Sub-cellular spectrochemical imaging of isolated human corneal cells employing synchrotron radiation-based Fourier-transform infrared microspectroscopy
Simon W.
Fogarty
ab,
Imran I.
Patel
b,
Júlio
Trevisan
b,
Takahiro
Nakamura
c,
Carol J.
Hirschmugl
d,
Nigel J.
Fullwood
a and
Francis L.
Martin
*b
aDivison of Biomedical and Life Sciences, Faculty of Health and Medicine, Lancaster University, Lancaster, UK
bCentre for Biophotonics, Lancaster Environment Centre, Lancaster University, Bailrigg, Lancaster LA1 4YQ, UK. E-mail: f.martin@lancaster.ac.uk; Fax: +44 (0)1524 510217; Tel: +44 (0)1524 510206
cDepartment of Opthalmology, Kyoto Perfectural University of Medicine, Kawaramachi Hirohoji, Kamigyo-ku, Kyoto, Japan
dSynchrotron Radiation Center, University of Wisconsin-Madison, Stoughton, Wisconsin, USA
First published on 1st November 2012
Abstract
Understanding stem cell (SC) biology remains challenging and one of the few human tissues within which their in situ location is well characterized is the cornea. Individual human corneal epithelial cells were isolated from biopsies of live tissues using fluorescence-activated cell sorting (FACS); these were divided into putative SCs, transit-amplifying (TA) cells and terminally-differentiated (TD) cells. Employing synchrotron radiation-based Fourier-transform infrared (SR-FTIR) microspectroscopy with a focal plane array (FPA), sub-cellular spatial resolution analysis of unstained isolated cells was achieved as a consequence of the brilliance of a 12 collimated beams arrangement allowing rapid spectral acquisition. Infrared (IR) spectra were extracted and pre-processed. Subsequent categorization with multivariate analysis of IR spectra derived from FPA images was used to investigate biomolecular changes between classes. A progressive segregation in cell-specific spectral categories with differentiation from SC to TA cell to TD cell was noted. Multiple different absorption peaks that discriminated putative SCs, TA cells and TD cells across DNA, protein and lipid spectral regions were identified. DNA regions (1080 and 1225 cm−1) and some protein regions (1443 cm−1) primarily segregated SCs from TA cells and TD cells, whilst amide regions and lipids (1,550, 1650 and 1740 cm−1) segregated TA cells and TD cells. Scanning electron microscopy images verified the external phenotypic characteristics of the different isolated cell types. These findings highlight the applicability of SR-FTIR microspectroscopy towards distinguishing SCs, TA cells and TD cells, and suggest that cellular classification via traditional methods of immunolabelling can be greatly aided by the use of spectral biomarkers.
Introduction
The importance of stem cell (SC) identification and classification has grown in recent years, particularly in the search for cancer SCs1 and for sites of tissue regenerative capacity (SC niches)2,3 useful for wound healing. Their apparent in situ location has been well characterized in cornea4,5 but not so in most tissues. In order to aid in the identification of SC regions, many different putative biomarkers have been applied, but it has proven very difficult to develop a biomarker that is conserved across different SC populations, meaning many remain elusive in their identification.Fluorescence-activated cell sorting (FACS) was developed in the 1960's as a means to increase the potential of flow cytometry for use on live viable cells. Since its development it has become a useful tool in biological research.6,7 FACS enables the identification of different cell classes through differential targeting using a fluorescent marker. This allows fluorescently labelled cells to be separated from non-labelled cells in a highly specific manner. Whilst traditional labelling techniques, such as immunolabelling can identify SCs based on a single biomolecule type, techniques like infrared (IR) spectroscopy have the ability to interrogate biological samples across a whole range of differing biomolecular regions. This is because cellular biomolecules absorb electromagnetic radiation in the mid-IR region (wavelengths 2–20 μm). These properties allow the generation of spectral fingerprints of interrogated biological material. This has given rise to the notion that IR spectroscopy may be applied to generate novel insights into SC biology.8,9
Fourier-transform IR (FTIR) spectroscopy is an emerging technique for biological analysis, and has been employed in cell characterization studies in malignant or pre-malignant tissue,10–14 cell type identification in tissue samples,9,15–17 and cell cycle studies.18,19 Bench-top FTIR spectrometers are most commonly used but can be limited in resolution due to their relatively weak light source, which results in poor signal-to-noise quality at high resolutions. A solution to this is to use synchrotron radiation (SR) as a light source via which the spatial resolution of FTIR spectroscopy is greatly enhanced, due to the high source brilliance. Recently, this has allowed high-resolution sub-cellular imaging of either isolated cells or tissues.8,20,21
