Fast and continuous-flow separation of DNA-complexes and topological DNA variants in microfluidic chip format†
Martina Viefhues*, Jan Regtmeier and Dario Anselmetti
Experimental Biophysics and Applied Nanoscience, Faculty of Physics, Bielefeld University, 33615 Bielefeld, Germany. E-mail: viefhues@physik.uni-bielefeld.de
First published on 24th October 2012
Abstract
The efficient detection, separation and purification of topological and (protein-)complexed DNA variants is mandatory for many state-of-the-art molecular medicine technologies, like medical diagnostics, gene- and cancer-therapy as well as plasmid vaccination. Here, we present the proof-of-concept of a novel micro-nanofluidic device for a fast and efficient, continuous-flow, and virtually label-free detection/purification protocol that goes beyond the standard methods of electrophoretic mobility shift assays, capillary electrophoresis and affinity chromatography. Based on dielectrophoretic trapping, analyte mixtures of small linear DNA-fragments (2.868 kbp and 6.0 kbp), topological DNA variants like plasmids (6.766 kbp) and minicircle-DNA (2.257 kbp), or cytostatic- and protein–DNA complexes were separated in the vicinity of a channel-spanning bowed ridge (creating a nanoslit). One analyte is continuously deflected due to dielectrophoretic trapping at the ridge whereas other species pass the nanoslit unhindered, resulting in two molecule specific pathways with baseline separated resolution. This offers one-step real-time separation of low analyte volumes on a one-minute timescale at low-costs. The underlying dielectrophoretic mechanism was quantified by determining the electrical polarizabilities of the molecules. Additionally, we compared the continuous-flow detection of DNA-complexes with well-established electrophoretic mobility shift assays. Future analytical and preparative applications, such as for plasmid pharmaceuticals as well as continuous sample harvesting in parallel microchip format, are discussed.
1. Introduction
The standard method for the separation of DNA compounds is gel electrophoresis, either in a slab gel or capillary format, or the different types of affinity chromatography. Although is widespread and used in many laboratories some intrinsic systematic disadvantages are obvious and remain unsolved: (1) preparation and separation takes at least 2 h;1 (2) the parameters of the separation cannot be adapted or optimized during the separation; (3) the considerable shear force effect during separation may affect analyte integrity and functionality (topology) and (4) sample elution and postprocessing is difficult and often relies on the use of toxic and mutagenic reagents. Especially the last two issues are increasingly important when considering future applications in modern medicine such as gene-therapy, plasmid vaccination, and cancer therapy where pharmaceutical grade DNA compounds (vectors) are compulsory for the medical needs as well as would reinforce the safety and regulatory concerns. Nevertheless, capillary electrophoresis and electrophoretic mobility shift assays (EMSAs) are standard analytical techniques for purification of small and large DNA fragments in plasmid vaccine production, for determination of binding affinities of new cytostatic drugs and rational drug design,2 and for protein–DNA interaction screenings3–5 like screening for specific antibody–DNA complexes, e.g. anti-double-stranded DNA antibodies, specific for the autoimmune disease systemic lupus erythematosus.6 Additionally, there is huge requirement of novel tools that allow fast processing of low analyte volumes in low cost devices suitable for real time process control. Microfluidics offers access to these requirements.A number of microfluidic devices have been demonstrated for the separation of long DNA fragments.7–15 Most of them operate in a batch processing mode (one sample volume after another)8–12 while few others operate continuously.13–15 In the latter case, the sample is continuously injected into the separation channel and subjected to a force at an angle to the direction of flow. This approach offers several advantages: continuous feeding of sample; continuous readout of separation efficiency; real-time adaption of separation parameters; the separated components can be collected at different outlets; potential for further integration and immediate postprocessing of separated species,16e.g. separation in a second dimension such as pH dependence. Despite all the remarkable efforts to separate DNA molecules, novel micro- and nanofluidic tools for detection, characterization and separation of protein– or drug–DNA complexes are missing. There are – commercially available – implementations of EMSAs in microfluidic chips,17 but as it is a miniaturization of the established method, both techniques, classical EMSA and the microfluidic variant, share most disadvantages. Moreover, there are flow cells for single-molecule studies of DNA–protein interactions offering no sample separation or preparation.18 So the natural next step is the development of a method that incorporates the advantages of miniaturization, especially the continuous sample processing.
Our method is based on dielectrophoresis at a bowed ridge generating a nanoslit over the full channel width (Fig. 1b and c). Dielectrophoresis (DEP) refers to the movement of a polarizable object in an inhomogeneous electric field.19 The separation criterion is the polarizability of the molecules.12,20 Due to the polarizability specific trajectories occur at the ridge, resulting in laterally separated analytes (Fig. 1). Most bioparticles (cells, DNA, and proteins) are addressable with DEP.16,21 For DNA molecules, it is assumed that the polarizability is dominated by the distortion of the surrounding ion cloud by an external electric field.21,22 The characteristic length of the ion cloud is given by the Debye length and depends on the electric potential of the molecule surface.23 In consequence, for DNA-complex formation with other (charged) molecules, e.g. proteins, a change of polarizability is assumed. This allows time-lapsed analysis of binding interactions. For example, DNA and a protein can be mixed up directly in the reservoir, and the complex-formation can be detected in real time, when the polarizability, and accordingly the trajectory, changes from the pure DNA to the protein–DNA complex. Further applications are determination of binding affinities in rational drug design or screening methods for diagnostics, e.g. mixing plasma of patients and DNA to detect anti-double-stranded DNA antibodies (specific for systemic lupus erythematosus6).
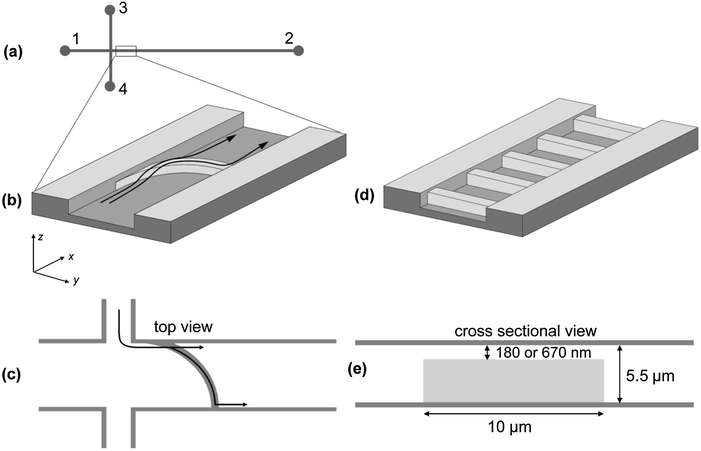 | ||
| Fig. 1 (a) Schematic layout of the microfluidic device (not to scale). All channels are 200 μm wide and 5.5 μm high, except for minicircle purification with width of 100 μm; channels 1, 3 and 4 are 3 mm long, channel 2 is 5 mm long. (b) Central element of the microfluidic separation device: a bowed ridge is placed in channel 2. The ridge reduces the flow through height to 180 or 670 nm. The black arrows demonstrate the working principle of the separation device. A mixture of two sorts of molecules is continuously injected as a narrow band, close to the left wall. At the ridge, one species passes the nanoslit unhindered, while the other one is deflected. The deflected species is transported along the ridge and continuously migrates further down the channel once it reached the opposite channel wall. (c) Top view of the bowed ridge. The two possible trajectories for separated species are indicated. (d) Scheme of the microstructure used for determining polarizabilities: instead of the bowed ridge, periodically arranged ridges with a period of 30 μm and a total number of 100 are fabricated in channel 2. (e) Cross-sectional view of a single ridge with a flow through height of 180 or 670 nm. | ||
Using dielectrophoresis, Hawkins et al.24 and Barrett et al.25 demonstrated the feasibility of continuously separating micro-meter sized objects at a ridge. Their systems incorporate slits of 100 μm and 5 μm in height, respectively excluding the manipulation of objects smaller than 1 μm, i.e. biomolecules.
