 (black curve), 4 μL min−1 (red curve), and 5 μL min−1 (blue curve). | ||
For the catalytic reduction of O2, laccase was preferentially used in this study because it catalyzes the reduction of O2 at a higher potential than bilirubin oxidase (BOD). Fig. 1B depicts the polarization curves at the laccase-based biocathode for O2 reduction in 0.16 M phosphate buffer (pH 6.0). O2 reduction commenced at +0.62 V, which was very close to the redox potential of laccase (i.e., 0.58 V vs. Ag/AgCl), showing that O2 was reduced into H2O under the bioelectrocatalysis of laccase through a direct electron transfer pathway (Fig. 1B, inset).25,47 Furthermore, the similarity between the onset potential for the O2 reduction at the biocathode (i.e., 0.62 V) with the thermodynamic equilibrium potential for the O2 reduction (i.e., 0.68 V vs. Ag/AgCl at pH 6.0) essentially indicates that a low overpotential was involved in the O2 reduction at the laccase-based biocathode used in the biogenerator in this study. The current density for the O2 reduction reached its maximum value of 250 μA cm−2 at 0.35 V under ambient atmosphere and ca. 350 μA cm−2 at the same potential under an O2-saturated atmosphere. The high current density was ascribed to the enhanced mass transport in the microchannel employed in this study, as described above.
As shown in Fig. 1C, the open circuit voltage (OCV) of the assembled biogenerator was as high as 0.78 V and the maximum power density reached 48 μW cm−2 at 0.40 V. These results substantially demonstrate that the utilization of the microfluidic technique actually makes it possible to enable the bioanode and the biocathodes to work efficiently in their individually favorable media. This is remarkable since, as reported in our early studies,47 laccase only works in weakly acidic media and losses almost all activity for the O2 reduction in neutral media. More importantly, we found that utilization of the microchip to develop biofuel cells eventually enables the performance of the as-prepared biogenerator to be dominated by that of the bioanode. This was evident by the lower effect of flow rate of the biocathodic stream on the performance of the microfluidic biogenerator within the range of the flow rate employed. In this case, the bioanodic stream was kept at a flow rate of 3 μL min−1, while the biocathodic stream was perfused at various flow rates. As displayed in Fig. 1C, the OCV (i.e., ca. 0.78 V) and the maximum power density (ca. 48 μW cm−2) of the assembled biogenerators remained almost unchanged when the flow rate of the biocathodic stream was varied, suggesting that the output of the assembled biogenerator would be dominated by that of the glucose bioanode, rather than that of the O2 biocathode, under the present conditions. This property essentially validates the assembled biogenerator based on the biofuel cell technology for neurochemical sensing.
Stability, linearity, and selectivity
The stability of the biofuel cell-based biogenerator was evaluated in vitro. As depicted in Fig. 2, the OCV values of the assembled biogenerator remained unchanged after continuously running the operation for at least 3 h. This result suggests that the uses of microfluidic technology eventually enables both the bioanode and biocathode to work efficiently and stably in their individual favorable media. This property substantially validates the assembled biogenerators for effective in vivo neurochemical sensing, as will be demonstrated below.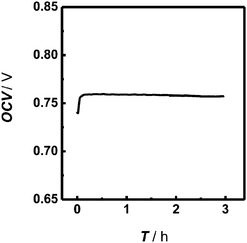 | ||
| Fig. 2 Open circuit voltage (OCV) continuously recorded with the assembled biofuel cell-based biogenerator as a function of time. The bioanodic stream was 0.16 M phosphate buffer (pH 7.0) containing 1 mM NAD+ and 2 mM glucose and the biocathodic stream was 0.16 M phosphate buffer (pH 6.0). Both streams were simultaneously and independently perfused into the microchip at 3 μL min−1. | ||
To quantitatively sense cerebral glucose, the biofuel cell-based biogenerator was integrated with in vivo microdialysis to form an online electrochemical detection system. In this case, aCSF was used as the bioanodic stream, instead of phosphate buffer which had been formerly used, whilst the biocathodic stream was unchanged. We found that the substitution of phosphate buffer with aCSF as the bioanodic stream did not result in an obvious change in the performance of the assembled biogenerator (data not shown). Under the conditions employed here, a well-defined voltage response generated on the external resistor was recorded for glucose, as displayed in Fig. 3. The voltage response showed a linear relationship with logarithmic glucose concentration within the concentration range of 0.2 to 1.0 mM (E/V = 0.17log Cglu/mM + 0.13, γ = 0.969), revealing that the microfluidic biofuel cell-based biogenerator is responsive towards glucose. Furthermore, the dynamic linear range for glucose with the assembled biofuel cell-based biogenerator covers the physiological levels of microdialysate glucose well, further validating the application of the self-powered biogenerators developed in this study for the continuous online monitoring of glucose in rat brain. It should be noted that the inner volume of the flow cell for the bioanode was ca. 2.5 μL, which was comparable to or much smaller than other kinds of self-powered biosensors reported previously.38–42 This property essentially endows the self-powered biogenerator developed in this study with a good temporal resolution for cerebral glucose sensing.
