 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic solid phase microextraction analysis for airborne methamphetamine: quantitation using isotopically substituted methamphetamine†
Elizabeth J.
McKenzie
ab,
Gordon M.
Miskelly
*a and
Paul A. G.
Butler
c
aForensic Science Programme, School of Chemical Sciences, The University of Auckland, Private Bag 92019, Auckland, New Zealand. E-mail: g.miskelly@auckland.ac.nz; Fax: +64-9-373-7422; Tel: +64-9-923-8338
bForensic & Industrial Science Ltd, PO Box 20103 Glen Eden, Auckland 0641, New Zealand
cSchool of Chemical Sciences, The University of Auckland, Private Bag 92019, Auckland, New Zealand
First published on 2nd July 2013
Abstract
A vapour dosing system was developed that gave constant concentrations of methamphetamine in the range 1 to 10 μg m−3. This was used to calibrate the response of solid phase microextraction (SPME) fibres in passive and dynamic sampling modes. Exposure of 100 μm polydimethylsiloxane SPME fibres to a constant concentration of 1–5 μg m−3 methamphetamine over 1–70 min followed by GC-MS analysis produced a curvilinear pre-steady state sorption curve sufficiently reproducible to enable calibration of the SPME absorption. There was no evidence for loss of methamphetamine from the polydimethylsiloxane fibre under static ambient conditions for 5 h or during exposure to 1 L min−1 airflow in a dynamic SPME sampler for 90 min at room temperature. Sequential exposure of SPME fibres to methamphetamine and d9-methamphetamine showed that both analytes were retained. These results demonstrate that solid phase microextraction can be used with pre-loaded isotopically substituted methamphetamine as an internal standard for accurate quantitation of airborne methamphetamine. The isotopic labeling experiments also showed that the dosing system had a small but significant reservoir of methamphetamine, even though it was constructed from mainly inert materials and much of it was at elevated temperature. We therefore recommend that separate vapour dosing units be used for labeled and unlabeled methamphetamine.
Introduction
We have been investigating the use of dynamic solid phase microextraction (SPME) sampling to detect airborne methamphetamine present in suspected or known former clandestine methamphetamine laboratories. The conversion of peak areas observed in gas chromatography-mass spectroscopy (GC-MS) chromatograms for the methamphetamine adsorbed or absorbed onto the SPME fibre into estimates of the airborne methamphetamine concentration in such studies required a controlled-concentration dosing system for methamphetamine vapour in the concentration range 1–10 μg m−3. The incorporation of an internal standard onto the SPME fibre to reduce variability during the analysis was also investigated.The measurement of methamphetamine concentrations is commonly performed when investigating former clandestine methamphetamine laboratories because methamphetamine is the primary analyte of concern and because it can be used as a surrogate for other potential contaminants at such a site. Most current methodologies for identifying whether a known or suspected former clandestine methamphetamine laboratory is contaminated, and whether the site has been adequately remediated, rely on surface-wipe sampling for methamphetamine.1,2 We are investigating whether measurement of airborne methamphetamine could complement surface-wipe sampling. Concentrations of airborne methamphetamine during the acts of smoking or manufacture of methamphetamine have been reported to be as high as 100–5000 μg m−3,3,4 but concentrations can decrease rapidly, with Van Dyke et al. reporting levels of 100–800 μg m−3 one day after a controlled manufacture,4 and the Minnesota Pollution Control Agency stating that concentrations of airborne methamphetamine were in the range 0.1–1 μg m−3 in former clandestine laboratories.5–7 Based on this information, the development of sampling systems for analyzing methamphetamine in air at former clandestine laboratories requires calibration methods that can deliver methamphetamine vapour at concentrations in the low μg m−3 range. We therefore developed a system that would generate methamphetamine vapour with a concentration near 1 μg m−3, since this is in the range expected for a former clandestine methamphetamine laboratory.