Since FTIR spectroscopy generates an integrated spectral fingerprint of the entire biomolecular composition, derived data is generally much more complex than that generated following analysis of a single targeted molecule. As a consequence, data handling for multiple generated spectra requires consideration, as category differences can frequently be subtle. Multivariate analysis reduces individual spectra into single points in scatter plots, segregated into clusters according to their spectral relatedness. This approach also allows identification of discriminating wavenumbers responsible for category segregation.22,23
Many studies which have sought to find unique SC biomarkers have used the cornea as a model tissue.15,24–27 The tissue morphology of the cornea has been well characterized,28,29 it is easily accessible for tissue excision, and unusually it is well understood where different cell populations are located in situ.5,25,30,31 SCs are located in the limbal region on the border between the cornea and sclera. As they divide and differentiate, they form transit-amplifying (TA) cells; these cells migrate inwards to the centre of the cornea to give rise to the basal layer of the tissue. These TA cells have a more limited ability to proliferate compared to SCs, but they still divide to generate terminally-differentiated (TD) cells, which constitute the topmost layers of the corneal epithelium. TD cells are the most specialized cell class, forming the uppermost layers of the corneal epithelium and lacking the apparent ability to proliferate. Currently however, there are no specific biomarkers of corneal epithelial SCs but experiments to determine their characteristics such as unlimited self-renewal, proliferative potential and phenotype expression have been conducted.26,32 There is differing levels of biomolecular expression from SCs to TA cells and TD cells which can lead to cell type identification.25 Various keratin molecules have been previously investigated as putative selective markers for limbal SCs.30 Following on from this, keratin 15 (K15) has been used as a putative biomarker in previous experiments to identify corneal SC populations.15,33–35 Our study aim was to isolate individual SCs, TA cells and TD cells from viable human corneas using FACS, and derive spectral images of these using high-resolution SR-based FTIR microspectroscopy.
Materials and methods
Tissue samples
Human donor corneal tissues (n = 4) were obtained from SightLife® (Seattle, WA) eye bank, and all corneas were stored at 4 °C in storage medium (Optisol; Chiron Ophthalmics, Inc., Irvine, CA). All experiments were performed in accordance with the tenets of the Declaration of Helsinki.Cell extraction and isolation
The three different cell types (SCs vs. TA cells vs. TD cells) were identified in situ using immunofluorescent staining (Fig. 1). Cell sections were counterstained with propidium iodide (PI) to identify total cell presence. For SC acquisition, a limbal biopsy was prepared (Fig. 1A) and SCs were identified in the limbal region as positively labelled for keratin 15 (K15) antibody (Fig. 1B). The corneal biopsy was dissipated using dispase enzyme and subsequent FACS Aria II (BD Biosciences) separated out the K15+ cells as a putative SC class.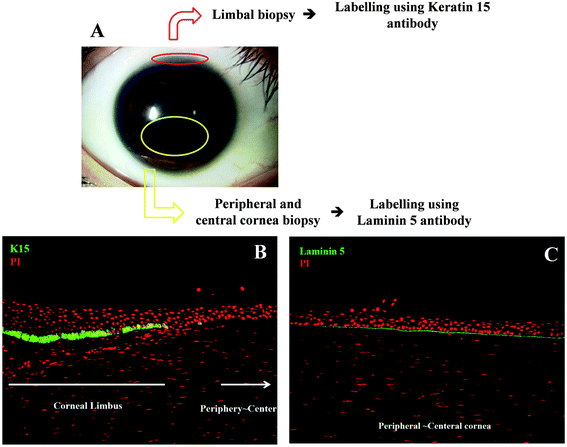 | ||
| Fig. 1 Immunofluorescent staining of corneal cell biopsies. Corneas were obtained from the US donor bank and regions isolated for cell staining (A) and identification (stained with propidium iodide to identify all cells and positive cells labelled green in B and C). K15+ cells in (B) are putative stem cells. In (C), Laminin 5+ (LAM5+) cells are putative transit-amplifying cells and Laminin 5− (LAM5−) cells are putative terminally-differentiated cells. | ||
For cell sorting of K15+ cells, human limbal tissues were first incubated at 37 °C for 1 h with 1.2 IU dispase to separate the epithelial cells. Cells from the limbal region were carefully separated from the underlying stroma using micro-forceps, and incubated at room temperature (RT) for 15 min with 1% foetal bovine serum (FBS). Limbal epithelial-cell suspensions were then incubated with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100-diluted mouse anti-human K15 (Lab Vision) at RT for 20 min, washed three times with PBS(−), and incubated with 1:1500-diluted Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). K15+ and K15− cells were isolated using FACS Aria II.
:100-diluted mouse anti-human K15 (Lab Vision) at RT for 20 min, washed three times with PBS(−), and incubated with 1:1500-diluted Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). K15+ and K15− cells were isolated using FACS Aria II.
For TA-cell and TD-cell acquisition, a peripheral and central corneal biopsy was performed (Fig. 1A). Cells were differentiated by expression of Laminin 5 (LAM5); TA cells were positively labelled for LAM5 whilst TD cells were negative for LAM5 (Fig. 1C). The corneal biopsy was dissipated once more using dispase enzyme. FACS was performed to separate LAM5+ cells as a putative TA cells and LAM5− cells as a putative TD-cell class.