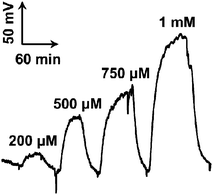 | ||
| Fig. 3 Typical voltage–time responses obtained with the online detecting system with the self-powered biogenerator as the detector toward glucose. The standard glucose solutions (concentrations are given in the figure) were online mixed with external aCSF containing 2.5 mM NAD+ in a T-joint and the resulting mixture was perfused into the microchip through inlet 1 at 3 μL min−1. Meanwhile, 0.16 M phosphate buffer (pH 6.0) used as the biocathodic stream was perfused into the microchip through inlet 2 with pump 3 at 3 μL min−1. A 600 kΩ external resistor was connected to the two electrodes of the biogenerator and the voltage generated on the resistor was continuously monitored by a home-made portable voltmeter. | ||
Since the microfluidic biofuel cell-based biogenerator was directly connected to an in vivo microdialysis system without sample collection and separation, it was essential to evaluate the selectivity of the biogenerator toward glucose. Among all the neurochemicals that may potentially interfere with glucose sensing, AA was first studied as AA presents in the cerebral systems at a high level and can be readily oxidized electrochemically. The latter feature of AA, unfortunately, means that it is a potential interferent through its oxidation at both the bioanode and the biocathode, resulting in the failure of the biogenerator for neurochemical sensing in cerebral systems.47,59 To study the interference from AA, 30 μM AA, 500 μM glucose and their mixture were separately perfused into the microfluidic biogenerator and the voltage generated at the resistor was recorded. As depicted in Fig. 4, the perfusion of 30 μM AA produced a small voltage response (ca. 3 mV), which is much lower compared with that for 500 μM glucose (ca. 33 mV). In addition, the voltage response for the mixture of 500 μM glucose and 30 μM AA was almost identical to that for 500 μM glucose. These results demonstrate that AA does not interfere with cerebral glucose sensing with the biogenerator under the present conditions.
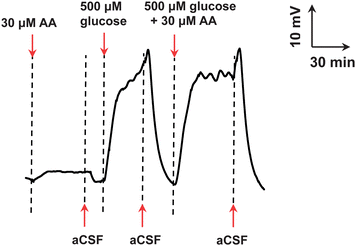 | ||
| Fig. 4 Voltage–time responses obtained with the online electrochemical detecting system with the assembled self-powered biogenerator as the detector for the standards of AA, glucose and their mixture (concentrations indicated in the figure). Other conditions were the same as those in Fig. 3. | ||
Moreover, other physiologically important species that commonly exist in the cerebral systems including 3,4-dihydroxyphenylacetic acid (DOPAC), dopamine (DA), uric acid (UA), 5-hydroxytryptamine (5-HT) and lactate also show no interference toward glucose sensing, as discussed below.