Here, we present a device with the smallest dimension of 180 nm, the method used for fabrication was kept simple anyway. A monolithic fabrication process of the microfluidic device via soft lithography26 allows an easy fabrication in most biology laboratories, as no clean room is required, once a masterwafer, the original negative relief of the actual fluidic network, was fabricated. A clean bench is sufficient for chip assembly.
Apart from interaction studies, our device allows fast/real-time purification/production control of minicircle-DNA. Minicircle-DNA is defined to contain almost exclusively the “Gene of interest” and its regulating sequence motifs without bacterial backbone sequences.27 Due to the production process the purification of minicircle-DNA is indispensable to remove the remaining parental DNA. Our device allows fast control of the purity with times of separation smaller than 1 min. Moreover, our microfluidic methodology does not rely on classical filtration, so the shear-force associated analyte deterioration could be reduced to a minimum. Together with the fact that our approach is inherently independent of toxic agents, future applications in molecular biotechnology and medicine can be implemented in a more compliant way with respect to the concerns of safety regulation in drug administration. So it becomes potentially interesting for applications in gene-therapy or plasmid vaccination.
In summary, the most prominent advantages of our technique are continuous-flow processing; fast separation time less than 1 min; accessibility to linear as well as circular DNA molecules; separation of small DNA-fragments and detection of DNA-complexes; baseline separated resolution; on-line adaptability of selectivity parameters via applied voltages. Thus it combines the advantages of three continuously operating microfluidic devices13–15 in a single device and additionally allows detection of DNA-complexes.
The paper is organized as follows: firstly theory and methods are explained, followed by the introduction of the device and a proof-of-concept for the separation of short DNA molecules. Then, the continuous flow detection of two different DNA-complexes and their separation from unbound DNA is demonstrated. Afterwards, the separation of minicircle-DNA and parental DNA is described. Additionally, the polarizabilities are quantified according to the DNA-complex separation. Finally, the separation results obtained in the micro-nanofluidic device are compared to well-established electrophoretic mobility shift assays and a brief discussion is given, focusing on practical aspects and future developments of the method.
2. Theory
Dielectrophoresis is the migration of a polarizable object in an inhomogeneous electric field. The necessary inhomogeneous electric field, denoted by![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) , is created by applying a voltage U(t) = UDC + UACsin(ωt) to a microfluidic channel in which an insulating ridge is placed (Fig. 1). The potential dielectrophoretic energy of a polarizable object can be written as16
, is created by applying a voltage U(t) = UDC + UACsin(ωt) to a microfluidic channel in which an insulating ridge is placed (Fig. 1). The potential dielectrophoretic energy of a polarizable object can be written as16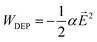 | (1) |
This phenomenological description assigns an effective polarizability α to the object, which quantifies the microscopic polarization effects by an effective induced dipole moment ![[p with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0070_20d1.gif) = α. In this model, α is assumed to be real-valued, i.e. polarization effects are isotropic and dissipative effects are neglected. The polarizability depends on the frequency of the electric field α = α(ω) so that only polarizabilities, determined for the same frequency, may be compared (see also the Results section). One can think of the DNA molecules to be effectively residing in a (quasi-static) dielectrophoretic energy landscape, whose “strength” is controlled by UAC, and which can be (independently) “tilted” with electrophoretic forces by applying UDC (for details refer to ref. 20).
= α. In this model, α is assumed to be real-valued, i.e. polarization effects are isotropic and dissipative effects are neglected. The polarizability depends on the frequency of the electric field α = α(ω) so that only polarizabilities, determined for the same frequency, may be compared (see also the Results section). One can think of the DNA molecules to be effectively residing in a (quasi-static) dielectrophoretic energy landscape, whose “strength” is controlled by UAC, and which can be (independently) “tilted” with electrophoretic forces by applying UDC (for details refer to ref. 20).
In Fig. 2a, the dielectrophoretic potential energy is shown in a cross-sectional view across the ridge. The DEP energy on the ridge is generally larger than the DEP energy in the rest of the channel. Especially at the edges of the ridge, the DEP energy is significantly increased. For positive α those regions act as dielectrophoretic traps, which is demonstrated in Fig. 2a. For a successful separation, i.e. deflection of one sort of molecules, it is necessary that the molecules, once trapped, can migrate along the ridge to the other channel wall. The electrophoretic field on top of the ridge was calculated using COMSOL 3.5 (Fig. 2b and c). It can be observed that the field lines do not cross the nanoslit strictly perpendicular but under an angle of maximal 15°. This means, there is a (small) tangential electrophoretic force component that drives the DNA, which is on top of the ridge, towards the other channel wall. The time-average velocity of the molecules in the microchannel and along the ridge could be described by electrophoretic, electroosmotic and dielectrophoretic forces, induced by DC and AC electric fields, DC and AC, and reads28
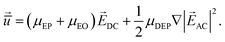 | (2) |
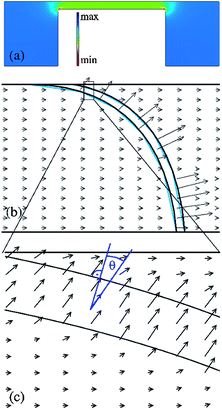 | ||
| Fig. 2 Numerical calculations of the dielectrophoretic potential and the electric field for a 3D model of the ridge (Comsol 3.5). (a) Cross-sectional view of the dielectrophoretic trapping potential near and on top of the ridge with a potential minimum (trap) on the ridge (for α > 0). (b) Electric field at the nanoslit, the arrow length is proportional to the electric field strength. (c) Detailed plot of the field lines on top of the ridge. The arrows cross the nanoslit with an angle of Θ ≈ 15° to the perpendicular. | ||
With μEP + μEO = ε(ζm − ζf)/ν being the sum of electrophoretic (EP) and electroosmotic (EO) mobilities μ, ε the permittivity, ζm the zeta potential of the molecule, ζf the zeta potential of the fluid, and ν the viscosity of the fluid. Escapes from the traps are mainly driven by Brownian motion, resulting in a distribution of escape times. This process can be best described by an inverse Kramers rate29
| τ ∝ exp(ΔWDEP/kT) | (3) |
| ∝ |UAC|), eqn (3) can be rewritten as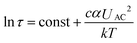 | (4) |
By modeling the electrical properties of the fluidic chip using an equivalent circuit diagram, we find c = 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 250. This constant represents the fraction of the applied voltage that drops over the nanoslit. The linear relationship between ln τ and UAC2 found in eqn (4) was used to determine α by a fit to the experimental data (see ESI Fig. 1–3†). Thus, experimentally, τ has to be determined from microscopy images showing how the DNA molecules migrate through the array of ridges for different values of UAC2 for the given UAC and ω (for details refer to ref. 12 and 20).
250. This constant represents the fraction of the applied voltage that drops over the nanoslit. The linear relationship between ln τ and UAC2 found in eqn (4) was used to determine α by a fit to the experimental data (see ESI Fig. 1–3†). Thus, experimentally, τ has to be determined from microscopy images showing how the DNA molecules migrate through the array of ridges for different values of UAC2 for the given UAC and ω (for details refer to ref. 12 and 20).
3. Methods
3.1. Masterwafer
Silicon wafers (CrysTec, Germany) were cleaned in caroic acid (Merck, Germany) for 5 min twice and rinsed with water. The first layer of SU8 photo resist (Microresist, USA, 12% or 17% solid fraction) was spin-coated (Delta 10, Ble-Laboratory Equipment GmbH, Germany) at 1000 rpm (12% solid fraction) or 900 rpm (17% solid fraction) for 30 s, respectively. Then the layer was exposed to UV light through a chromium mask (Delta Mask, Netherland) in a mask aligner MJB3 (Süss MicroTec, Germany) for 2 s. The height of the layer was determined, 180 nm (12% solid fraction) or 670 nm (17% solid fraction), using a DekTak-Profilometer 3030 ST (Stanford Nanofabrication Facility Equipment, USA). The second layer of SU8 (52% solid fraction) was spin-coated (3000 rpm for 30 s). The wafer was again exposed to UV light for 7 s, carefully aligned to the first layer in the mask aligner. Baking and development were performed according to supplier's information. Finally, the wafer was silanized with tridecafluor-1,1,2,2-tetra-hydrooctyl-trichlorsilane (Merck, Germany) in an exsiccator.3.2. Chip fabrication
Two layers of PDMS were used for replica molding. First, a layer of h-PDMS (3.4 g vinylmethylsiloxane-dimethylsiloxane trimethylsiloxy terminated copolymer (ABCR GmbH & Co KG, Germany), 18 μl platinum–divinyltetramethylsiloxane complex (ABCR GmbH & Co KG, Germany), 1 droplet of 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcycltetrasiloxane (Sigma Aldrich, Germany) and 1 ml (25–35% methylhydrosiloxane)-dimethylsiloxane copolymer (ABCR GmbH & Co KG, Germany)) was spincoated onto the wafer at 1500 rpm for 10 s followed by 3 min on a hot plate HAT-303D (AVT-Technonolgie, Germany) at 65 °C.30 This layer of h-PDMS had a thickness of about 40 μm and thus completely contained the microstructured channel formed by the masterwafer. For the second layer 6.7 g polydimethylsiloxane (PDMS) Sylgard 184 and 0.67 g silicone elastomer curing agent (PDMS-linker, both Dow Corning GmbH, Germany) were poured on the top and cured on a hot plate at 65 °C for 40 min.The PDMS double-layer was peeled off and the microstructured part of the slab was cut out. Reservoirs were punched into it at the channel ends. The channel was closed with PDMS spincoated cover slips. Before chip assembly the coated cover slips and the PDMS slabs were cleaned in acetone, ethanol, and deionized water in each case for 10 s in an ultrasonic bath (Transsonic460, Digitana AG, Germany) and oxidized in an oxygen plasma (home built plasma chamber31) for 30 s. Afterwards, the channels were filled with working buffer, 1 mM phosphate buffer (pH 7.4, 0.2 mM NaCl) (Fluka, Germany) containing 1 mM ethylenediaminetetraacetic acid (EDTA) (Fluka, Germany), 2 mM 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox, Fluka, Germany), 0.25% N-dodecyl-β-D-maltoside (DDM) (Sigma Aldrich, Germany) and 0.03% methyl cellulose (MC) (Sigma Aldrich, Germany), and incubated for at least 30 min before starting the experiments to reduce unspecific adsorption of DNA molecules onto the surfaces.32 A poly(methyl methacrylate) (PMMA) block with integrated reservoirs and electrodes was placed on top of the chip to enlarge the reservoirs and simplify handling.
3.3. Sample preparation
Linear 6.0 kbp DNA (derivative of pUC 19 DH 5α) was purchased from MBBL GmbH, Germany. Circular pUC 18 DNA (2.686 kbp) was extracted from E. coli DH 5 cells using the QIAprep Miniprep Kit (Qiagen, USA). The 2.686 kbp DNA was digested with NdeI and purified using the QIAquick PCR Purification Kit (Qiagen, Germany). For the experiments the DNA was stained with YOYO-1 (Molecular Probes, USA), the YOYO-1 to DNA base pair ratio was 1:10. For the DNA cytostatics complex YOYO-1 and actinomycin D (Mw 384 kDa, ratios of 1:5, 1:10 or 1:20 ACTD per DNA base pair) were added and incubated on a Vortexer (Vortexer-Genie 2, Scientific Industries, USA, 0-level) for 2 h. For the DNA polymerase complex, YOYO-1 and E. coli RNA polymerase core enzyme (Epicentre Biotechnologies, USA, MW 389 kDa) were added (0.1 μg DNA and 0.5 μg polymerase) and incubated on a Vortexer (0-level) for 2 h. The storage buffer of the E. coli RNA polymerase core enzyme contained 50% glycerol. The DNA stock solution was diluted to 10 pM with 1 mM phosphate buffer, containing 1 mM EDTA, 2 mM Trolox, 0.25% DDM and 0.03% MC.323.4. Setup
The measurements were performed on an inverted microscope (Axiovert 200, Zeiss, Germany) with a motorized x/y-stage (99S008, Ludl Electronic Products, USA). A 100× oil-immersion objective (Plan Neofluar, NA 1.3, Zeiss, Germany) was used together with a mercury arc lamp (HBO 50) and a fluorescence filter set (BP 450-490, FT 510, BP 515-565, Zeiss, Germany). A grey filter (25% transmittance) reduced the incident light. A CCD interline-transfer camera (Imager3LS, LaVision, Germany) with the corresponding grabber card and software (DaVis 6.2) was used for data acquisition with an 8 by 8 binning and 10 frames per second (fps). The voltage signal was created via a LabView 6i program and function generator DS345 (Stanford Systems, USA). The output signal was amplified by a high voltage amplifier (AMT-1B60-L Matsusada, Japan). Three power supplies (HCL 14-12500, FUG, Germany) generated additional DC-voltages.3.5. Chip experiments
The DNA solution was pipetted into reservoir 3 (Fig. 1). The specific voltages used in the separation experiments were chosen as follows. First, the DC-voltages at reservoirs 1 to 4 were chosen such that a continuous molecule flow, parallel to the channel wall, from channel 3 into channel 2 was achieved with DC-voltages between −1 V and −4 V (Fig. 1). It was assured that the inflowing molecules occupy less than a quarter of the total width of channel 2. Then the AC-voltage was switched on to reasonably small amplitudes (50 V) and its effect on the particle motion was observed while varying the frequency over the interval from 50 to 1000 Hz. If no deflection was observed the amplitude was increased, followed by varying the frequencies until a complete deflection of the DNA at the ridge was observed for the first time. The frequency was thus optimized to the smallest voltages necessary.The nanoslit was inspected after separation experiments due to unspecific adsorption of DNA molecules onto the nanoslit surface. The inspection reveals that a negligible amount of DNA adsorb onto the surface of the nanoslit.
For quantifying the polarizabilities, pinched injection protocol was used,33i.e. DC-voltages at reservoir 1, 2 and 3 were set such that the DNA molecules migrate from reservoir 3 to reservoir 4 without leaking into channels 1 and 2. Then an AC-voltage was superimposed by a DC-voltage, both applied at reservoir 1, to drive a defined volume of DNA solution into channel 2 and to create selective dielectrophoretic traps at the ridges simultaneously. Fluorescence image sequences of molecules passing several ridges were taken for different UAC.
3.6. Image acquisition and processing
The region of interest (ROI) captured 86 μm in the x-direction and 68.6 μm in the y-direction, i.e. only about one third of the total channel width. In order to collect intensity data of the fluorescent molecules over the whole channel width, the channel was scanned during the running separation experiments with a constant speed of 10 μm s−1 along the y-direction at different x-positions of interest (Fig. 3a and c). The y-positions of the recorded images within the channel were determined from scanning speed and time. Raw image data were processed in DaVis 6.2 with a nonlinear filter. To obtain the intensity distributions of molecules within the channel at specific x-positions upstream and downstream of the ridge, as shown in Fig. 3, we proceeded as follows: in the series of images recorded during the scan in the y-direction at a fixed x-position, each image was partitioned into six slices of 11.4 μm height (in y-direction), in order to reduce convolution effects that would occur due to the y-extension of the ROI. Then, the total fluorescence intensity within these slices was determined. Successively recorded images are shifted by 1 μm in the y-direction (due to the frame rate of 10 fps and the scanning speed of 10 μm s−1), so that in every eleventh image those slices overlap within a precision of less than 1 μm. The measured fluorescence intensity was averaged over (up to six) such overlapping slices and plotted as a function of the y-position. These data were fitted with Gaussian curves and the resolution was calculated according to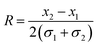 with x2, x1 being the position of peak maximum and σ1, σ2 the corresponding peak width.34 Therefore, values of R larger than 1 indicate the baseline separated resolution.
with x2, x1 being the position of peak maximum and σ1, σ2 the corresponding peak width.34 Therefore, values of R larger than 1 indicate the baseline separated resolution.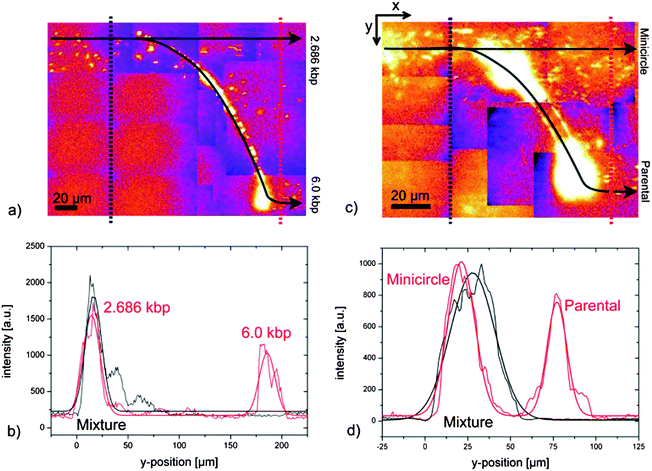 | ||
| Fig. 3 Separation results. Top: fluorescence image (collage) at the nanoslit. From the left, a mixture of DNA is continuously injected (yellow spots correspond to single DNA molecules). The black arrows are guides to the eye demonstrating the two possible trajectories. The black and red dotted lines show along which paths the fluorescence intensities were determined. Bottom: fluorescence intensity plots of separation. The black and red curves represent fluorescence intensities measured at the black and red dotted lines, respectively. The curves were fitted with Gaussian functions. (a) 2.686 kbp DNA pass the nanoslit unhindered, whereas 6.0 kbp DNA are trapped on the ridge and migrate towards the opposite channel wall before they escape due to Brownian motion (channel width 200 μm; height of nanoslit 670 nm). (b) Separation of 2.686 kbp and 6.0 kbp DNA molecules (resolution 2.66; UAC = 650 V, and ω = 350 Hz). (c) Minicircle-DNA (2.247 kbp) pass the nanoslit unhindered, whereas parental DNA (6.766 kbp) migrate on the ridge towards the opposite channel wall (channel width 100 μm and height of nanoslit 180 nm). (d) Separation of minicircle- and parental DNA (resolution 1.24; UAC = 200 V, and ω = 350 Hz). | ||
3.7. EMSA
The sample preparation was the same as for the chip experiments, excluding the sample dilution. The gel electrophoresis was performed in a 1.2% agarose gel with a 1 fold TAE buffer (40 mM Tris (99%, Roth, Germany), 10 mM NaCl (Riedel-de-Haen, Germany) and 1 mM EDTA). The samples (0.1 μg DNA) were pipetted with the same volume of loading buffer (TE/G buffer, MBBL GmbH, Germany) into the wells and 333 V m−1 (Power PAC 3000, BIO RAD, USA) were applied for 3 h. The gel, thereafter, was placed into an ethidium bromide (Merck, Germany) bath for 5 min and then in a water bath for 10 min. Finally the gel was photographed under UV light (3UV-38 3UV LAMP, UVP, Cambridge, UK).4. Results
4.1. Design of the device
When designing the device some preliminary conditions had to be considered. The device has to allow (a) controlled continuous injection, (b) continuous processing, and (c) be capable to process objects in the submicrometer range, such as short DNA fragments or macromolecules.Our microfluidic device consists of a cross-injector with one structured channel (Fig. 1a) to allow controlled continuous flow injection of the analyte. The device works in continuous flow mode, so a selective force had to be applied perpendicular to the direction of flow. Here, a bowed insulating ridge (Fig. 1b and c), reducing the flow through height, is the central element of the device for continuous detection of DNA interactions and separation. The range of the addressable analyte(-size) is selectable due to the height of the nanoslit and was optimized for the separation of DNA-complexes.
Sample molecules are led towards the ridge as a narrow band (Fig. 1b and c), by applying appropriate DC-voltages (see also the Chip experiments section). At the ridge, two possible pathways exist: either a molecule passes over the ridge unhindered (first pathway), or it is deflected along the ridge and travels further down the channel once it reached – laterally separated from the first pathway – the opposite wall of the channel. For the second pathway the interplay of electrophoretic and dielectrophoretic forces is most important. Therefore, two voltages were applied, a static voltage UDC was responsible for electrophoretic transport in the channel network and an alternating voltage UAC of frequency ω was used for dielectrophoretic trapping of molecules on the bowed ridge (cf.Eqn (2)). Since UAC induces (on average) no net transport, superposition of DC- and AC-voltage allows for the independent control of transport of molecules (UDC) and trapping (UAC), respectively (see the Methods section for details).
The central selection criterion for trapping is the polarizability α. The potential energy of an object of polarizability α in an external electric field is given by eqn (1). DNA molecules are attracted towards the regions of the strongest electric field (for positive α which is confirmed in many studies22,35). The strongest field is generated in the nanoslit resulting in a region of the deepest potential well on the ridge (Fig. 2a). DNA molecules are trapped when the dielectrophoretic potential energy is higher than the potential energy of the electrophoretic motion and the thermal energy.
For sufficient continuous-flow separation the selectively trapped DNA had to be transported perpendicular to the direction of flow, i.e. from the left to the right channel side wall, Fig. 1b. As already mentioned in the Theory section the electric field lines do not cross the nanoslit strictly perpendicular. At the left end, where the ridge starts almost parallel to the main channel axis, an angle of Θ = 15° to the perpendicular is found resulting in a small tangential electrophoretic force component (Fig. 2c). This angle converges towards zero towards the right end of the ridge, where it hits the channel wall perpendicularly. The escape process from the ridge is driven by Brownian motion and steric effects of the DNA accumulating at the end of the ridge. Therefore, it is a statistical process resulting in a distribution of escape times τ. This has important practical implications for separation: UDC and UAC have to be chosen such that one analyte species can pass the ridge unhindered while the other is deflected along the ridge but can escape within a not too long time span by Brownian motion, i.e. the dielectrophoretic energy must not be much larger than the thermal energy. The frequency and amplitude of AC-voltage are the main parameters to control selectivity (see the Chip experiments section). To estimate the appropriate AC-voltage the electric field at the nanoslit could be calculated via an equivalent circuit model.20
The device was realized via poly(dimethylsiloxane) (PDMS) soft lithography: a photolithographically structured masterwafer was used as a mold, over which the liquid PDMS was cast and finally cured resulting in a flexible elastomer.26 The masterwafer was fabricated in a two-layer contact lithography process with final heights of the nanoslit of 670 nm or 180 nm. In order to achieve a stable mold, two layers of PDMS were necessary: a 40 μm layer of hard PDMS (h-PDMS)30 and a 1 mm layer of Sylgard 184 PDMS.36,37 For the single use of PDMS, the nanoslit collapses. Whereas for using h-PDMS solely, peel-off of the masterwafer is impossible, when the h-PDMS is too brittle. The masterwafer was silanized (see Section 3.1), thus the PDMS did not adhere to the SU-8 structures and a high reliability and reproducibility could be achieved. Potential bending of the ridge was checked with fluorescent particles of appropriate size, which easily passed the nanoslit over the whole channel width for 95% of the devices. Thus, we entered the regime of nanofluidics, which allows the generation of very high electric field inhomogeneities and the manipulation of small DNA fragments.
4.2. Separation of 2.686 kbp and 6.0 kbp DNA
As a proof-of-concept, a mixture of linear DNA molecules (2.686 kbp and 6.0 kbp DNA) was injected (Fig. 3a). Two distinct pathways of DNA molecules were identified downstream of the ridge for an AC-voltage of 650 V at 350 Hz: the 2.686 kbp molecules passed the nanoslit unhindered, whereas the 6.0 kbp fragments were deflected along the ridge. Identification was achieved by testing the two species separately under identical conditions (see ESI Fig. 4†). For a quantitative analysis, the fluorescence signals over the full channel width were determined up- and downstream of the ridge (Fig. 3b). Upstream of the ridge, only one narrow peak was observed (the narrow band used for continuous DNA injection); downstream of the ridge, two baseline separated peaks were found, relating to the two separated DNA species. Because of the continuous operation, no actual separation time exists. The separation starts as soon as the first injected molecules reach the ridge (about 15 s). For migration along the ridge a molecule needed another 30 s. So, the first results were obtained within less than 1 min. Despite the fast response time the resolution of 2.66 is very competitive compared to other continuously operating gel free techniques.13–154.3. Detection and separation of DNA-complexes
In order to address molecular binding analysis, a mixture of 6.0 kbp DNA fragments and 6.0 kbp DNA actinomycin D (ACTD) complexes (an intercalating cancer drug, ratio 1:5 ACTD per DNA base pair) were continuously separated. The DNA-complexes were deflected (Fig. 4) at the ridge for an AC-voltage of 600 V at 650 Hz, i.e. exhibit larger polarizability. Again one distinct peak was detected upstream of the ridge depicting the mixture of the pure DNA and the DNA–ACTD-complex whereas two peaks were observed downstream of the ridge with a resolution of 2.0. The two peaks were identified by testing the two species separately under identical conditions (see ESI Fig. 4†). The peak downstream of the ridge is sharper than upstream because of self-focusing. Directly behind the ridge at the left end the electric field has a component pointing towards the channel wall (Fig. 2b and c).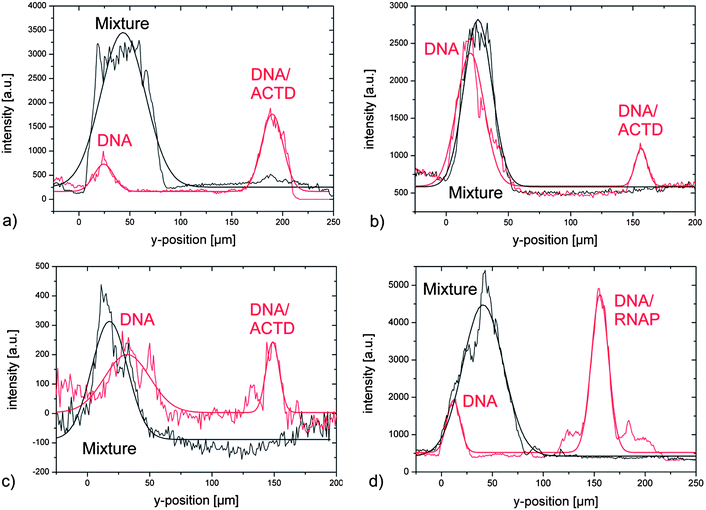 | ||
| Fig. 4 Detection of complex formation. Fluorescence intensity plots of separation of DNA and DNA-complexes up- and downstream of the ridge (cf.Fig. 3a). (a) DNA–ACTD-complex with 1 actinomycin D per 5 DNA base pairs (ω = 550 Hz at UAC = 600 V and resolution R = 2.0). (b) DNA–ACTD-complex with 1 actinomycin D per 10 DNA base pairs (ω = 550 Hz at UAC = 650 V and resolution R = 2.14). (c) DNA–ACTD-complex with 1 actinomycin D per 20 DNA base pairs (ω = 500 Hz at UAC = 600 V and resolution R = 1.36). (d) Separation of 6.0 kbp DNA from 6.0 kbp DNA–RNAP-complex (resolution 2.4; about 15 polymerase per DNA, UAC = 650 V, and ω = 300 Hz). | ||
In additional experiments mixtures of 6.0 kbp DNA and DNA–ACTD-complexes with reduced ACTD concentration were tested to check the sensitivity of the system. For the reduction to half amount of ACTD (ratio of 1:10 ACTD per DNA base pair, UAC = 650 V and ω = 550 Hz) a clear separation with a resolution of 2.14 was achieved (Fig. 4b). By adapting the parameters, UAC = 600 V and ω = 500 Hz, two distinct peaks could be detected (Fig. 4c) even for further reduction of ACTD to a ratio of 1:20 ACTD per DNA base pair, representing fourth of the starting concentration. A baseline separated resolution of 1.36 demonstrates the very high sensitivity to molecular recognition.
As a second example DNA-complexes with Escherichia coli (E. coli) RNA polymerase core enzyme (RNAP) were investigated. The mixture of 6.0 kbp DNA and DNA–RNAP-complexes was continuously injected and separated at the ridge for an AC-voltage of 300 Hz at 650 V (Fig. 4d), with a resolution of 2.4. Noticeable shoulders to the left and right of the DNA-complex peak were observed. The reasons were further analyzed by means of atomic force microscopy. A heterogeneous distribution of the proteins was found giving a hint for the observed shoulders (see ESI Fig. 5†).
4.4. Enhanced device: separation of minicircle-DNA and parental DNA
The device proved to be efficient for the separation of small linear DNA-fragments and detection of protein–DNA and drug–DNA complexes. The device might be enhanced to separate/detect proteins. According to the small size of proteins the necessary electric fields are even stronger. Therefore, the nanoslit height was reduced to 180 nm over a channel width of 100 μm.In this device a continuous-flow separation of circular DNA, minicircle-DNA (2.247 kbp) and its parental DNA (6.766 kbp), was performed to demonstrate the general applicability of the miniaturized system. The larger parental DNA was fully deflected at the ridge for an AC-voltage of 200 V at 350 Hz, whereas the smaller minicircle-DNA passed the nanoslit unhindered (Fig. 3c and d). So the necessary AC-voltages for DNA separation could be reduced by a factor of about 3, significantly reducing joule heating. For downstream applications a baseline separated resolution is indispensable. Here, we demonstrate, even for a reduced channel width of 100 μm, a baseline separated resolution of 1.24.
The miniaturization offers two advantages, the necessary voltages could be reduced, and the range of manipulable molecules is extended to smaller DNA molecules. Thus for applying 600 V AC-voltages, DNA molecules with polarizability of 4.4 × 10−31 Fm2 could be manipulated. This corresponds to very small DNA fragments of about 3 bp to 80 bp (see ESI†).
4.5. Polarizability of DNA and DNA-complexes
A critical point for dielectrophoretic manipulation is the difficulty of obtaining quantitative values for the polarizability of the objects of interest. For the continuous separation itself, an a priori knowledge is not necessary as the separation parameters can be adapted in real-time allowing an on-line feedback loop. But the polarizability is an interesting metric for the electrical properties of the bulk material of the sample molecules and their ion clouds20 that allows us to verify and understand the mechanism in more detail. Therefore, we also determined the polarizabilities in an array of linear ridges perpendicular to long channel axis (Fig. 1d). The molecules were injected via a pinched injection protocol33 over the full width of channel 2 (see the Methods section). Fluorescence image sequences of molecules crossing several ridges were acquired for different applied UAC. The distribution of escape times was extracted and the polarizabilities were determined automatically (for details refer to ref. 20). The results are summarized in Table 1.| Probe | ω [Hz] | α [10−30 Fm2] |
|---|---|---|
| a 0.022% glycerine was added to the solution, in order to obtain the same viscosity as for the 6.0 kbp DNA–RNAP-complex. | ||
| 2.686 kbp DNA | 350 | 2.55 ± 0.38 |
| 6.0 kbp DNA | 350 | 4.30 ± 0.37 |
| 6.0 kbp DNA | 550 | 2.68 ± 0.28 |
| 6.0 kbp DNA–ACTD-complex | 550 | 3.48 ± 0.10 |
| 6.0 kbp DNAa | 300 | 1.41 ± 0.46 |
| 6.0 kbp DNA–RNAP-complex | 300 | 1.95 ± 0.39 |
The obtained data agree very well with the separation results: objects with a larger polarizability were deflected, whereas objects with smaller polarizability passed the ridge unhindered. It is important to notice that the polarizability of the objects depends on the frequency of the applied UAC and on the buffer, as was observed when adding glycerine, thus viscosity changed. These results are in agreement with previously published findings.22 Additionally, the polarizability depends strongly on details of the technique and buffer conditions. The values obtained agree with previously published data: 5.5 × 10−31 Fm2 for 4.4 kbp DNA fragments using transient electric birefringence (TEB) in Tris buffer (0.2 mM, pH 8);38 2.3 × 10−30 Fm2 for 5 kbp DNA fragments using TEB in sodium phosphate (1 mM Na, pH 7.2);39 3.2 × 10−28 Fm2 for 8 kbp DNA fragments using conductivity dispersion in 1 mM NaCl;40 dielectrophoretic trapping in a microelectrode array 6 × 10−31 Fm2 for 8 kbp DNA fragments in 3 mM Hepes, 2 mM NaOH, pH 6.9.35 For DNA–protein complexes, to the best of our knowledge, no values have been published yet.
4.6. Comparison to electrophoretic mobility shift assay (EMSA)
When a novel technique is introduced, a comparison to the well established method is required. EMSAs have been widely used to determine whether a molecule binds to DNA fragments. Typically, two lanes in an agarose gel are used, one with the DNA only, and in the other lane the mixture of DNA and the anticipated binding partner. If a shift is observed, i.e. one analyte migrates more slowly, it is assumed that the binding partner is bound to the DNA molecules.1First, we separated 2.686 kbp DNA from 6.0 kbp DNA fragments in an agarose gel with and without YOYO-1 labeling (Fig. 5A–D). Then the shift was analyzed, which was induced by the binding of YOYO-1 to DNA molecules (Fig. 5B and C). Additional EMSA experiments verified that ACTD as well as the E. coli RNAP bind to fluorescent stained DNA fragments (Fig. 5E–H). Since a shift was observed, a successful binding was assumed. The built complexes are stable due to the high binding constants of YOYO-1, 6 × 108 mol−1,41 and actinomycin D, 106 mol−1,42 the binding constant of E. coli RNA polymerase core enzyme is 1.16 × 106 mol−1.43 Resolutions of 0.29 and 0.46 were achieved for complex separation. The run time for a gel was 3 h.
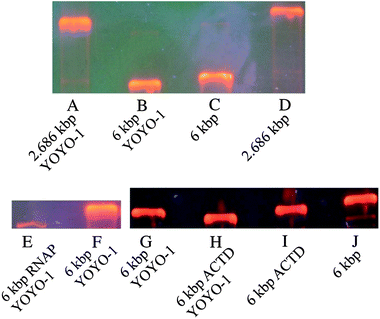 | ||
| Fig. 5 Electrophoretic mobility shift assays. Three shift assays are shown (A–D), (E and F) and (G–J). (A) 2.686 kbp DNA labeled with YOYO-1. (B) 6.0 kbp DNA labeled with YOYO-1. (C) 6.0 kbp DNA. (D) 2.686 kbp DNA. (E) 6.0 kbp DNA with E.coli RNAP and YOYO-1. (F) 6.0 kbp DNA labeled with YOYO-1. (G) 6.0 kbp DNA labeled with YOYO-1. (H) 6.0 kbp DNA with ACTD and YOYO-1. (I) 6.0 kbp DNA with ACTD. (J) 6.0 kbp DNA. The resolution between (A) and (B) is 1.2. The resolution between (C) and (D) is 1.77. The resolution between (E) and (F), polymerase induced shift, is 0.46. The resolution between (G) and (H), ACTD induced shift, is 0.29. The resolution between (B) and (C), indicating the YOYO-1 induced shift, is 0.22 and is therefore smaller than the other shifts. | ||
5. Discussion and conclusions
In summary, we demonstrated sufficient continuous-flow detection and separation of small DNA fragments and DNA-complexes by means of dielectrophoresis at an insulating ridge. We separated: (1) 2.686 kbp and 6.0 kbp DNA fragments (linear) and (2) parental and minicircle-DNA (circular). Additionally we detected and separated DNA-complexes: (1) cancer drug-DNA complexes for three different ratios of drug per DNA base pair and (2) protein–DNA complexes. All separations were with baseline separated resolution that proves the high sensitivity of the system. To verify and understand the mechanism in more detail the polarizabilities were also determined.There are some practical aspects that one might consider before applying this new microseparation method. Due to the polarization mechanism the buffer has considerable influence on separation efficiency and the parameters, which have to be applied. For example, the optimal frequency (a frequency that allows for the usage of low AC-voltages reducing joule heating and electrochemical side effects) depends on the viscosity of the solution, a fact that the determined polarizabilities reflect for the 6.0 kbp DNA fragments for different amounts of glycerin. Furthermore, the polarizability depends on the ionic strength21,44–46 (see also ESI Fig. 6† showing the presented technique works for 10 mM phosphate buffer). But due to the continuous operating mode, different parameters such as frequency, UDC and UAC can quickly be tested with real-time response allowing an on-line optimization. In gel electrophoresis this would be equivalent to changing the applied voltage (analogous to UDC) and gel concentration (analogous to UAC) during the experiment.
The results of polarizabilities of 2.686 kbp and 6.0 kbp DNA agree with previous published data. Whereas the change in polarizability for the DNA-complexes might have two reasons. (1) The ion cloud that surrounds the molecule could be changed due to the charge of the complexed molecule. (2) The radius of gyration could be changed by intercalation or alignment of ACTD or RNAP to DNA.47,48 But no significant change could be determined for the radius of gyration of the DNA-complexes relative to pure DNA (see ESI Fig. 7†) so a change of polarizability based on the ion cloud is more probable.
Since fluorescent staining is not mandatory for the presented method, although as demonstrated above, it did not interfere with the binding of ACTD or E. coli RNAP. As a label-free variant, one could think e.g. of spatially resolved optical adsorption. So the device allows new screening methods in medicine, e.g. detection of anti-double-stranded DNA antibodies.
The method demonstrated proved to be very robust concerning the separation of analyte mixtures composed of two different species. As soon as the experimenter observed a splitting of the injected sample molecules, i.e. one fraction passes the nanoslit while another fraction is deflected, for appropriate parameters, we could prove a successful separation afterwards (by injecting samples consisting of only one species under identical conditions). The ability to separate more complex mixtures would further broaden the applicability of the method. We found that the dielectrophoretic potential depends on the y-position at the ridge, a fact formerly noticed and theoretically analyzed by Hawkins et al.24 This implies that objects differing in polarizability should leave the ridge at a different position. But neither they nor we could experimentally observe such a behavior. Therefore, we propose a slightly different device layout for multiple separation: a channel branching to several channels with different ridge layouts and consequently different dielectrophoretic potentials.
In conclusion, the most prominent advantages of our technique are: continuous-flow processing; fast separation time less than 1 min; accessibility to linear as well as circular DNA molecules; separation of small DNA-fragments and detection of DNA-complexes; baseline separated resolution; on-line adaptability of selectivity parameters via applied voltages. Thus it combines the advantages of three continuously operating microfluidic devices13–15 in a single device and additionally allows detection of DNA-complexes.
Up to now the throughput of 2 nl min−1 for one single device was achieved in continuous processing mode. But for several parallelized devices the throughput could be increased easily, with promising applications in purification of minicircle-DNA for gene-therapy or plasmid vaccination.
Especially, this last point is of central interest since our microfluidic separation methodology does not rely on classical filtration steps and therefore reduces the shear-force associated analyte deterioration to a minimum. Together with the fact that our microfluidic approach is inherently independent of toxic or mutagenic elution or preparation reagents, a potential use with respect to applications in medical genetics can be implemented in a much smoother way considering the rigorous concerns of safety regulation in drug administration.
Furthermore and in analogy to EMSA, the presented method can be used to study association and/or dissociation kinetics. Here, miniaturization offers another advantage. For automated handling of fluids in the device, time scales much shorter than the limit of about 1 min for manual solution handling could be addressed. Since operation of the device is stable for 180 nm nanoslit height it would allow also separation of proteins.
Acknowledgements
We thank Ralf Eichhorn for very fruitful discussion throughout the whole process from conception of the experiments over analyzing the experimental data to the written manuscript. This work was supported by the German Science Foundation (DFG) in the collaborative research center (SFB 613) project D2. We especially thank Eugenie Fredrich and Verena Leder for assistance in the laboratory. We thank Anja Rischmüller and Martin Schleef from Plasmid Factory for production and supply of minicircle-DNA. We thank Katja Tönsing for support concerning the biochemical background.References
- L. M. Hellman and M. G. Fried, Nat. Protoc., 2007, 2, 1849–1861 CrossRef CAS.
- S. Mandal, M. Moudgil and S. K. Mandal, Eur. J. Pharmacol., 2009, 625, 90–100 CrossRef CAS.
- K. Hens, J.-D. Feuz, A. Isakova, A. Iagovitina, A. Massouras, J. Bryois, P. Callaerts, S. E. Celniker and B. Deplancke, Nat. Methods, 2011, 8, 1065–1070 CrossRef CAS.
- S. Lalonde, D. W. Ehrhardt, D. Loqué, J. Chen, S. Y. Rhee and W. B. Frommer, Plant J., 2008, 53, 610–635 CrossRef CAS.
- L.-P. Yu, Y.-Z. Sun and Z.-X. Zhao, Curr. Pharm. Anal., 2009, 5, 112–119 CrossRef CAS.
- A. Rahman and D. A. Isenberg, N. Engl. J. Med., 2008, 358, 929–939 CrossRef CAS.
- J. Tegenfeldt, C. Prinz, H. Cao, R. Huang, R. Austin, S. Chou, E. Cox and J. Sturm, Anal. Bioanal. Chem., 2004, 378, 1678 CrossRef CAS.
- B. Li, X. Fang, H. Luo, Y.-S. Seo, E. Petersen, Y. Ji, M. Rafailovich, J. Sokolov, D. Gersappe and B. Chu, Anal. Chem., 2006, 78, 4743–4751 CrossRef CAS.
- O. Bakajin, T. A. Duke, J. Tegenfeldt, C. F. Chou, S. S. Chan, R. H. Austin and E. C. Cox, Anal. Chem., 2001, 73, 6053–6056 CrossRef CAS.
- J. Han and H. G. Craighead, Science, 2000, 288, 1026–1029 CrossRef CAS.
- P. Doyle, J. Bibette, A. Bancaud and J.-L. Viovy, Science, 2002, 295, 2237 CrossRef CAS.
- J. Regtmeier, T. T. Duong, R. Eichhorn, D. Anselmetti and A. Ros, Anal. Chem., 2007, 79, 3925–3932 CrossRef CAS.
- J. Fu, R. B. Schoch, A. L. Stevens, S. R. Tannenbaum and J. Han, Nat. Nanotechnol., 2007, 2, 121–128 CrossRef CAS.
- L. Huang, J. Tegenfeldt, J. Kraeft, J. Sturm, R. Austin and E. Cox, Nat. Biotechnol., 2002, 20, 1048–1051 CrossRef CAS.
- L. Huang, E. Cox, R. Austin and J. Sturm, Science, 2004, 304, 987–990 CrossRef CAS.
- J. Regtmeier, R. Eichhorn, M. Viefhues, L. Bogunovic and D. Anselmetti, Electrophoresis, 2011, 32, 2253–2273 CrossRef CAS.
- J. Clark, T. Shevchuk, P. M. Swiderski, R. Dabur, L. E. Crocitto, Y. I. Buryanov and S. S. Smith, BioTechniques, 2003, 35, 548–554 CAS.
- L. R. Brewer and P. R. Bianco, Nat. Methods, 2008, 5, 517–525 CrossRef CAS.
- H. Pohl, Dielectrophoresis: The Behavior of Neutral Matter in Nonuniform Electric Fields, Cambridge University Press, 1978 Search PubMed.
- J. Regtmeier, R. Eichhorn, L. Bogunovic, A. Ros and D. Anselmetti, Anal. Chem., 2010, 82, 7141–7149 CrossRef CAS.
- D. Porschke, J. Biophys. Chem., 1997, 66, 241–257 CrossRef CAS.
- C. Chou, J. Tegenfeldt, O. Bakajin, S. Chan, E. Cox, N. Darnton, T. Duke and R. Austin, Biophys. J., 2002, 83, 2170–2179 CrossRef CAS.
- B. Kirby and E. Hasselbrink, Electrophoresis, 2004, 25, 187–202 CrossRef CAS.
- B. G. Hawkins, A. E. Smith, Y. A. Syed and B. J. Kirby, Anal. Chem., 2007, 79, 7291–7300 CrossRef CAS.
- L. M. Barrett, A. J. Skulan, A. K. Singh, E. B. Cummings and G. J. Fiechtner, Anal. Chem., 2005, 77, 6798–6804 CrossRef CAS.
- Y. Xia and G. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- P. Mayrhofer, M. Blaesen, M. Schleef and W. Jechlinger, J. Gene Med., 2008, 10, 1253–1269 CrossRef CAS.
- M. Viefhues, R. Eichhorn, E. Fredrich, J. Regtmeier and D. Anselmetti, Lab Chip, 2012, 12, 485–494 RSC.
- P. Hänggi, P. Talkner and M. Borkovec, Rev. Mod. Phys., 1990, 62, 251–341 CrossRef.
- T. W. Odom, J. C. Love, D. B. Wolfe, K. E. Paul and G. M. Whitesides, Langmuir, 2002, 18, 5314–5320 CrossRef CAS.
- W. Hellmich, J. Regtmeier, T. Duong, D. Anselmetti and A. Ros, Langmuir, 2005, 21, 7551–7557 CrossRef CAS.
- M. Viefhues, S. Manchanda, T.-C. Chao, D. Anselmetti, J. Regtmeier and A. Ros, Anal. Bioanal. Chem., 2011, 401, 2113–2122 CrossRef CAS.
- S. V. Ermakov, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 2000, 72, 3512–3517 CrossRef CAS.
- J. Giddings, Unified Separation Science, John Wiley Sons: New York, 1991 Search PubMed.
- S. Tuukkanen, A. Kuzyk, J. Toppari, H. Häkkinen, V. Hytönen, E. Niskanen, M. Rinkiö and P. Törma, Nanotechnology, 2007, 18, 295204 CrossRef.
- K. M. Choi and J. A. Rogers, J. Am. Chem. Soc., 2003, 125, 4060–4061 CrossRef CAS.
- M. Viefhues, J. Regtmeier and D. Anselmetti, J. Micromech. Microeng., 2012, 22, 115024 CrossRef.
- N. C. Stellwagen, Biopolymers, 1981, 20, 399–434 CrossRef CAS.
- J. Elias and D. Eden, Macromolecules, 1981, 14, 410–419 CrossRef CAS.
- M. Hanss and J. C. Bernengo, Biopolymers, 1973, 12, 2151–2159 CrossRef CAS.
- A. Larsson, C. Carlsson and M. Jonsson, Biopolymers, 1995, 36, 153–167 CrossRef CAS.
- F.-M. Chen, F. Sha, K.-H. Chin and S.-H. Chou, Biophys. J., 2003, 84, 432–439 CrossRef CAS.
- P. Bonarek, S. Kedracka-Krok, B. Kepys and Z. Wasylewski, Acta Biochim. Pol., 2008, 55, 537–547 CAS.
- D. C. Rau and E. Charney, Biophys. Chem., 1983, 17, 35–50 CrossRef CAS.
- D. C. Rau and E. Charney, Biophys. Chem., 1981, 14, 1–9 CrossRef CAS.
- D. C. Rau and V. A. Bloomfield, Biopolymers, 1979, 18, 2783–2805 CrossRef CAS.
- T. P. Hunt and B. Magasanik, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 8453–8457 CrossRef CAS.
- H. Chen, X. Liu and D. J. Patel, J. Mol. Biol., 1996, 258, 457–479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2an36056j |
| This journal is © The Royal Society of Chemistry 2013 |