Previous systems for generating methamphetamine vapour have been designed to test integrating samplers such as acid-treated filters, and so deliver a known amount of methamphetamine, although the actual concentration in such a system will vary with time.8 Most controlled-concentration vapour generation systems have been designed for volatile or semivolatile liquids. Such systems can introduce the analytes into the gas stream via permeation tubes, bubblers, or syringe pumps.9 Semivolatile solids have been equilibrated with a volume of air and then this air has been diluted in a gas stream.10 However, this approach did not seem appropriate for methamphetamine given its low vapour pressure and tendency to adsorb on surfaces. Instead, an alternative approach reported by Edmiston et al. where the analyte was dissolved in a carrier solvent and then injected into a gas stream was investigated,10 using a syringe-pump sample introduction into a system based on that reported by Koziel et al.9 but modified based on recommendations by Johnson et al.11
Experimental
Methamphetamine hydrochloride (>98%, Australian Government National Measurement Institute) was obtained from the Institute for Environmental Science and Research (ESR) and DL-methamphetamine-d9 (100 μg mL−1 ISOTEC 99%) was obtained from Sigma Aldrich. All concentrations below are on a methamphetamine free base basis.A solution of methamphetamine free base was prepared in the following manner: approximately 1 mL of a 4% mass/volume aqueous solution of sodium hydroxide (99%, ECP) was added to 1 mL of 1 mg mL−1 methamphetamine hydrochloride in dichloromethane (99.9% Environmental Control Products) in a glass screw-cap 16 mm diameter by 100 mm long culture tube (Kimax, Thermofisher). The two-phase solution was vortex-mixed for 3 min and centrifuged at ∼990 rpm (rcf ∼ 87 g) for 5 min. The bottom layer containing dichloromethane was transferred to another culture tube. Dichloromethane (1 mL) was added to the remaining sodium hydroxide solution and the process repeated. The combined dichloromethane extracts were passed through a drying column of anhydrous sodium sulfate (99%, Environmental Control Products), supported on pesticide-grade glass wool (Supelco, Sigma Aldrich), and were evaporated at 26 °C under a gentle flow of nitrogen to ∼1 mL, then transferred to a 10 mL volumetric flask and made up to volume with acetonitrile (AR, Univar, Ajax), giving a final concentration of 100 μg mL−1 methamphetamine base.
The internal standard d9-methamphetamine solution was prepared in a similar way. However, as the d9-methamphetamine was supplied as a 100 μg mL−1 solution in methanol, 250 μL of this solution was added to dichloromethane at the base extraction stage and the final evaporation was carried out in a GC vial insert almost to dryness, after which 250 μL acetonitrile was added to make ∼100 μg mL−1 d9-methamphetamine base. Recovery over the extraction process was tested by liquid injection GC-MS by comparison of peak area with the non-deuterated methamphetamine acetonitrile solution.
Apparatus
A diagram and photographs of the apparatus used to produce constant concentrations of methamphetamine vapour are shown in Fig. 1 and 2.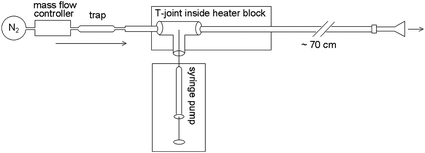
 | ||
| Fig. 2 Vapour-generation system, comprising mass flow controller, vaporisation unit and syringe pump. Image a shows the generator connected to a 1.5 L gas sampling tube into which the dynamic SPME sampler is inserted, while image (b) shows the generator connected to a glass funnel. | ||
The nitrogen carrier gas (99.998%, oxygen-free, BOC) flow was controlled with an Alicat mass flow controller MC-5SLPM-D (Instrumatics), and a contaminant trap (Anasorb/Tenax 226-171 SKC sorbent tube, Thermofisher), was installed downstream of the mass flow controller. Components were coupled together using stainless steel ¼′′ adapters, male ¼′′ to male NPT connectors, ¼′′ to ¼′′ unions, ⅜′′ polytetrafluoroethylene (PTFE) ferrules, and perfluoroalkoxy (PFA) tubing ¼′′ outer diameter (OD), obtained from Swagelok. The gas flow rate was set at 2 L min−1 and was checked independently with a TSI 4100 series flow meter (Thermofisher).