Online measurement of striatum glucose in the brain microdialysate
Fig. 5 depicts typical voltage responses for the microdialysate continuously sampled from the striatum of the rat brain. On the basis of the demonstrations mentioned above, the voltage response was ascribed to extracellular glucose in the cerebral system. The basal level of the microdialysate glucose in the striatum of rat brain was measured to be ca. 530 μM, which was almost consistent with the values reported previously.61,62 To ensure that the power output of the self-powered biogenerator had originally resulted from glucose in the brain microdialysate, the bioanode without GDH modification (i.e., MG/SWNT-modified electrode) was used as the anode to assemble a biofuel cell by the same procedures employed for the assembly of the glucose/O2 biofuel cell. Unlike the results obtained with the glucose/O2 biofuel cell-based biogenerator, a very slight power output (ca. 5 mV) of the prepared biogenerator was recorded, which mainly generates from AA and other kinds of electroactive neurochemicals including DOPAC, DA, UA, and 5-HT in the brain microdialysate. These results eventually demonstrate that the assembled microfluidic self-powered biogenerator presented here could be used for the selective in vivo sensing of glucose in cerebral systems.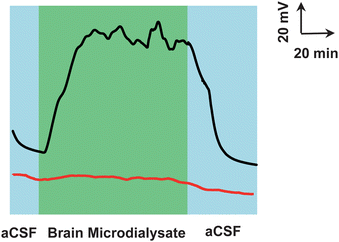 | ||
| Fig. 5 Typical voltage–time response recorded for brain microdialysate continuously sampled from living rats with GDH/MG/SWNT-based (black curve) and MG/SWNT-based (red curve) biogenerators as the detector. The microdialysates were continuously sampled from the brain striatum of the rat, online mixed with external aCSF containing 2.5 mM NAD+ and the resulting mixture was perfused into the microchip through inlet 1 at 3 μL min−1. Other conditions were the same as those in Fig. 3. | ||
Conclusions
By efficiently combining in vivo microdialysis with biofuel cell technology, we have successfully developed a new method for effective in vivo neurochemical sensing with self-powered biogenerators as the biosensors. The use of microfluidic technology essentially enables the bioanode and biocathode to effectively work in their individually favorable conditions and validates the biofuel cell-based self-powered biogenerators for glucose sensing in a simple way. The assembled biogenerator exhibits a high stability and a good selectivity and can thus be developed to continuously monitor brain chemistry based on a galvanic cell mechanism, rather than electrolyte cell mechanisms that have already been used in in vivo electrochemistry. This study essentially offers a new approach to probing brain chemistry with simplicity both in theoretical aspects and experimental procedures.Acknowledgements
This work is financially supported by NSF of China (Grant no. 91213305, 20975104, 20935005, 21127901, and 21210007 for L. Mao, 91132708 for P. Yu, and 20905071 for Y. Lin.), National Basic Research Program of China (973 Program, 2010CB933502) and Chinese Academy of Sciences (KJCX2-YW-W25 and Y2010015).References
- J. A. Stamford and J. B. Justice, Anal. Chem., 1996, 68, 359–363 CrossRef.
- J. N. Stuart, A. B. Hummon and J. V. Sweedler, Anal. Chem., 2004, 76, 120–128 CrossRef.
- C. J. Watson, B. J. Venton and R. T. Kennedy, Anal. Chem., 2006, 78, 1391–1399 CrossRef.
- D. L. Robinson, A. Hermans, A. T. Seipel and R. M. Wightman, Chem. Rev., 2008, 108, 2554 CrossRef CAS.
- M. Zhang, P. Yu and L. Mao, Acc. Chem. Res., 2012, 45, 533–543 CrossRef CAS.
- O. Niwa, T. Horiuchi, R. Kurita and K. Torimitsu, Anal. Chem., 1998, 70, 1126–1132 CrossRef CAS.
- O. Niwa, R. Kurita, T. Horiuchi and K. Torimitsu, Anal. Chem., 1998, 70, 89–93 CrossRef CAS.
- S. S. Park, M. Hong, C. K. Song, G. J. Jhon, Y. Lee and M. Suh, Anal. Chem., 2010, 82, 7618–7624 CrossRef CAS.
- Y. S. Singh, L. E. Sawarynski, P. D. Dabiri, W. R. Choi and A. M. Andrews, Anal. Chem., 2011, 83, 6658–6666 CrossRef CAS.