For cell sorting of LAM5+ cells, human corneal tissues (without the limbal region) were first incubated at 37 °C for 1 h with 1.2 IU dispase to separate the epithelial cells. Cells from the peripheral and central corneal region were carefully separated from the underlying stroma using micro-forceps, and incubated at RT for 15 min with 1% FBS. The corneal epithelial cells were then incubated with 1:100-diluted mouse anti-human LAM5 (Chemicon) at RT for 20 min, washed three times with PBS(−), and incubated with 1:1500-diluted Alexa Fluor 488-conjugated goat anti-mouse IgG. LAM5+ and LAM5− cells were isolated by using FACS Aria II.
Isolated individual cells were fixed in 70% ethanol. Whilst using any fixative will change cell morphology somewhat, 70% ethanol is believed to preserve overall cell integrity better than other fixatives such as formalin; it also allows better visualization of the fixed cell architecture.36 Cells were then concentrated and purified by centrifugation and washing with distilled water before the resultant cell suspension was pipetted onto barium fluoride (BaF2) slides and allowed to dry. This was repeated to give a greater amount of the cell suspension on each BaF2 slide to maximize the number of cells for analysis. The cells were also prepared for scanning electron microscopy (SEM) by air-drying, followed by gold coating in an Edwards 150 sputter coater. Individual cells were examined on a JEOL 5600 scanning electron microscope (Fig. 2).
 | ||
| Fig. 2 Scanning electron microscopy images of cells from each category; (A) typical putative stem cell (scale bar = 5 μm), (B) typical putative transit-amplifying cell (scale bar = 5 μm), (C) typical putative terminally-differentiated (TD) cell (scale bar = 5 μm), (D) area of TD cell within white box in (C) enlarged (scale bar = 2 μm). | ||
Spectral acquisition
Data was obtained at the IRENI (IR Environmental Imaging) beamline at the Synchrotron Radiation Center (SRC), Wisconsin, USA. Spectra were taken using a focal plane array (FPA), illuminated by 12 radiation beams arranged in a 3 × 4 matrix, which was attached to a FTIR spectrometer (Bruker Optics Inc. USA).21 These were acquired in transmission mode with 32 scans covering the target area. Before each FPA image, a background was taken from a cell-free area. Each FPA image consists of 9216 spectra in a 96 × 96 pixel image. Each spectrum was acquired over a geometric effective area at the sample plane of 0.54 × 0.54 μm2, with a spectral resolution of 4 cm−1.Data processing
All FPA images generated were processed using MATLAB r2010a (Mathworks Inc., Natick, USA) coupled with our in-house toolbox (http://bioph.lancs.ac.uk/iroot) and additional scripts (freely available). The full processing protocol can be seen in Fig. 3. To compare spectra of cells from different FPA images, spectra were initially extracted and recombined within MATLAB for multivariate computational analysis. Initially the full FPA images underwent Pre-processing 1 (Fig. 3A) and wavelet de-noising before hierarchical cluster analysis (HCA) was implemented. HCA was used to distinguish between spectral points from biological regions and those from the background alone.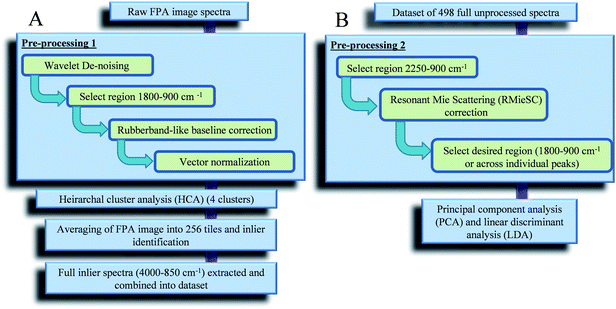 | ||
| Fig. 3 Protocol for the analysis of focal plane array images in MATLAB: (A) generation of an inlier dataset; and, (B) inlier dataset analysis. | ||
Each FPA image was averaged into 256 tiles (giving a new pixel size of 3.2 μm × 3.2 μm) with those identified as biological extracted for use in analysis. Tiles with less than 30% classed as biological spectra were discarded. This generated a group of averaged spectra for each FPA image, which were combined in Microsoft Excel to form a full dataset.
Due to the differences in cell size within each category (SCs vs. TA cells vs. TD cells), the number of spectra generated through the isolation and extraction method from HCA differed between categories. Five SC FPA images were generated (derived from n = 4 tissue samples), each separated into 256 average tiles to give a total of 1280 tiles. Of these 1280 tiles, only 129 were determined as biological for analysis. Furthermore, five TA-cell FPA images gave a total of 1280 tiles, of which only 215 were determined as biological whilst four TD-cell FPA images gave a total of 1024 tiles, of which only 154 were determined as biological. This led to a total dataset of 498 inlier spectra for analysis using principal component analysis and linear discriminant analysis (PCA-LDA).