The vapour generation unit in the vaporisation system was constructed from two lengths of Restek Silcosteel®-CR treated ⅜′′ OD, 0.277′′ inner diameter (ID) 316L grade stainless steel tubing (Shimadzu Scientific Instruments) connected to opposing ends of a ⅜′′ by ⅜′′ by ¼′′ reducing union tee (Swagelok™) containing a glass dispersion rod with a machined flat based on a design by Johnson et al.11 The vapour generation unit was connected to PFA tubing or a glass funnel with ⅜′′ to ¼′′ reducing unions and PTFE ferrules. A trimmed Agilent BTO septum (Analytical Consumables) in the small tube of the union tee acted as the syringe needle port. The vapour generation unit was contained within an aluminium block (Fig. 2) that was heated by a hotplate (IKA RCT basic) while the temperature in the Al block was monitored independently with a Greisinger GTH 175/Pt digital thermometer probe. The Al block was held at 120 °C for most of the experiments with the glass mixing chamber and short outlet tube, and was held at 185 °C for the final set of experiments including those involving pre-dosing fibres with d9-methamphetamine. Methamphetamine solutions were dispensed using 50 μL SGE gastight syringes (Phenomenex) in a Multi-phaser NE-1000 syringe pump (New Era Pump Systems Inc.).
Experiments comparing dynamic and passive SPME configurations were carried out using a custom-made ∼1.5 L glass mixing chamber (Fig. 2, left photograph), coupled to the end of the vapour generation unit with PFA tubing and Swagelok™ fittings. The dynamic SPME sampler (see ESI,† for construction and operation details) could be inserted so that its inlet was in the center of this mixing chamber. The mixing chamber also had septa on its periphery for the insertion of standard SPME fibres for passive sampling. All SPME experiments used PDMS fibres (100 μm PDMS 57342-U, Supelco). Due to carryover in this system, for the experiments involving dosing with both methamphetamine and d9-methamphetamine, the PFA tubing downstream of the volatiliser and the mixing chamber were replaced with a small glass funnel with a ∼⅜′′ OD neck, fitted to the end of the vapour generation assembly using a Swagelok™ ⅜′′ adapter and PTFE ferrules. In this case, the dynamic sampler inlet was inserted as far inside the funnel as possible, without making a seal (Fig. 2, right photograph). A duplicate vapour generator ending in a small glass funnel was constructed for use with d9-methamphetamine for the internal standard experiments. This abbreviated form of the vapour generation unit initially resulted in elevated gas temperatures at the sampling point (41 °C under the above conditions), so the outlet Silcosteel® tubing was lengthened from 15 cm to 70 cm, resulting in an exit gas temperature of 26 °C.
Procedures
The temperature of the vapour generator was allowed to stabilize, then solutions of either 24 μg mL−1 or 100 μg mL−1 methamphetamine base in acetonitrile were injected at a rate of 5 μL h−1 into nitrogen flowing at 2 L min−1, producing a concentration of 1 μg m−3 or 4.2 μg m−3. The system was allowed to stabilise for ∼3 h and the achievement of a steady-state methamphetamine concentration was confirmed by SPME sampling and GC-MS analysis at 20 min intervals. A typical run of 8 h passed 40 μL of acetonitrile and either ca. 1 or 4 μg of methamphetamine through the system.The dynamic SPME sampler was inserted into either the mixing chamber or the funnel outlet and gas was drawn through the sampler at 1 L min−1, giving an average flow velocity of 0.433 m s−1 inside the Silcosteel® tube. A SPME fibre (conditioned in a GC inlet at 250 °C prior to sampling) was inserted into the sampler and exposed to the vapour stream. Exposure was timed to the second using a digital wristwatch. The SPME fibre was retracted into its needle, then the fibre holder was decoupled from the sampler and taken to the GC-MS for analysis.