- M. A. Rahman, A. Kothalam, E. S. Choe, M. S. Won and Y. B. Shim, Anal. Chem., 2012, 84, 6654–6660 CrossRef CAS.
- M. Zhang, K. Liu, K. Gong, L. Su, Y. Chen and L. Mao, Anal. Chem., 2005, 77, 6234–6242 CrossRef CAS.
- Z. Zhang, L. Zhao, Y. Lin, P. Yu and L. Mao, Anal. Chem., 2010, 82, 9885–9891 CrossRef CAS.
- Y. Jiang, H. Zhao, Y. Lin, N. Zhu, Y. Ma and L. Mao, Angew. Chem., Int. Ed., 2010, 49, 4800–4804 CrossRef CAS.
- B. Kong, A. Zhu, Y. Luo, Y. Tian, Y. Yu and G. Shi, Angew. Chem., Int. Ed., 2011, 50, 1837–1840 CrossRef CAS.
- G. Bazzu, G. G. M. Puggioni, S. Dedola, G. Calia, G. Rocchitta, R. Migheli, M. S. Desole, J. P. Lowry, R. D. O'Neill and P. A. Serra, Anal. Chem., 2009, 81, 2235–2241 CrossRef CAS.
- G. Rocchitta, O. Secchi, M. D. Alvau, R. Migheli, G. Calia, G. Bazzu, D. Farina, M. S. Desole, R. D. O'Neill and P. A. Serra, Anal. Chem., 2012, 84, 7072–7079 CrossRef CAS.
- Z. Wang and J. Song, Science, 2006, 312, 242–246 CrossRef CAS.
- B. J. Hansen, Y. Liu, R. Yang and Z. Wang, ACS Nano, 2010, 4, 3647–3652 CrossRef CAS.
- I. Willner, Science, 2002, 298, 2407 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS.
- A. Heller, Phys. Chem. Chem. Phys., 2004, 6, 209–216 RSC.
- V. Soukharev, N. Mano and A. Heller, J. Am. Chem. Soc., 2004, 126, 8368–8369 CrossRef CAS.
- E. Katz and I. Willner, J. Am. Chem. Soc., 2003, 125, 6803–6813 CrossRef CAS.
- Y. Yan, W. Zheng, L. Su and L. Mao, Adv. Mater., 2006, 18, 2639–2643 CrossRef CAS.
- F. Gao, Y. Yan, L. Su, L. Wang and L. Mao, Electrochem. Commun., 2007, 9, 989–996 CrossRef CAS.
- G. T. R. Palmore, H. Bertschy, S. H. Bergens and G. M. Whitesides, J. Electroanal. Chem., 1998, 443, 155–161 CrossRef CAS.
- L. Amir, T. K. Tam, M. Pita, M. M. Meijler, L. Alfonta and E. Katz, J. Am. Chem. Soc., 2008, 131, 826–832 CrossRef.
- P. Scodeller, R. Carballo, R. Szamocki, L. Levin, F. Forchiassin and E. J. Calvo, J. Am. Chem. Soc., 2010, 132, 11132–11140 CrossRef CAS.
- M. J. Moehlenbrock, T. K. Toby, A. Waheed and S. D. Minteer, J. Am. Chem. Soc., 2010, 132, 6288–6289 CrossRef CAS.
- C. M. Moore, S. D. Minteer and R. S. Martin, Lab Chip, 2005, 5, 218–225 RSC.
- M. Togo, A. Takamura, T. Asai, H. Kaji and M. Nishizawa, J. Power Sources, 2008, 178, 53–58 CrossRef CAS.
- F. S. Saleh, L. Mao and T. Ohsaka, Analyst, 2012, 137, 2233–2238 RSC.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS.
- P. Cinquin, C. Gondran, F. Giroud, S. Mazabrard, A. Pellissier, F. Boucher, J. P. Alcaraz, K. Gorgy, F. Lenouvel, S. Mathe, P. Porcu and S. Cosnier, PLoS One, 2010, 5, e10476 Search PubMed.
- T. Miyake, K. Haneda, N. Nagai, Y. Yatagawa, H. Onami, S. Yoshino, T. Abe and M. Nishizawa, Energy Environ. Sci., 2011, 4, 5008–5012 CAS.