The high resolution of spectral acquisition employed in this study means that discrete scattering processes become more apparent and relevant. Resonant Mie scattering can be very problematic since the wavelengths of the probing radiation are the same size as heterogeneities in the samples and can introduce artefacts into the acquired IR spectra, making observations unreliable.37 This leads to spectral features that are not strictly absorption bands and appear to have shifted peak positions, but absorption profiles can be recovered with established methods.38 Thus, to accurately represent the underlying biochemical structure, iterative programs are applied to extract absorption spectra from the collected data that has contributions from both absorption and scattering.38 Therefore the original, binned and selected biological spectral dataset was put through Pre-processing 2 (Fig. 3B) in MATLAB using resonant Mie Scattering (RMieSc) scripts37,38 before undergoing PCA-LDA.
PCA reduces spectra individually to single points which are separated by principal components (PCs); as a consequence, similar spectra can be visualized within clusters in a scatter plot. LDA was applied to the output from PCA; LDA separates the data based by category rather than individual spectra. LDA reduces each class to an average point to maximize between-category variation and minimize within-class variation.23
Results and discussion
The biochemical-cell fingerprint spectrum (1800–900 cm−1) was analyzed to identify the predominant between-category segregating factors (Fig. 4). In Fig. 4A, the mean spectra for each category are very similar and it's quite difficult to interpret the extent of discriminating wavenumbers. Differences become much more pronounced following PCA-LDA clustering, and Fig. 4B shows clear cluster separation by linear discriminant (LD)1 and LD2 (factor 1 and 2 in Fig. 4C, respectively). The cluster vectors (Fig. 4D–F) show the relationship between the separate class scatter plots in Fig. 4B and, it appears that the TA-cell and TD-cell categories are much more closely related than they are individually to the SC category. This is especially observable in Fig. 4F, where the cluster vectors for TA cells and TD cells are very similar in shape and are markedly discriminated away from the SC category set as the origin.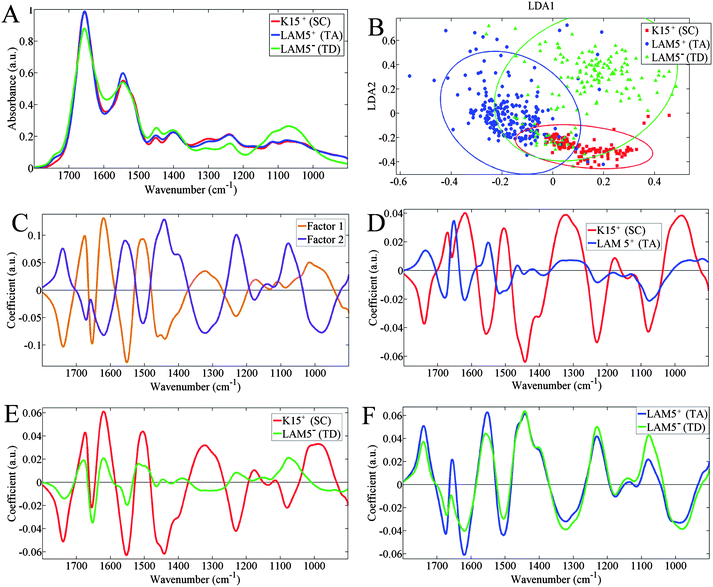 | ||
| Fig. 4 Spectral data of extracted inlier spectra: (A) mean spectra per class; (B) two-dimensional principal component analysis-linear discriminant analysis (PCA-LDA) scores plot; (C) PCA-LDA loadings plots for LD1 (orange) and LD2 (purple); (D) cluster vectors plot with terminally-differentiated-cell category set as origin; (E) cluster vector plot with transit amplifying-cell category set as origin; and origin; and, (F) cluster vector plot with stem cell category set as origin. | ||
Further PCA-LDA data displayed in Fig. 5 focuses the analysis on individual peaks taken from the full spectrum in Fig. 4A. This highlights the between-category differences at each biomolecular region, which may be missed when analyzing the entire spectrum. The spectral peaks can be split into three broad categories: DNA/RNA, proteins and lipids. Fig. 5A and B correspond to phosphate bending of DNA/RNA, while Fig. 5C–F correspond to various protein regions and Fig. 5G corresponds to lipid alterations. SCs are primarily separated from TA cells and TD cells by DNA/RNA spectral regions, which could be due to restructuring and alterations in DNA strands compared to more specialized cells.
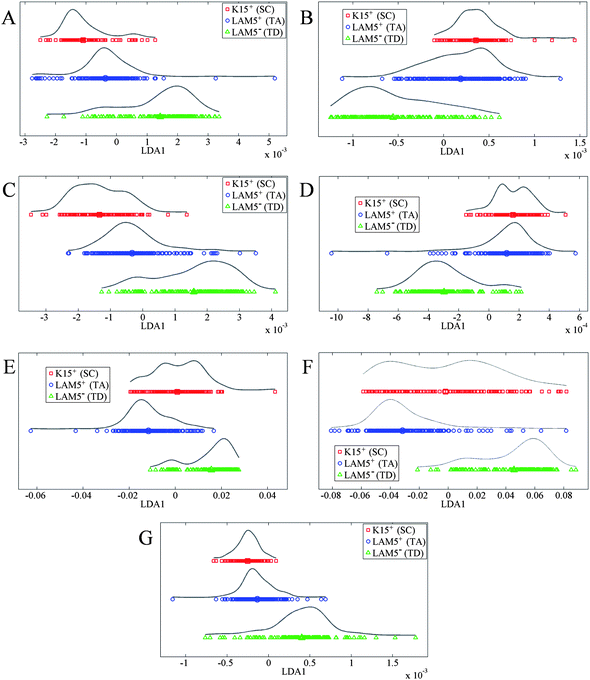 | ||
| Fig. 5 One-dimensional principal component analysis-linear discriminant analysis scatter plots of individual peaks: (A) 1020–1100 cm−1 corresponding to symmetric phosphate stretching vibrations (νsPO2−); (B) 1210–1260 cm−1 corresponding to asymmetric phosphate stretching vibrations (νsPO2−); (C) 1360–1430 cm−1 corresponding to symmetric CH3 deformations (protein); (D) 1430–1470 cm−1 corresponding to asymmetric CH3 deformations (protein); (E) 1490–1590 cm−1 corresponding to the Amide II; (F) 1610–1700 cm−1 corresponding to the Amide I; and, (G) 1720–1780 cm−1 corresponding to lipid. | ||
FTIR microspectroscopy has been used in previous studies to identify putative SC, TA cell and TD cell categories in situ,15 but never on individual corneal cells differentially isolated by FACS. Isolated cells allow greater interrogation and analysis of specific cell types that have been putatively identified as SCs, TA cells or TD cells. Gradual differences may arise due to cellular differentiation processes of multipotent SCs giving rise to TD cells. Consequently, both Fig. 4 and 5 show a degree of overlap between spectra from different categories. This overlap is not unexpected as the cells are separated by FACS, which is based on a non-specific biomarker very likely capable of labelling more than one cell type. However, it can be observed that there is a distinct progression in alterations from SCs to TA cells and then to TD cells; TD cells are typically not as structurally closely related to SCs as TA cells are. This is to be expected as it probably reflects the on-going changes in biomolecular content and structure as cell types become more differentiated.
To aid cell identification and characterization, the main differing between-category factors can be analyzed. Between-category differences are evident within the IR spectra themselves (Fig. 4A), but it is hard to quantify which biomolecular regions differ most. As shown in Fig. 4B, the SC category is predominantly separated from the TA cell and TD categories by LD2, whilst those of the TA cell and TD cell are predominantly separated by LD1. These LDs are shown to be based on different contributing spectral peaks in Fig. 4C. From these loadings plots, it appears that LD1 is mostly influenced by Amide I and Amide II peaks (1650 cm−1 and 1550 cm−1, respectively). Although LD2 is also influenced by these amide regions, it is mostly influenced by regions lower down the spectrum, notably asymmetric CH3 deformations in proteins (1443 cm−1) and, both symmetric (νsPO2−; 1080 cm−1) and asymmetric (νasPO2−; 1230 cm−1) phosphate stretching vibrations. Therefore, SCs are clustered away from other cell types via DNA/RNA alterations and protein CH3 deformations, whilst TA cells and TD cells are segregated mainly by amide alterations.
Also evident from Fig. 4B is that the spectra of the SC category clusters are much tighter together than the other categories, indicating there is less within-category variance in these spectra. This is an important point as within-category variance is contributed to by heterogeneity within the same cell types, i.e., cells within each category being at different stages of their respective life cycle. Therefore, the biochemical structures of TD cells vary a lot more than the SC category. TD cells can change a lot biochemically as they vary from newly-produced cells arising from TA-cell division up to the superficial layers of the corneal epithelium where they are likely to begin undergoing apoptosis and are being constantly lost.39 Meanwhile, it's important for SCs to maintain their structure in the process of self-renewal to prevent cell loss and genomic alteration which could lead to mutation or impaired function in differentiated cells.26
The amide profiles, shown by peaks for Amide II in Fig. 5E and Amide I in Fig. 5F, become more conserved as the cells become increasingly differentiated. This could be due to specialization of cells and differential expression of proteins, especially those necessary for differentiated-cell function. As shown in Fig. 1C, a thin layer of LAM5+ (putative TA cell) cells rest on top of the basement membrane, and will be associated with the basement membrane through numerous intracellular and membrane factors.5,26,30 As TA cells differentiate and move upwards in the epithelium becoming TD cells, they become dissociated from the basement membrane. This will involve major biomolecular modifications, notably in protein structure, to limit the factors which keep TA cells associated with the basement membrane.27 Interestingly, a key differentiating factor between SCs from TA cells and TD cells (in Fig. 4D and E) is at 1443 cm−1, which corresponds to the protein region of asymmetric CH3 deformations. In Fig. 4C, 1443 cm−1 is shown to be a major contributor to LD2. Whilst it is a differentiating factor for SCs there is little difference in 1443 cm−1 absorbance between TA cells and TD cells, displayed in Fig. 4F. This suggests important protein characteristics associated with SCs alone.
Lipid alterations also occur as cells differentiate. The lipid head group-associated peak at 1740 cm−1 (Fig. 4A) shows increasing intensity from SCs to TA cells and then TD cells. This indicates higher levels of lipid content in TD cells compared to SCs. Increases in lipid expression have been reported to occur due to differentiation of cells40 and such alterations would mostly likely be associated with the plasma membranes, e.g., phospholipids. Also, it was found that lipid composition becomes more diverse as cells become more differentiated (Fig. 5G); lipid composition is highly conserved in SCs whilst it is less so in TD cells. This could be a consequence of apoptotic processes altering the biochemical structure of cells;39 however, this remains to be fully investigated.41 TD cells are late-stage cells, which regularly undergo apoptosis due to being located in the superficial corneal epithelial-cell layers where cells are damaged by exposure to the external environment; this requires their constant replenishment with fresh cells. SCs are less likely to undergo apoptosis, being protected in the limbal niche region where they perpetuate their small population through their ability for self-renewal.
The SEM images in Fig. 2 give a visual ultrastructural representation of external cell structure. They lend support to the spectral data in showing progressive cellular changes from SCs through to TA cells to TD cells. These progressive alterations are linked to the structural changes involved in cellular differentiation as biomolecules are rearranged and are differentially expressed. In Fig. 2 it appears that as the cells differentiate they become less smooth and change shape; SCs are elongated and columnar (Fig. 2A), TA cells are spherical (Fig. 2B) and, TD cells are flat and squamous (Fig. 2C). Microvilli also appear across the entire surface of the TD cell (Fig. 2C, magnified in Fig. 2D). The FACS process, as expected, will have disrupted the cellular morphologies to some degree but it is still possible to observe the progression in cell structural changes as cells differentiate. Although SEM images are not in themselves definitive evidence, when coupled with the observations of differential immunolabelling and spectral changes, a definite progression in cell structural changes can be seen from the pluripotent SC to non-proliferative TD cells with TA cells as an intermediate.
Whilst it's useful to generate isolated spectra from cellular samples for each category, it's also possible to generate specific image maps of each cell category (Fig. 6). Comparing the cells from the different categories it is clear to see that there are numerous differences. For the SC category there is a high degree of protein expression throughout the cell; this is shown by the high intensity of Amide I and Amide II across the whole cell. Amide I especially seems almost ubiquitously expressed throughout the cell. Meanwhile DNA expression is spread out in the SC, to accommodate the large size of the nucleus relative to the cytoplasm. Within this nuclear region there are numerous sites where there is high expression of DNA, and this could correspond to regions of dense chromatin indicative of a typically quiescent cell; this is a typical feature of SCs.
 | ||
| Fig. 6 Spectral focal plane array intensity image maps at specific wavenumbers that show distribution of key biomolecules; amide image maps correspond to protein distribution and phosphate image maps correspond to DNA distribution. Each image map is at a single wavelength detailed above the images, baseline at 0 absorption; figures were made in MATLAB. Scale bar = 5 μm. | ||
Differences in biomolecular distributions are evident from SC to TA cells. In the TA cell compared to SCs, protein expression appears more localized to certain regions of the cell. This compartmentalization is not seen for DNA which seems to be more widely expressed than in the SC category. This could reflect increased proliferation of TA cells to give rise to TD cells. The TD-cell category shows a cell that is approaching the end of its lifetime. The cell has become much more compact in order to prepare for cell death via apoptosis. The DNA and the protein spectral regions do not seem to be too different from each other and have been condensed together; this is unlike in the previous two cell categories where there is differential location of DNA and protein. Overall, it is clear to see from the spectral image maps how progression in morphology from SC to that of TD cell causes significant alterations in the biochemical distribution of protein and DNA (Fig. 6).