SPME-GC-MS analysis
Gas chromatography-mass spectrometry (GC-MS) was used for quantitation of analyte absorbed on the SPME fibres. The GC-MS instrument parameters were based on published guidelines for SPME-GC-MS.12 Samples were analysed on an HP 6890 gas chromatograph with a HP 5973 mass spectrometer or on an Agilent 7890 gas chromatograph with an Agilent 5975C XL MSD with a triple-axis detector in positive-ion electron-impact (EI) ionisation mode, at 70 eV electron energy, with electron multiplier (EM) voltage +12 V relative to autotune value. Samples were injected manually through an Agilent BTO septum at 250 °C in splitless mode using a 0.75 mm internal diameter deactivated glass SPME liner (Supelco), with a purge flow of 30 mL min−1 after 1.5 min. The column was a 30 m HP-5MS 0.25 mm internal diameter, 0.25 μm stationary phase with instrument-grade helium (BOC) as the carrier gas, with a constant flow rate of 1 mL min−1 and initial pressure of 7.5 psi, giving a linear velocity of 36 cm s−1. The GC oven temperature program started at 40 °C held isothermal for 2.5 min, increased 40 °C min−1 to 300 °C and held 3 min. The transfer line to the MSD was 300 °C, with source at 230 °C and quadrupole at 150 °C. Mass spectra were acquired in scan mode from 38–300 amu, with detection threshold set to zero. After SPME fibre desorption in the GC inlet, the fibre was left in the inlet during the chromatographic separation to ensure it was clean for the next analysis.Peak integration and identification was carried out using MSD Chemstation D01.02 (Agilent) and the NIST Mass Spectral Library (2008). Peak areas for quantitation were measured from extracted ion chromatograms for the main ion fragments of underivatised methamphetamine (58 amu) and d9-methamphetamine (65 amu).
Results and discussion
Previous methods to generate methamphetamine vapour for analytical analysis have focused on volatilisation of a known amount of methamphetamine and have looked at an integrated collected amount.8 In contrast, calibration of an SPME or similar sampler requires constant concentrations of methamphetamine vapour over extended periods of time. We modeled our dosing system on a recently-reported system for generating trinitrotoluene (TNT) vapour, in which the analyte was dissolved in an organic solvent and then slowly injected into a gas stream.10 The reported advantage of this approach was that it could generate steady-state concentrations of TNT vapour, even though TNT is a solid at room temperature. This vapour generation method requires having a solvent present at concentrations approximately a thousand-fold higher (by mass) than the analyte of interest at the sampling point, but as long as this does not interfere with the analysis then it is a pragmatic approach to obtaining steady state vapour concentrations of a semi-volatile solid. We incorporated a vapour generation unit based on a Swagelok™ T adapter containing a cylinder insert with a machined flat as reported by Johnson et al., with the expectation that this would increase the effectiveness of the volatilisation step.11 We modified the reported design by using a glass insert that was free-moving in the adapter.For most experiments reported, methamphetamine base was prepared by basification of methamphetamine hydrochloride followed by transfer into acetonitrile, a solvent that is compatible with the PDMS coating on the SPME fibre.13 We also performed a limited number of experiments using heptane and water as carrier solvents.
The behaviour of the vapour generation unit was first characterized using solutions of tetradecane in heptane. These showed that stable concentrations of tetradecane could be obtained after a short equilibration time, and that there was little retention of hydrocarbon analytes within the vapour generation unit or the downstream apparatus. A GC-MS peak due to diphenylsulfide was identified as originating from the mass flow controller, so a hydrocarbon absorber was placed immediately upstream of the vapour generator.
The time dependence of methamphetamine absorption onto PDMS SPME fibres was investigated by exposing fibres in the dynamic air sampler to a gas stream containing 4.2 μg m−3 methamphetamine for 1–120 min. The increase in methamphetamine peak area for this concentration over time was curvilinear, with near-linear behaviour between 5–60 min (Fig. 3).
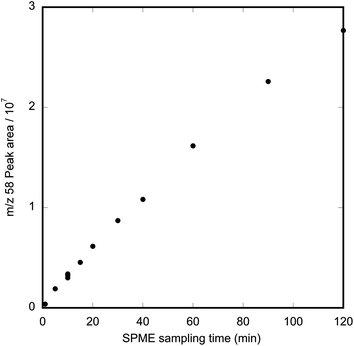 | ||
| Fig. 3 Graph of peak area for methamphetamine (m/z 58) as a function of exposure time when a PDMS SPME fibre was exposed to a gas stream containing 4.2 μg m−3 methamphetamine. | ||
The absorption of methamphetamine to PDMS did not reach steady state within 120 min. The almost linear relationship is typical of pre-equilibrium SPME behaviour and can be exploited as a means of calibration.14 Similar pre-equilibrium behaviour has been reported previously for sampling with the SPME fibre parallel to the airflow.15 The fact that methamphetamine on the fibre is well below equilibrium concentrations for sampling times less than 2 h means that, under our sampling regime, there is little danger of analyte competition or displacement; the PDMS is acting as an infinite sink for methamphetamine.
A solution of 24 μg mL−1 methamphetamine in acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.2) was prepared from the 100 μg mL−1 methamphetamine in acetonitrile solution and dispensed under the same conditions as the previous solution, to produce a methamphetamine vapour concentration of 1 μg m−3 in nitrogen. A linear increase in methamphetamine absorbed on the SPME fibre was observed over 90 min, indicating that vapour generation and SPME absorption were not affected by the use of an aqueous solution (Fig. 4).
:3.2) was prepared from the 100 μg mL−1 methamphetamine in acetonitrile solution and dispensed under the same conditions as the previous solution, to produce a methamphetamine vapour concentration of 1 μg m−3 in nitrogen. A linear increase in methamphetamine absorbed on the SPME fibre was observed over 90 min, indicating that vapour generation and SPME absorption were not affected by the use of an aqueous solution (Fig. 4).
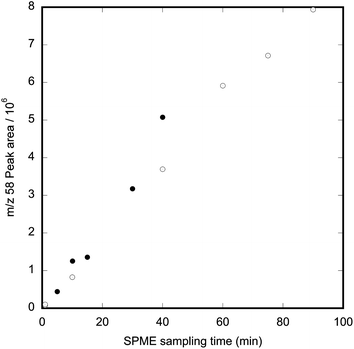 | ||
| Fig. 4 Relationship between exposure time and methamphetamine ion-fragment abundance when a PDMS fibre is exposed to 1 μg m−3 methamphetamine vapour using an injection solvent of 1:3.2 acetonitrile:water. The different symbols show data collected on sequential days. | ||
Repeated 20 min exposures of two PDMS SPME fibres in the dynamic air sampler to methamphetamine vapour at 4.2 μg m−3 methamphetamine resulted in intra-fibre relative standard deviations of 9% (n = 3) and 6% (n = 3). An estimate of inter-fibre variability obtained by measuring the difference in mean response between these two fibres during the same experimental run gave a relative deviation of 7% (n = 2). The inherent variability in SPME makes internal standard methods preferable to external calibration, especially in situations where multiple fibres are to be used for field sampling.
The stability and retention of methamphetamine on PDMS SPME fibres was characterised by pre-loading fibres with methamphetamine in the vapour dosing system (4.2 μg m−3 methamphetamine at 2 L min−1, sampled with the dynamic sampler at 1 L min−1 for 40 min), then exposing them to a flow of uncontaminated air at 1 L min−1 in the dynamic sampler for 5–40 min. Control samples were obtained by exposing SPME fibres in the dynamic sampler to an 4.2 μg m−3 methamphetamine for 40 min and then analyzing them immediately. Before conducting these desorption tests, a control SPME sample using the dynamic sampler within the fume hood being used for these experiments showed there was no detectable background methamphetamine contamination.
The results in Fig. 5 show that methamphetamine is not lost from PDMS SPME fibres when they are exposed to a 1 L min−1 airstream devoid of methamphetamine for up to 90 min. This observation suggests that the methamphetamine on the fibre represents an integration of the concentrations of methamphetamine to which the fibre has been exposed. Experiments in which the SPME fibres were removed from the sampler and stored in Silcosteel® field sample tubes showed that under these conditions methamphetamine was retained at initial levels for at least 5 h.
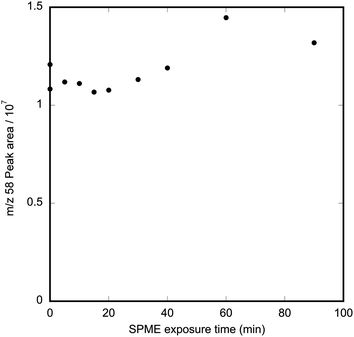 | ||
| Fig. 5 Graph showing effect of length of time of exposure to laboratory air at 1 L min−1 on methamphetamine retained on PDMS SPME fibres. Fibres were pre-loaded by exposure to 4.2 μg m−3 methamphetamine at 1 L min−1 for 40 min. | ||
Based on the above results with the methamphetamine vapour generation apparatus, we were able to estimate the airborne methamphetamine vapour concentrations at former clandestine laboratories that had been noted to have detectable airborne methamphetamine using the dynamic SPME sampler prior to construction of the vapour generator. If we assumed that the SPME fibres used had similar absorption behaviour to those used to characterize the vapour generator and similar response from the GC-MS, then the m/z 58 peak areas obtained after 10 min dynamic sampling corresponded to methamphetamine concentrations of ca. 3 and 0.2 μg m−3 at one site before and after cleaning respectively, and 0.6 μg m−3 at another site after cleaning.
Deuterated methamphetamine as an internal standard
The observed absorption and desorption characteristics of methamphetamine on PDMS SPME fibres suggested that isotopically substituted methamphetamine could be used as an internal standard when sampling for airborne methamphetamine using SPME. This would be advantageous, because as noted above there can be differences in the adsorption capacities of SPME fibres especially once they have been in use. We investigated this use of an isotopically-substituted internal standard by using vapour generation systems to load SPME fibres first with d9-methamphetamine and then with methamphetamine and also with the isotopologues loaded in the reverse order.The first attempts at introducing d9-methamphetamine into the vapour generation apparatus with the glass sampling reservoir showed that there was retention of a small amount of strongly but reversibly bound methamphetamine within the system. Thus, if methamphetamine was dosed into the system until steady state was reached and then the syringe was removed from the injection block, the amount of methamphetamine absorbed on an SPME fibre inserted into the outlet of the vapour generator decreased, with a half life of about 3 h. However, if d9-methamphetamine was then injected into the injection block, increased levels of unlabeled methamphetamine were detectable by SPME. Indeed, with the glass reservoir the unlabeled methamphetamine could exceed the amount of d9-methamphetamine on the SPME fibres in the first hour after changeover. This retention was greatly reduced by changing to a straight pipe outlet ending in a funnel, for which the half-time for a step change in methamphetamine concentration was about 10 min. However, some retention still occurred as shown in Fig. 6, where introduction of d9-methamphetamine is causing an increase in the signal at m/z 58 due to unlabeled methamphetamine. It should be noted that the mass spectrum for d9-methamphetamine has no significant peak at m/z 58. Because of this retention of methamphetamine within the system, the results for pre-loading d9-methamphetamine shown below were obtained using separate vapour generators for d9-methamphetamine and methamphetamine.
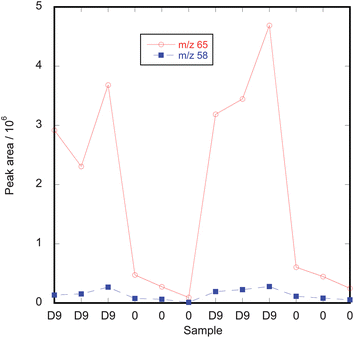 | ||
| Fig. 6 Effect of injecting d9-methamphetamine to give a concentration of 4.2 μg m−3 into a vapour generation system that had been previously used to generate 4.2 μg m−3 methamphetamine. Each data point corresponds to a sequential 20 min SPME measurement. The m/z 58 and m/z 65 peaks correspond to unlabeled methamphetamine and d9-methamphetamine respectively. “D9” on the Sample axis refers to times when d9-methamphetamine was being introduced to the system while “0” refers to times when the syringe was removed from the vapour generator. | ||
SPME fibres were exposed to 4.2 μg m−3 methamphetamine in the dynamic sampler for 20 min, retracted and kept at room temperature for 3–5 h while the dosing system was equilibrated with 4.2 μg m−3 d9-methamphetamine, then the fibres were exposed to the vapour stream for 20 min before being analysed by GC-MS. Controls were also collected to confirm peak areas for the 20 min exposure times. Repeated 20 min exposures of two PDMS SPME fibres in one of the funnel-terminated vapour generation systems resulted in intra-fibre relative standard deviations of 6% (n = 10) and 6% (n = 10). These two fibres had very similar absorption behavior for methamphetamine, with the means of the two data sets only differing by 2%.
The results in Table 1 show that when a 100 μm PDMS fibre is exposed to 4.2 μg m−3 methamphetamine for 20 min, then exposed to 4.2 μg m−3 d9-methamphetamine for 20 min, the peak areas corresponding to each analyte detected by GC-MS are very similar. These results show that in a dynamic pre-equilibrium system, 100 μm PDMS SPME fibres can be spiked with a controlled amount of isotopically substituted methamphetamine internal standard before field sampling, without loss of standard and without competitive displacement.
| Peak area ion 65/106 | Peak area ion 58/106 | Ratio | |
|---|---|---|---|
| Methamphetamine then d9-methamphetamine | 2.05 | 1.84 | 1.1 |
| Methamphetamine then d9-methamphetamine | 2.73 | 2.49 | 1.1 |
| Methamphetamine then d9-methamphetamine | 2.84 | 3.11 | 0.9 |
| d9-Methamphetamine then methamphetamine | 1.52 | 1.56 | 1.0 |
| d9-Methamphetamine then methamphetamine | 1.80 | 1.79 | 1.0 |
| d9-Methamphetamine then methamphetamine | 1.37 | 1.38 | 1.0 |
One minor problem with our choice of internal standard is that there is a small amount of interference by the GC-MS response of unlabeled methamphetamine on d9-methamphetamine. The chromatographic peaks for methamphetamine and d9-methamphetamine are close together (0.02 min apart) and overlap at their base. The mass spectrum of unlabelled methamphetamine includes an ion fragment at m/z 65 of sufficient intensity to produce a shoulder on the m/z 65 peak for d9-methamphetamine. This phenomenon has been reported previously for methamphetamine and its deuterated isotopologues.16 The ratio of the m/z 65 and 58 peaks for methamphetamine was constant (0.045, with a standard deviation of 0.001; n = 27) so the m/z 65 peak areas for d9-methamphetamine could be corrected for the small contribution from methamphetamine.