- M. Rasmussen, R. E. Ritzmann, I. Lee, A. J. Pollack and D. Scherson, J. Am. Chem. Soc., 2012, 134, 1458–1460 CrossRef CAS.
- H. Cheng, Q. Qian, X. Wang, P. Yu and L. Mao, Electrochim. Acta, 2012, 82, 203–207 CrossRef CAS.
- E. Katz, A. F. Bückmann and I. Willner, J. Am. Chem. Soc., 2001, 123, 10752–10753 CrossRef CAS.
- L. Deng, C. Chen, M. Zhou, S. Guo, E. Wang and S. Dong, Anal. Chem., 2010, 82, 4283–4287 CrossRef CAS.
- D. Wen, L. Deng, S. Guo and S. Dong, Anal. Chem., 2011, 83, 3968–3972 CrossRef CAS.
- M. N. Germain, R. L. Arechederra and S. D. Minteer, J. Am. Chem. Soc., 2008, 130, 15272–15273 CrossRef CAS.
- M. T. Meredith and S. D. Minteer, Anal. Chem., 2011, 83, 5436–5441 CrossRef CAS.
- R. L. Arechederra, A. Waheed, W. S. Sly and S. D. Minteer, Analyst, 2011, 136, 3747–3752 RSC.
- F. Xu, J. Biol. Chem., 1997, 272, 924–928 CAS.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2002, 124, 12962–12963 CrossRef CAS.
- Q. Qian, L. Su, P. Yu, H. Cheng, Y. Lin, X. Jin and L. Mao, J. Phys. Chem. B, 2012, 116, 5185–5191 CrossRef CAS.
- J. P. Lowry, M. G. Boutelle and M. Fillenz, J. Neurosci. Methods, 1997, 71, 177–182 CrossRef CAS.
- L. Persson and L. Hillered, J. Neurosurg., 1992, 76, 72–80 CrossRef CAS.
- M. I. Sweeney, Neurosci. Biobehav. Rev., 1997, 21, 207–217 CrossRef CAS.
- T. Back, M. Hoehn, G. Mies, E. Busch, B. Schmitz, K. Kohno and K. A. Hossmann, Ann. Neurol., 2000, 47, 485–492 CrossRef CAS.
- S. Liu, H. Shi, W. Liu, T. Furuichi, G. S. Timmins and K. J. Liu, J. Cereb. Blood Flow Metab., 2004, 24, 343–349 CrossRef.
- W. Zheng, Q. Li, L. Su, Y. Yan, J. Zhang and L. Mao, Electroanalysis, 2006, 18, 587–594 CrossRef CAS.
- X. Fang, H. Chen, S. Yu, X. Jiang and J. Kong, Anal. Chem., 2011, 83, 690–695 CrossRef CAS.
- M. Zhang, K. Liu, L. Xiang, Y. Lin, L. Su and L. Mao, Anal. Chem., 2007, 79, 6559–6565 CrossRef CAS.
- Y. Lin, K. Liu, P. Yu, L. Xiang, X. Li and L. Mao, Anal. Chem., 2007, 79, 9577–9583 CrossRef CAS.
- K. Liu, Y. Lin, P. Yu and L. Mao, Brain Res., 2009, 1253, 161–168 CrossRef CAS.
- X. Li, H. Zhou, P. Yu, L. Su, T. Ohsaka and L. Mao, Electrochem. Commun., 2008, 10, 851–854 CrossRef CAS.
- X. Li, L. Zhang, L. Su, T. Ohsaka and L. Mao, Fuel Cells, 2009, 9, 85–91 CrossRef CAS.
- X. Wang, J. Wang, H. Cheng, P. Yu, J. Ye and L. Mao, Langmuir, 2011, 27, 11180–11186 CrossRef CAS.
- Y. Lin, N. Zhu, P. Yu, L. Su and L. Mao, Anal. Chem., 2009, 81, 2067–2074 CrossRef CAS.
- X. Zhuang, D. Wang, Y. Lin, L. Yang, P. Yu, W. Jiang and L. Mao, Anal. Chem., 2012, 84, 1900–1906 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |