 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUrine adulteration: can bleach be used to mask MDMA use?
Annie Q. N.
Pham
a,
Tamsin
Kelly
b and
Shanlin
Fu
*a
aCentre for Forensic Science, School of Chemistry and Forensic Science, University of Technology, Sydney (UTS), Ultimo, NSW, Australia 2007. E-mail: shanlin.fu@uts.edu.au
bNational Centre for Forensic Studies, Faculty of Education, Science, Technology and Mathematics, University of Canberra (UC), Bruce, ACT, Australia 2601
First published on 14th June 2013
Abstract
Concerns regarding specimen integrity have long been a major issue of urine drug testing due to acts of urine adulteration. At a high concentration, in vitro urine adulteration using sodium hypochlorite (bleach) produced false-negative results for 3,4-methylenedioxymethamphetamine (MDMA) in CEDIA® immunoassay screening with strong negative readings. However, these strong negative readings may act as a warning sign for further investigation of the sample where the detection of a unique marker in the form of N-chloroMDMA will suggest urine adulteration via bleach. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) identified N-chloroMDMA is a major product formed between hypochlorite and MDMA in urine. N-ChloroMDMA was found stable at 4 °C for at least 10 h, but decomposed over time at room temperature (20 °C) with MDMA being identified as one of its main decomposition products.
1 Introduction
According to the Australian Crime Commission Illicit Drug Data Report 2010–11,1 MDMA (3,4-methylenedioxymethamphetamine or ‘ecstasy’) was and remained the second most commonly used illicit drug in Australia, after cannabis.The act of adulteration has long been the primary challenge for urine drug testing. Such methods of urine adulteration (in vivo, in vitro and substitution2–6) are carried out by individuals in an effort to “beat” a urine drug test, and thus, avoid detection of their drug use. Chemical (in vitro) adulteration—the primary focus of this study—involves the addition of foreign substances or chemicals (e.g.sodium chloride, liquid hand soap, vinegar, Visine® eye drops,4–8 ‘Urine Luck’, ‘Stealth’, ‘Klear’ and ‘UrineAid’2,6) to a urine specimen after it is collected. The presence of these foreign substances acts to either interfere with the screening and/or confirmatory methods,2 or convert the targeted drug to compounds which are not detected in the screening or confirmatory methods.5
Currently, the existing methods used to determine the integrity of a urine sample only allow for the detection of adulterated samples (e.g. commercial adulterant dipstick indicators2 and sample integrity tests2,4,6). However, there are no means to reveal what drug(s) of abuse was masked via adulteration. There is limited literature regarding chemical adulterants and their effects on the screening and/or confirmatory methods for amphetamine-type stimulants (ATS), let alone a study purely focused on the chemical adulteration of MDMA in urine. In this study, research was conducted into the chemical adulteration of MDMA in urine using sodium hypochlorite (NaOCl) – the active ingredient of bleach and a popular chemical adulterant. Of primary focus, the chemical reaction between NaOCl and MDMA was investigated to identify stable reaction products. The potential of using these identified stable reaction products as markers of MDMA abuse when urine specimens have been adulterated with bleach takes on an alternative and novel approach in the fight against urine adulteration. The effect of bleach on the routine drug analysis of MDMA in urine was also investigated.
2 Experimental
2.1 Materials and reagents
All reagents were of analytical grade unless indicated otherwise. MDMA hydrochloride (1 mg mL−1 in methanol) was sourced from Alltech-Applied Sciences (State College, PA, USA). Deuterated MDMA (MDMA-d5, 1 mg mL−1 in methanol) was supplied from Cerilliant (Austin, TX, USA). Working standard solutions of MDMA (100, 10 and 5 μg mL−1) and MDMA-d5 (10 μg mL−1) were prepared by appropriate dilution with methanol. Pentafluoropropionic acid (PFPA, 99%) and two strengths of sodium hypochlorite (NaOCl) were purchased from Sigma-Aldrich (St Louis, MO, USA). The stronger NaOCl reagent (referred to as “strong NaOCl”) contained 10–15% available chlorine, while the other (referred to as “weak NaOCl”) contained 5–10% available chlorine. NaOCl concentration was determined spectrophotometrically at 0.319 M (“strong NaOCl”) and 0.029 M (“weak NaOCl”) using a molar absorbance coefficient (ε) of 350 M−1 cm−1 for the absorbance of −OCl at 292 nm.9 A stock solution of 1 M ammonium formate was prepared and stored in a refrigerator (4 °C) before use.Blank urine samples were sourced from healthy individuals. Real urine samples positive for MDMA were obtained from the Toxicology Unit, Pacific Medicine Laboratory Services (PaLMS), and the Toxicology and Forensic Unit, ACT Government Analytical Laboratory (ACTGAL) following removal of sample identification. All urine samples, along with all standards and solutions when not in use, were stored in a refrigerator (4 °C).
2.2 LC-MS/MS
The Agilent MassHunter Data Acquisition Software (version B.02.00, Agilent Technologies Inc., Palo Alto, CA, USA) was used to operate the LC-MS/MS instrumentation, consisting of the Agilent 1200 series LC system connected to the Agilent 6460 Triple Quad LC/MS via an electrospray ionisation (ESI) interface. Chromatographic separation was achieved on an XBridge (Waters Corporation, Ireland) C18 column (150 mm × 4.6 mm, 3.5 μm) maintained at 25 °C at a flow rate of 0.3 mL min−1. Gradient elution was employed using the mobile phases of 2 mM ammonium formate solution in water (solvent A) and acetonitrile (solvent B). The gradient elution profile was as follows: solvent B increased from 40% to 70% in 10 min and maintained at 70% for 4 min; followed by 95% solvent B for 6 min before equilibrating with 40% solvent B for 10 min totalling a run time of 30 min. The following MS conditions were used; fragmentor voltage 90 V; gas flow 10 L min−1 for both drying gas (300 °C) and sheath gas (350 °C), nebulizer gas pressure 35 psi; capillary voltage 3500 V and nozzle voltage 1000 V. The injection volume was 1 μL and samples were analysed in full scan mode (m/z 50–450) or selected reaction monitoring (SRM) mode using positive ionisation. Three SRM transitions (Table 1) were optimised and monitored for MDMA and the reaction product, which was later identified (refer to Section 3.2).| Product | Precursor ion m/z | Product ion m/z | Collision energy (eV) |
|---|---|---|---|
| MDMA | 194 | 163 | 6 |
| 194 | 135 | 20 | |
| 194 | 105 | 20 | |
| Reaction product (N-chloroMDMA) | 228 | 163 | 6 |
| 228 | 135 | 20 | |
| 228 | 105 | 25 |
| Kinetic experiment | Composition of stock reaction mixturesb | Final MDMA conc. (μg mL−1) | Final NaOCl conc. (×10−2 M) | Auto sampler temp. (°C) |
|---|---|---|---|---|
| a Each experiment was performed for 5 h, except Kinetic Experiment 4 which was performed over 10 h. b Weak NaOCl: 5–10% chlorine; strong NaOCl: 10–15%. | ||||
| 1 | 10 μL of 10 μg mL−1 MDMA reacted with 49.5 μL weak NaOCl in 40.5 μL of urine | 1 | 1.4 | 20 |
| 2 | 10 μL of 10 μg mL−1 MDMA reacted with 30 μL strong NaOCl in 60 μL of urine | 1 | 9.6 | 20 |
| 3 | 10 μL of 100 μg mL−1 MDMA reacted with 30 μL strong NaOCl in 60 μL of urine | 10 | 9.6 | 20 |
| 4 | 10 μL of 100 μg mL−1 MDMA reacted with 30 μL strong NaOCl in 60 μL of urine | 10 | 9.6 | 4 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50% B) was continuously infused through a ‘tee’ using a Harvard Apparatus (South Natick, Massachusetts, USA) syringe pump set at a flow rate of 25 μL min−1 and syringe diameter of 4.3 mm. The following 1 μL injections were injected into the LC system: mobile phase, blank urine, blank urine containing 9.6 × 10−2 M NaOCl (i.e. 30 μL strong NaOCl reacted with 70 μL blank urine) and blank urine containing 4.5 × 10−2 M NaOH (i.e. 30 μL of 0.15 M NaOH reacted with 70 μL blank urine). Blank urine from two donors was used for the matrix effects experiment. All samples were analysed in SRM mode.
:50% B) was continuously infused through a ‘tee’ using a Harvard Apparatus (South Natick, Massachusetts, USA) syringe pump set at a flow rate of 25 μL min−1 and syringe diameter of 4.3 mm. The following 1 μL injections were injected into the LC system: mobile phase, blank urine, blank urine containing 9.6 × 10−2 M NaOCl (i.e. 30 μL strong NaOCl reacted with 70 μL blank urine) and blank urine containing 4.5 × 10−2 M NaOH (i.e. 30 μL of 0.15 M NaOH reacted with 70 μL blank urine). Blank urine from two donors was used for the matrix effects experiment. All samples were analysed in SRM mode.
2.3 Immunoassay screening testing
Immunoassay screening testing was performed using the CEDIA®Amphetamine/Ecstasy Assay test kit together with 0.3 and 0.5 μg mL−1 calibrators supplied by Thermo Fisher Scientific (Scoresby, Vic, Australia). The CEDIA® screening was performed according to the manufacturer's instructions, on an Olympus AU 2700 analyser (Olympus America Inc., Melville, NY). The test samples consisted of blank urine spiked with MDMA and NaOCl. Three concentrations of MDMA (0.3, 1.0 and 10 μg mL−1) were tested using two concentrations of NaOCl (1.4 × 10−2 and 9.6 × 10−2 M). Additionally, a real urine sample containing MDMA obtained from PaLMS was adulterated with two lower concentrations of NaOCl (1.3 × 10−2 and 7.4 × 10−2 M). Appropriate control samples were prepared and differed from the reaction samples with the addition of 0.15 M NaOH in place of NaOCl. Each sample was prepared in triplicate, apart from blanks which were prepared in duplicate (Table 3).| Exp. # | Sample type | Urine vol. (μL) | Final MDMA conc. (μg mL−1) | Final NaOCl conc. (×10−2 M) | Final NaOH conc. (×10−2 M)b | Ave. assay reading (% RSD, n = 3) | Result (Pos or Neg) |
|---|---|---|---|---|---|---|---|
| a Sample produced a false-negative result. b All control samples were prepared by the addition of 0.15 M NaOH in place of NaOCl. i.e. for Experiments 1–3, 88 μL NaOCl or NaOH was added to samples ‘Reaction 1’ or ‘NaOH control 1’, respectively. Similarly, 600 μL NaOCl or NaOH was added to samples ‘Reaction 2’ or ‘NaOH control 2’, respectively. c For Experiment 4, an authentic urine sample containing 0.203 μg mL−1 MDMA (determined by the testing laboratory at the Toxicology Unit, PaLMS, NSW, Australia) was used. Samples ‘Reaction 1’ or ‘NaOH control 1’ had 22 μL NaOCl or NaOH added, respectively. Similarly, samples ‘Reaction 2’ or ‘NaOH control 2’ had 150 μL NaOCl or NaOH added, respectively. | |||||||
| 1 | Reaction 1 | 1852 | 0.30 | 1.4 | 0 | 1.12 (8.78) | Pos |
| Reaction 2 | 1340 | 0.30 | 9.6 | 0 | −0.83 (−4.89) | Nega | |
| NaOH control 1 | 1852 | 0.30 | 0 | 0.7 | 1.27 (1.98) | Pos | |
| NaOH control 2 | 1340 | 0.30 | 0 | 4.5 | 1.29 (4.32) | Pos | |
| MDMA control | 1940 | 0.30 | 0 | 0 | 1.22 (4.50) | Pos | |
| 2 | Reaction 1 | 1712 | 1.00 | 1.4 | 0 | 2.53 (4.68) | Pos |
| Reaction 2 | 1200 | 1.00 | 9.6 | 0 | −0.85 (0.00) | Nega | |
| NaOH control 1 | 1712 | 1.00 | 0 | 0.7 | 2.60 (0.54) | Pos | |
| NaOH control 2 | 1200 | 1.00 | 0 | 4.5 | 2.70 (1.61) | Pos | |
| MDMA control | 1800 | 1.00 | 0 | 0 | 2.62 (2.50) | Pos | |
| 3 | Reaction 1 | 1712 | 10.00 | 1.4 | 0 | 3.32 (1.98) | Pos |
| Reaction 2 | 1200 | 10.00 | 9.6 | 0 | −0.85 (0.00) | Nega | |
| NaOH control 1 | 1712 | 10.00 | 0 | 0.7 | 3.50 (1.29) | Pos | |
| NaOH control 2 | 1200 | 10.00 | 0 | 4.5 | 3.60 (0.98) | Pos | |
| MDMA control | 1800 | 10.00 | 0 | 0 | 3.48 (1.16) | Pos | |
| 4c | Reaction 1 | 500 | 0.19 | 1.3 | 0 | 0.59 (3.39) | Pos |
| Reaction 2 | 500 | 0.16 | 7.4 | 0 | −0.85 (0.00) | Nega | |
| NaOH control 1 | 500 | 0.19 | 0 | 0.6 | 1.70 (0.00) | Pos | |
| NaOH control 2 | 500 | 0.19 | 0 | 3.5 | 0.60 (0.00) | Pos | |
| Blanks | NaOCl 1 | 1912 | 0 | 1.4 | 0 | −0.05 (−141.42) | Neg |
| NaOCl 2 | 1400 | 0 | 9.6 | 0 | −0.85 (0.00) | Neg | |
| Urine | 2000 | 0 | 0 | 0 | 0.04 (0.00) | Neg | |
2.4 GC-MS confirmatory testing
3 Results and discussion
3.1 Reaction profile between MDMA and hypochlorite
Water provided a relatively matrix-free medium for studying the reaction. Hence, a greater concentration of NaOCl was required in its urinary reaction with MDMA. Comparing the LC-MS full scan chromatograms obtained for the relevant blanks and NaOCl reacted MDMA standards in both water and urine (Fig. 1) revealed there was one obvious major reaction product (Fig. 1c and d), eluting at approximately 18.5 min. Both reactions carried out in water and in urine appeared to completely oxidise MDMA in the sample, as indicated by the absence of a peak at approximately 9.7 min – the time at which MDMA eluted in both water (Fig. 1a) and in urine (Fig. 1b). A similar result was obtained when a real urine specimen containing MDMA was adulterated with NaOCl (data not shown).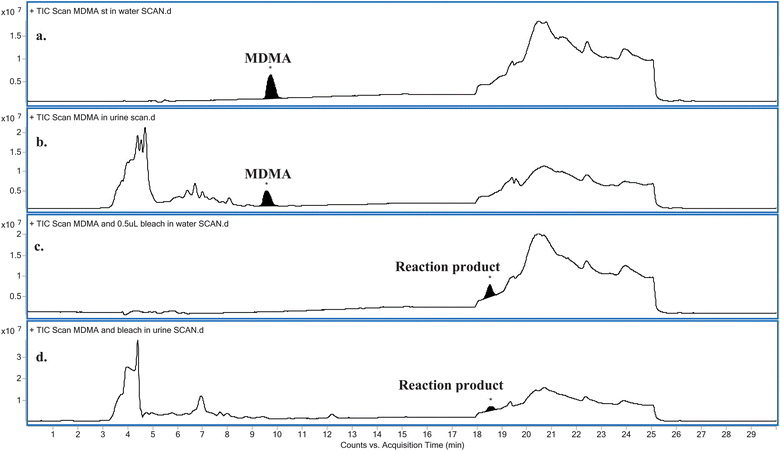 | ||
| Fig. 1 Comparison between the LC-MS full scan chromatograms obtained for: (a) MDMA in water, (b) MDMA in urine, (c) MDMA reacted with NaOCl in water and (d) MDMA reacted with NaOCl in urine. | ||
3.2 Identification of major reaction product(s)
Comparisons between the mass spectra obtained for the unknown reaction product from the reaction between MDMA and NaOCl performed in the two different matrices (i.e.water and urine), showed that they were the same product. The mass spectra obtained for MDMA and the unknown reaction product (Fig. 2) reveal similarities in terms of common fragmentation ions (m/z 58, 105, 135 and 163) found in both mass spectra. With regards to the unknown reaction product structural elucidation, the common fragmentation ions suggest it has a structure similar to MDMA. Apart from the fragmentation ions of m/z 58, 105, 135 (base peak) and 163, closer inspection of the mass spectrum also reveals an additional and hypothesised molecular ion [M + H]+ peak occurring at m/z 228 and a characteristic [M + H + 2]+ chlorine isotopic cluster observed (m/z 228 and m/z 230, respectively; Fig. 2c) indicative of a mono-chlorinated MDMA reaction product. There are only two possible positions at which the chlorine atom could be substituted onto the MDMA molecule: (i) either on the benzene ring or (ii) on the amine moiety of MDMA. Lewis et al.10 confirmed the identity of a chlorinated analog of MDMA as 2-chloroMDMA. In the study, the mass spectrum of 2-chloroMDMA exhibited the two ions of m/z 169 and 196, which showed the characteristic A + 2 chlorine isotopic cluster, both of which are absent in the mass spectrum results of this study. In a more recent study conducted by Maresova et al.,11 a chlorinated MDMA product, hypothesised as 6-chloroMDMA, was detected and similarly, showed the ion of m/z 169 exhibiting the characteristic A + 2 chlorine isotopic cluster. Thus, it was concluded that the unknown reaction product was most likely a chloramine species in the form of N-chloroMDMA (i.e. the chlorine substitution took place on the amine moiety of MDMA). The proposed structure and detailed fragmentation pattern of the reaction product are illustrated in Fig. 3. Product ion scan and precursor ion scan experiments (data not shown) supported the proposed fragmentation pathways.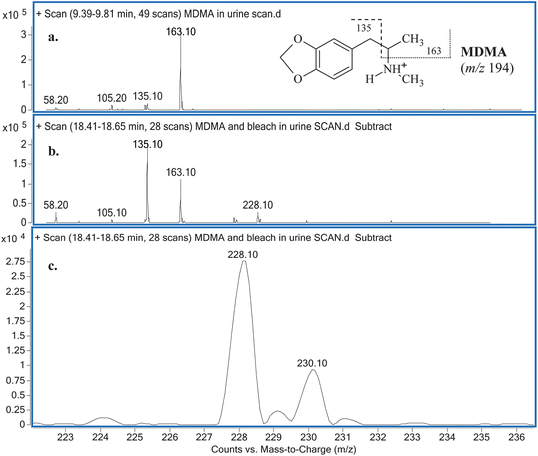 | ||
| Fig. 2 (a) MSspectrum obtained for MDMA in urine with its detailed fragmentation pattern (inset) and (b) MSspectrum obtained for the unknown reaction product with (c) an emphasis on ions m/z 228 and 230. | ||
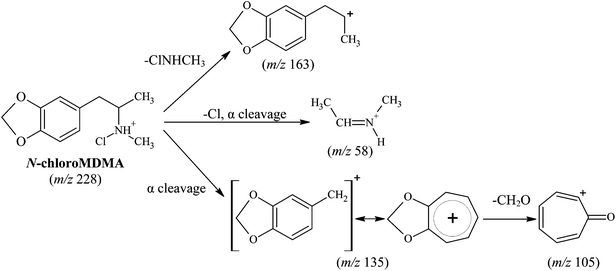 | ||
| Fig. 3 Proposed structure and fragmentation pattern for reaction product (N-chloroMDMA). | ||
3.3 Stability of N-chloroMDMA
The calibration obtained for the range of 0.15–1.00 μg mL−1 and 0.15–10.00 μg mL−1 achieved coefficient of determination (r2) values of 0.9697–0.9883 and 0.9961–0.9998, respectively. Across all kinetic studies, the reaction between NaOCl and MDMA was fast with N-chloroMDMA being formed after two minutes. The effects of temperature, MDMA concentration and NaOCl concentration were investigated. Results from Kinetic Experiment 3 (20 °C, i.e. room temperature) showed two evident trends: the peak area of N-chloroMDMA slowly decreased over time, while the concentration of MDMA slowly increased over time (Fig. 4). Approximately 41% of MDMA was detected and remained at the end of the 5 h kinetic experiment. Kinetic Experiment 4 (4 °C, i.e. refrigeration temperature) involved using blank urine from two different donors. The results were consistent with each other and collectively showed remarkably different findings from Kinetic Experiment 3. It was found that MDMA was destroyed and unquantifiable, but the peak area for N-chloroMDMA remained relatively stable (7–9% RSD) throughout both of the 10 h monitored periods (data not shown). Thus, this shows that N-chloroMDMA was unstable at room temperature and possibly reverted back into MDMA, but was relatively stable at refrigeration temperature. Potentially, this shows that N-chloroMDMA could be used as a marker of MDMA abuse when urine specimens have been adulterated with bleach, if the collected urine sample is stored at 4 °C immediately and up until analysis via LC-MS/MS.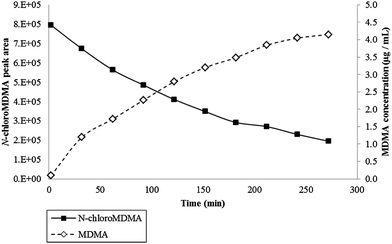 | ||
| Fig. 4 Data obtained for Kinetic Experiment 3 (i.e. 10 μL of the 100 μg mL−1 MDMA was reacted with 30 μL strong NaOCl in 60 μL of urine) performed at 20 °C for 5 h. Primary axis plots N-chloroMDMA peak area over time. Secondary axis plots MDMA concentration (μg mL−1) over time. | ||
In Kinetic Experiment 3, NaOCl was found to oxidise approximately 59% of the MDMA starting concentration (over a 5 h period). Although N-chloroMDMA was shown to decompose and possibly revert back into MDMA, this process cannot possibly account for the 59% alone. It is well documented in literature that many chloramine species are unstable and undergo various secondary reactions including decomposition and disproportionation.12–14 Thus, it is hypothesised that N-chloroMDMA decomposes to other products such as an aldehyde in addition to MDMA. However, no other products were detected under the experimental conditions employed. LC-MS/MS was also performed in negative ionisation mode, but failed to detect any additional reaction products from the reaction mixture. More research is required to investigate this issue.
Kinetic Experiments 1 and 2 involved a relatively low starting MDMA concentration of 1 μg mL−1. The results of Kinetic Experiment 1 (using a low NaOCl concentration) were similar to Kinetic Experiment 3 where the peak area of N-chloroMDMA slowly decreased over the 5 h period. For Kinetic Experiment 2 (using a high NaOCl concentration), N-chloroMDMA was detected in the first injection (i.e. two minutes into the reaction), but was not detected in the second injection (i.e. 32 minutes into the reaction) nor in any subsequent injections. In both experiments, MDMA was not quantifiable and NaOCl effectively oxidised MDMA to below the cut-off concentration for confirmatory testing (0.15 μg mL−1).15 However, N-chloroMDMA was detected in both experiments and this shows the potential of N-chloroMDMA being used as a marker of MDMA abuse in bleach adulterated urine specimens.
3.4 Matrix effects
Using continuous post-column infusion, it was clear that there was ion suppression encountered for the quantitation of MDMA. Although the post-column infusion method does not fully quantitate matrix effects compared to the method utilised by Matuszewski et al.,16,17 approximate ion suppression percentages were calculated as 8% for urine and 9.5% for both the addition of NaOH and NaOCl in urine. The agreement of the ion suppression percentages encountered for NaOH and NaOCl in urine supports the addition of NaOH in calibration standards in this study to calculate the concentration of MDMA remaining in reaction samples. Ion suppression was also evident at the retention time of N-chloroMDMA, but was not calculated as N-chloroMDMA was not quantified throughout this study.3.5 Effect of bleach on the routine drug analysis of MDMA in urine
In a majority of laboratories and for routine urine drug testing, immunoassays are often employed for screening tests, followed by GC-MS confirmatory testing.18,19 The lowest concentration utilised with the CEDIA® immunoassays in this study was 0.30 μg mL−1 MDMA as this is the recommended concentration cut-off value for the ATS screening tests.15 Only the use of the higher concentration of NaOCl effectively produced false-negative results across all samples (Table 3). Interestingly, the CEDIA® immunoassay readings for all the false-negative samples are similar to each other at approximately −0.85. Thus, a reading of −0.85 may potentially indicate a sample has been adulterated with a high concentration of bleach to conceal MDMA use and act as a warning sign for further investigation of the sample. The false-negative results produced in this study are consistent with findings from other similar studies. Bleach has previously been demonstrated to produce false-negative results across the EMIT7 (enzyme multiplied immunoassay technique) and FPIA20 (fluorescence polarisation immunoassay) systems. The primary mechanism of action of bleach is oxidation,2,4,5 and Mikkelson and Ash7 and Wu2 postulated that bleach interferes with EMIT through oxidising NADH, which decreases its absorbance at 340 nm. Additionally, the alkaline pH changes caused by bleach can alter binding and reaction rates,2,7 producing false-negative results for EMIT, CEDIA® and FPIA.2 The study conducted by Mikkelson and Ash7 also showed that bleach caused concentration-dependent interference for the EMIT for amphetamines. In a more recent study by Chou and Giang,20 bleach was evaluated as having a moderate to high potential to produce a false-negative result for FPIA for amphetamines.For GC-MS analysis, the calibration obtained in the range of 0.05–10.0 μg mL−1 achieved a r2 values of 0.999. The results summarised in Table 4 show that the time the samples were left to react did not have a significant effect on the final percentage loss of MDMA, with a reported 11% and 10% MDMA loss in GC-MS Experiments 1 and 2, and 24% and 20% loss in GC-MS Experiments 3 and 4. Although no false-negative results were obtained from these tests, a false negative result might be possible if the initial MDMA concentration is low and around the cut-off concentration at 0.15 μg mL−1. To test this, blank urine samples spiked with MDMA at 0.15 μg mL−1 were exposed to NaOCl at concentrations of 1.16 × 10−2 and 1.90 × 10−2 M. GC-MS analysis showed that NaOCl oxidised the starting MDMA concentrations significantly below the cut-off concentration value (data not shown), with a reported MDMA loss ranging from 53–64%. These oxidation percentages are similar to those reported by Chou et al.,21 who observed a 36–63% decrease in a sample's initial amphetamines (methamphetamine, amphetamine, MDMA and 3,4-methylenedioxyamphetamine) concentration with the use of bleach as an adulterant. In the study, the amphetamine concentrations in the urine samples were approximately 0.625 μg mL−1, representing 125% of the Taiwanese cut-off concentration value. Bleach effectively produced false-negative results for all MDMA samples and Chou and Giang20 attributed the effectiveness of bleach as an adulterant to its ability to degrade analytes and/or deactivate the derivatising agent through oxidation before and/or during sample preparation, in addition to the dilution effect from adding excess liquid to a sample.
| Initial MDMA conc. (μg mL−1) | Final NaOCl conc. (×10−2 M) | Exp. # | Time sample was left to react (min) | Final MDMA conc. (μg mL−1) | Percentage MDMA oxidised (%) |
|---|---|---|---|---|---|
| a All GC-MS experimental results (average, n = 2) returned positive as the cut-off concentration for confirmatory testing is 0.15 μg mL−1.15 | |||||
| 1.0 | 1.4 | 1 | 10 | 0.89 | 11 |
| 2 | 240 | 0.88 | 12 | ||
| 10.0 | 9.6 | 3 | 10 | 7.58 | 24 |
| 4 | 240 | 8.04 | 20 | ||
It is worth noting that in our study the net loss of MDMA in urine following NaOCl exposure was not always comparable between results obtained from the LC-MS/MS and the GC-MS analysis even though the reaction conditions were maintained as close as possible. This may be attributed to the different sample preparation procedures used by the two analytical methods and the elevated temperatures (i.e. injection port and column temperatures) applied in GC-MS analysis. In LC-MS/MS, samples were analysed immediately without any further sample preparation, while in GC-MS, sample analysis was only possible following a sequence of sample preparation steps including LLE extraction, solvent evaporation and derivatisation.
4 Conclusion
The findings of this study identified one major reaction product, N-chloroMDMA, arising from the oxidation reaction of MDMA in urine after being exposed to bleach. N-ChloroMDMA was found to be relatively stable at 4 °C and can not only potentially indicate MDMA use, but also urine adulteration via bleach. However, N-chloroMDMA was unstable at 20 °C and decomposed into MDMA as well as possibly other degradation products. At a high concentration, bleach was shown to be an effective adulterant producing false-negative results across CEDIA® immunoassay testing with strong negative readings. However, these readings were all quite similar with a value of −0.85, which may act as a warning sign for further investigation of the sample. Confirmatory analysis of these false-negative specimens for the presence of MDMA by LC-MS/MS or GC-MS may reveal MDMA abuse.Acknowledgements
The authors wish to thank the Toxicology Unit, PaLMS for the supply of positive urine samples and for the use of their laboratory equipment and facilities required for the initial immunoassay screening tests. They would also wish to thank the Toxicology and Forensic Unit, ACTGAL for access to their urine samples.References
-
C. Claydon, 2010 National Drug Strategy Household Survey Report, Australian Institute of Health and Welfare, Canberra, 2011 Search PubMed
.
- A. H. B. Wu, J. Clin. Ligand Assay, 2003, 26, 11–18 Search PubMed
-
R. L. McIntire and J. E. Lessenger, in Drug Courts: A New Approach to Treatment and Rehabilitation, ed. J. E. Lessenger and G. F. Roper, Springer, New York, 1st edn, 2007, pp. 247–254 Search PubMed
-
O. A. Katz, N. B. Katz, S. Mandel and J. E. Lessenger, in Drug Courts: A New Approach to Treatment and Rehabilitation, ed. J. E. Lessenger and G. F. Roper, SpringerNew York, 1st edn, 2007, pp. 255–262 Search PubMed
- W. B. Jaffee, E. Trucco, S. Levy and R. D. Weiss, J. Subst. Abuse Treat., 2007, 33, 33–42 CrossRef
-
A. Dasgupta, in Drugs of Abuse: Body Fluid Testing, ed. R. C. Wong and H. Y. Tse, Humana Press, 1st edn, 2005, pp. 217–232 Search PubMed
- S. L. Mikkelsen and K. O. Ash, Clin. Chem., 1988, 34, 2333–2336 CAS
-
L. Broussard, in Handbook of Drug Monitoring Methods: Therapeutics and Drugs of Abuse, ed. A. Dasgupta, Humana Press, 1st edn, 2008, pp. 317–335 Search PubMed
- J. C. Morris, J. Phys. Chem., 1966, 70, 3798–3805 CrossRef CAS
- R. J. Lewis, D. Reed, A. G. Service and A. M. Langford, J. Forensic Sci., 2000, 45, 1119–1125 CAS
- V. Maresova, J. Hampl, Z. Chundela, F. Zrcek, M. Polasek and J. Chadt, J. Anal. Toxicol., 2005, 29, 353–358 CrossRef CAS
- C. L. Hawkins, D. I. Pattison and M. J. Davies, Amino Acids, 2003, 3–4, 259–274 CrossRef
- J. M. Antelo, F. Arce, M. C. Castro, J. Crugeiras, J. C. Pérez-Moure and P. Rodríguez, Int. J. Chem. Kinet., 1995, 27, 703–717 CrossRef CAS
- J. M. Antelo, F. Arce, J. L. Armesto, A. Garcia-Verdugo, F. J. Penedo and A. Varela, Int. J. Chem. Kinet., 1985, 17, 1231–1245 CrossRef CAS
- AS/NZS 4308:2008 Procedures for specimen collection and the detection and quantitation of drugs of abuse in urine, Standards Australia/Standards New Zealand, 2008.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS
- F. T. Peters, O. H. Drummer and F. Musshoff, Forensic Sci. Int., 2007, 165, 216–224 CrossRef CAS
- D. Catlin, D. Cowan, M. Donike, D. Fraisse, H. Oftebro and S. Rendic, Clin. Chim. Acta, 1992, 207, S13–S26 CrossRef CAS
-
Handbook of Forensic Drug Analysis, ed. F. Smith and J. Siegel, Elsevier, New York, USA, 2005 Search PubMed
- S.-L. Chou and Y.-S. Giang, J. Forensic Sci., 2008, 7, 1–12 Search PubMed
- S.-L. Chou, Y.-C. Ling, M.-H. Yang and Y.-S. Giang, J. Chin. Chem. Soc., 2008, 55, 682–693 CAS
| This journal is © The Royal Society of Chemistry 2013 |