In conclusion, SR-FTIR microspectroscopy can be used in order to differentiate between isolated putative SCs, TA cells and TD cells. The differences expressed between these cell categories can be thought of as corresponding to biochemical-cell alterations associated with differentiation, thus providing vital mechanistic insights. This could potentially allow visualization of the spatial location of characterized spectral biomarkers, conserved across different cell populations from multiple tissues. Also, there is the potential for the identification of differing mechanistic insights which may not be entirely observable with standard microscopy.42 However, whilst the data shown provides strong evidence for the benefits of SR-FTIR microspectroscopy in its ability to distinguish cell categories, it does not currently provide information about specific biomolecular distributions such as of particular proteins as in immunolabelling. Further experiments focussing on isolated cellular components, e.g., nuclei, mitochondria,43 coupled with novel computational approaches44 will allow more detailed investigations into the molecular changes that occur during the cellular differentiation process.
Acknowledgements
This work is based on research conducted at the IRENI beamline whose construction and development was supported by NSF under award #0619759. This work is funded by the Biotechnology and Biological Sciences Research Council (BBSRC). We would also like to thank Ralf Wehlitz for his assistance during the beamtime experiments.References
- C. T. Jordan, M. L. Guzman and M. Noble, N. Engl. J. Med., 2006, 355, 1253–1261 CrossRef CAS.
- L. Li and T. Xie, Annu. Rev. Cell Dev. Biol., 2005, 21, 605–631 CrossRef CAS.
- T. Tumbar, G. Guasch, V. Greco, C. Blanpain, W. E. Lowry, M. Rendl and E. Fuchs, Science, 2004, 303, 359–363 CrossRef CAS.
- P. Carrier, A. Deschambeault, M. Talbot, C. J. Giasson, F. A. Auger, S. L. Guérin and L. Germain, Invest. Ophthalmol. Visual Sci., 2008, 49, 1376–1385 Search PubMed.
- J. T. Daniels, J. K. G. Dart, S. J. Tuft and P. T. Khaw, Wound Repair Regen., 2001, 9, 483–494 CrossRef CAS.
- L. A. Herzenberg, D. Parks, B. Sahaf, O. Perez, M. Roederer and L. A. Herzenberg, Clin. Chem., 2002, 48, 1819–1827 CAS.
- F. L. Battye, A. Light and D. M. Tarlinton, J. Immunol. Methods, 2000, 243, 25–32 CrossRef CAS.
- M. J. Walsh, M. J. German, M. Singh, H. M. Pollock, A. Hammiche, M. Kyrgiou, H. F. Stringfellow, E. Paraskevaidis, P. L. Martin-Hirsch and F. L. Martin, Cancer Lett., 2007, 246, 1–11 CrossRef CAS.
- M. J. Walsh, T. G. Fellows, A. Hammiche, W.-R. Lin, N. J. Fullwood, O. Grude, F. Bahrami, J. M. Nicholson, M. Cotte, J. Susini, H. M. Pollock, M. Brittan, P. L. Martin-Hirsch, M. R. Alison and F. L. Martin, Stem Cells, 2008, 26, 108–118 CrossRef CAS.
- S. Mordechai, R. K. Sahu, Z. Hammody, S. Mark, K. Kantarovich, H. Guterman, A. Podshyvalov, J. Goldstein and S. Argov, J. Microsc., 2004, 215, 86–91 CrossRef CAS.
- B. R. Wood, M. A. Quinn, F. R. Burden and D. McNaughton, Biospectroscopy, 1996, 2, 143–153 CrossRef CAS.
- J. G. Kelly, T. Nakamura, S. Kinoshita, N. J. Fullwood and F. L. Martin, Analyst, 2010, 135, 3120–3125 RSC.
- D. C. Malins, N. L. Polissar, K. Nishikida, E. H. Holmes, H. S. Gardner and S. J. Gunselman, Cancer, 1995, 75, 503–517 CrossRef CAS.
- I. I. Patel, J. Trevisan, P. B. Singh, C. M. Nicholson, R. K. Krishnan, S. S. Matanhelia and F. L. Martin, Anal. Bioanal. Chem., 2011, 401, 969–982 CrossRef CAS.
- T. Nakamura, J. G. Kelly, J. Trevisan, L. J. Cooper, A. J. Bentley, P. L. Carmichael, A. D. Scott, M. Cotte, J. Susini, P. L. Martin-Hirsch, S. Kinoshita, N. J. Fullwood and F. L. Martin, Mol. Vision, 2010, 16, 359–368 CAS.
- M. J. German, H. M. Pollock, B. Zhao, M. J. Tobin, A. Hammiche, A. Bentley, L. J. Cooper, F. L. Martin and N. J. Fullwood, Invest. Ophthalmol. Visual Sci., 2006, 47, 2417–2421 Search PubMed.
- A. J. Bentley, T. Nakamura, A. Hammiche, H. M. Pollock, F. L. Martin, S. Kinoshita and N. J. Fullwood, Mol. Vision, 2007, 13, 237–242 CAS.
- H. Y. Holman, M. C. Martin, E. A. Blakely, K. Bjornstad and W. R. McKinney, Biopolymers, 2000, 57, 329–335 CrossRef CAS.
- J. R. Mourant, Y. R. Yamada, S. Carpenter, L. R. Dominique and J. P. Freyer, Biophys. J., 2003, 85, 1938–1947 CrossRef CAS.
- P. Dumas, G. D. Sockalingum and J. Sulé-Suso, Trends Biotechnol., 2007, 25, 40–44 CrossRef CAS.
- M. J. Nasse, R. Reininger, T. Kubala, S. Janowski and C. Hirschmugl, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 582, 107–110 CrossRef CAS.
- F. L. Martin, J. G. Kelly, V. Llabjani, P. L. Martin-Hirsch, I. I. Patel, J. Trevisan, N. J. Fullwood and M. J. Walsh, Nat. Protoc., 2010, 5, 1748–1760 CrossRef CAS.
- J. G. Kelly, J. Trevisan, A. D. Scott, P. L. Carmichael, H. M. Pollock, P. L. Martin-Hirsch and F. L. Martin, J. Proteome Res., 2011, 10, 1437–1448 CrossRef CAS.
- J. D. Zieske, G. Bukusoglu and M. A. Yankauckas, Invest. Ophthalmol. Visual Sci., 1992, 33, 143–152 CAS.
- A. Schermer, S. Galvin and T. T. Sun, J. Cell Biol., 1986, 103, 49–62 CrossRef CAS.
- U. Schlötzer-Schrehardt and F. E. Kruse, Exp. Eye Res., 2005, 81, 247–264 CrossRef.
- Z. Chen, C. S. de Paiva, L. Luo, F. L. Kretzer, S. C. Pflugfelder and D.-Q. Li, Stem Cells, 2004, 22, 355–366 CrossRef CAS.
- R. M. Lavker, S. C. G. Tseng and T. T. Sun, Exp. Eye Res., 2004, 78, 433–446 CrossRef CAS.
- W. Li, Y. Hayashida, Y. T. Chen and S. C. G. Tseng, Cell Res., 2007, 17, 26–36 CrossRef CAS.
- A. Pajoohesh-Ganji and M. A. Stepp, Biol. Cell, 2005, 97, 265–276 CrossRef CAS.
- L. Takács, E. Tóth, A. Berta and G. Vereb, Cytometry, Part A, 2009, 75, 54–66 CrossRef.
- H. S. Dua and A. Azuara-Blanco, Surv. Ophthalmol., 2000, 44, 415–425 CrossRef CAS.
- S. Yoshida, S. Shimmura, T. Kawakita, H. Miyashita, S. Den, J. Shimazaki and K. Tsubota, Invest. Ophthalmol. Visual Sci., 2006, 47, 4780–4786 Search PubMed.
- M. Lyngholm, H. Vorum, K. Nielsen, M. Østergaard, B. Honoré and N. Ehlers, Exp. Eye Res., 2008, 87, 96–105 CrossRef CAS.
- S. Merjava, A. Neuwirth, M. Tanzerova and K. Jirsova, Histol. Histopathol., 2011, 26, 323–331 Search PubMed.
- A. Hammiche, M. J. German, R. Hewitt, H. M. Pollock and F. L. Martin, Biophys. J., 2005, 88, 3699–3706 CrossRef CAS.
- P. Bassan, H. J. Byrne, F. Bonnier, J. Lee, P. Dumas and P. Gardner, Analyst, 2009, 134, 1586–1593 RSC.
- P. Bassan, A. Kohler, H. Martens, J. Lee, E. Jackson, N. Lockyer, P. Dumas, M. Brown, N. Clarke and P. Gardner, J. Biophotonics, 2010, 3, 609–620 CrossRef CAS.
- H. Ren and G. Wilson, Invest. Ophthalmol. Visual Sci., 1996, 37, 1017–1025 CAS.
- W. Tanthanuch, K. Thumanu, C. Lorthongpanich, R. Parnpai and P. Heraud, J. Mol. Struct., 2010, 967, 189–195 CrossRef CAS.
- J. K. Pijanka, D. Kumar, T. Dale, I. Yousef, G. Parkes, V. Untereiner, Y. Yang, P. Dumas, D. Collins, M. Manfait, G. D. Sockalingum, N. R. Forsyth and J. Sulé-Suso, Analyst, 2010, 135, 3126–3132 RSC.
- W. Pang, A. A. Ahmadzai, I. I. Patel, X. Qiu, M. Liles, A. J. Quantock and F. L. Martin, Invest. Ophthalmol. Visual Sci., 2012, 53, 1162–1168 CAS.
- J. K. Pijanka, A. Kohler, Y. Yang, P. Dumas, S. Chio-Srichan, M. Manfait, G. D. Sockalingum and J. Sulé-Suso, Analyst, 2009, 134, 1176–1181 RSC.
- J. Trevisan, P. P. Angelov, P. L. Carmichael, A. D. Scott and F. L. Martin, Analyst, 2012, 137, 3202–3215 RSC.
| This journal is © The Royal Society of Chemistry 2013 |