Conclusions
Using a modified vapour-dosing system based on systems reported by Johnson et al.11 and Koziel et al.,9 together with a dynamic SPME air sampler, we have demonstrated that SPME can be used successfully for the quantitation of methamphetamine vapour at sub-ppb levels. Methamphetamine is sufficiently strongly absorbed onto PDMS fibres that it is possible to pre-load fibres with isotopically substituted methamphetamine at a known vapour concentration and then use that as an internal standard for dynamic SPME sampling performed up to 5 h afterwards. The strong but reversible adsorption of methamphetamine inside the vapour generator meant that it was impractical to dose SPME fibres with unlabeled and isotopically substituted methamphetamine in the same vapour dosing block, so duplicate dosing systems were constructed. For methamphetamine air concentrations below 1 μg m−3, it should be possible to increase the sensitivity of the dynamic SPME sampler by increasing the air sampling pump rate (for the same sampling time) or by increasing the length of time that vapour is sampled.15Acknowledgements
The authors would like to thank N. Lloyd (University of Auckland), for advice on experimental design, and C. Hughes, M. J. Wadsworth, and A. J. Mead (University of Auckland), for fabrication of apparatus.References
- Ministry of Health, Guidelines for the Remediation of Clandestine Methamphetamine Laboratory Sites, Ministry of Health, Wellington, 2010 Search PubMed.
- M. V. Van Dyke, K. A. Serrano, S. Kofford, J. Contreras and J. W. Martyny, J. Occup. Environ. Hyg., 2011, 8, 636–641 CrossRef CAS.
- J. W. Martyny, S. L. Arbuckle, C. S. McCammon Jr, E. J. Esswein, N. Erb and M. Van Dyke, J. Chem. Health Saf., 2007, 14, 40–52 CrossRef CAS.
- M. Van Dyke, N. Erb, S. Arbuckle and J. Martyny, J. Occup. Environ. Hyg., 2009, 6, 82–89 CrossRef CAS.
- Minnesota Pollution Control Agency, Data Review – Delavan Meth in Air Sampling, Minnesota Pollution Control Agency, 2007, accessed from, http://proteus.pca.state.mn.us/cleanup/meth.html on 29 April 2008 Search PubMed.
- Minnesota Pollution Control Agency, Personal Air Sampling, Minnesota Pollution Control Agency, 2007, accessed from http://proteus.pca.state.mn.us/cleanup/meth.html on 29 April 2008 Search PubMed.
- Minnesota Pollution Control Agency, Data Review – St. Peter Meth in Air Sampling, Minnesota Pollution Control Agency, 2007, accessed from http://proteus.pca.state.mn.us/cleanup/meth.html on 29 April 2008 Search PubMed.
- P. C. Raynor and T. Carmody, Final Report: Meth Labs Sampling: Air and HVAC Systems. Minnesota Pollution Control Agency CFMS No. A-79651, University of Minnesota, Minneapolis, 2006 Search PubMed.
- J. A. Koziel, P. A. Martos and J. Pawliszyn, J. Chromatogr., A, 2004, 1025, 3–9 CrossRef CAS.
- P. L. Edmiston, D. P. Campbell, D. S. Gottfried, J. Baughman and M. M. Timmers, Sens. Actuators, B, 2010, 143, 574–582 CrossRef CAS.
- T. J. Johnson, S. W. Sharpe and M. A. Covert, Rev. Sci. Instrum., 2006, 77, 094103 CrossRef.
- J. J. Langenfeld, S. B. Hawthorne and D. J. Miller, J. Chromatogr., A, 1996, 740, 139–145 CrossRef CAS.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS.
- J. Ai, in Applications of Solid Phase Microextraction, ed. J. Pawliszyn, The Royal Society of Chemistry, Cambridge, UK, 1999, pp. 22–37 Search PubMed.
- E. Gómez Alvarez, M. V. Moreno, S. Gligorovski, H. Wortham and M. V. Cases, Talanta, 2012, 88, 252–258 CrossRef.
- R. H. Liu, S.-M. Wang and D. V. Canfield, Quantitation and Mass Spectrometric Data of Drugs and Isotopically Labeled Analogs, CRC Press, Boca Raton, Florida, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of construction and operation of dynamic SPME sampler. See DOI: 10.1039/c3ay40536b |
| This journal is © The Royal Society of Chemistry 2013 |