Progress and perspectives in developing polymeric vectors for in vitro gene delivery
Yanan
Yue
*a and
Chi
Wu
*ab
aDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong. E-mail: yananyue.linda@gmail.com; chiwu@cuhk.edu.hk; Fax: +852 2603 5057; Tel: +852 2609 6266
bThe Hefei National Laboratory of Physical Science at Microscale, Department of Chemical physics, The University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 1st October 2012
Abstract
The development of safe, efficient and controllable gene-delivery vectors has become a bottleneck to human gene therapy. Synthetic polymeric vectors, although safer than viral carriers, generally do not possess the required efficacy, apparently due to a lack of functionality to overcome at least one of many intracellular gene-delivery obstacles. Currently, the exact mechanisms of how these polymeric vectors navigate each intracellular obstacle (“slit”), as well as their particular physical/chemical properties that contribute to efficient intracellular trafficking remain largely unknown, making it rather difficult to further improve the efficacy of non-viral polymeric vectors in vitro and in vivo. In this review, we first give a brief overview of synthetic polymeric vectors that have been designed and developed for gene delivery and highlight some promising candidates for clinical applications. Our main focus is on discussing the intracellular trafficking mechanisms of the DNA–polymer complexes (“polyplexes”), with less effort on the DNA–polymer complexation in the extracellular space as well as the in vivo systemic administration of genes in animal models and human clinical trials. In particular, we identified and discussed four critical, but often over-looked issues for successful DNA–polymer intracellular trafficking, especially our recent confirmation that it is free cationic polymer chains in the solution mixture of DNA and polymer that actually promote gene transfection and the polycationic chains within the polyplexes mainly play a protective role. Instead of the previously proposed and widely used escape model from late endolysosomes, the current hypothesis is that free polycationic chains with a sufficient length (∼20 nm) can block the initial endocytic-vesicle-to-endolysosome pathway.
 Yanan Yue Yanan Yue | Yanan YUE received her B.S. degree in Polymer Materials and Engineering from Zhejiang University in 2007 and her Ph.D. in Chemistry at the Chinese University of Hong Kong, under the supervision of Professor Chi WU. Her research interests mainly focus on the development of non-viral vectors for molecular medicines, especially the effect of free polycationic chains on the intracellular trafficking of DNA–polymer complexes in gene transfection. She is now working in the Department of Chemistry, the University of Chicago, under the supervision of Professor Chuan HE. |
 Chi Wu Chi Wu | Chi WU graduated from the Department of Chemical Physics at the University of Science and Technology of China (USTC) in 1982. After obtaining his Ph.D. in 1987 followed by two-year postdoctoral experience under the supervision of Professor Benjamin Chu in the State University of New York at Stony Brook, he moved to BASF (Ludwigshafen, Germany) in 1989; first as an Alexander von Humboldt Fellow for one year with Dr. Dieter Horn and then as the supervisor of laser light-scattering laboratory. In 1992, he resigned from BASF and joined the Department of Chemistry at the Chinese University of Hong Kong (CUHK) as a Lecturer. He underwent a double promotion directly to Reader in 1996; and became a Chair Professor of Chemistry and Honorary Professor of Physics in 1999. Recently, he has been appointed as a Wei Lun Chair Professor of Chemistry since 2010. |
Introduction
Gene therapy, considered to be treating genetically-related diseases by transferring exogenous nucleic acids into specific cells of patients, has attracted great interest over the past few decades.1 Advances in molecular biology and biotechnology as well as the completion of the Human Genome Project have led to the recognition of numerous disease-relating genes.2 It has been gradually and generally realized that the development of safe, efficient and controllable gene-delivery vectors is now a bottleneck in clinical applications.2,3Gene delivery vectors can generally be divided into viral and non-viral ones. Viruses, such as adenovirus, adeno-associated virus, lentivirus and retrovirus, have evolved as a sophisticated gene-delivery vehicle and can be readily transformed into a viral vector by replacing part of its genome with a therapeutic gene.4–6 Such recombinant viral vectors are efficient but at the same time potentially dangerous, previously leading to severe immune/inflammatory reactions or even cancer in patients.7–9 Non-viral vectors, including synthetic polymers10 and lipids,11–14 offer several advantages over their viral counterparts, such as low immune toxicity, construction flexibility and facile fabrication.2,15 In particular, cationic polymers have attracted much interest because it is relatively easy to tune their chemical and physical properties through polymer chemistry so that they can acquire multiple functions for gene delivery. However, at this moment, their low gene transfection efficiency greatly limits their clinical applications. For instance, polyethylenimine (PEI), one of the few most effective and versatile polymeric vectors, remains ∼105 times less efficient than its viral counterpart.16 Since the first demonstration of polycation-mediated gene transfection in 1987,17 hundreds, if not thousands, of cationic polymers with different chain lengths and topologies have been synthesized and explored as non-viral vectors for gene delivery.
Among them, PEI is the most intensively studied example and hitherto exhibits nearly the highest in-vitro transfection efficiency in the absence of any exogenous endosomolytic agent.18–20 The optimal efficacy of PEI has been attributed to its unique ability to navigate through a series of intracellular “slits”, including the escape from lysosomes (acidic vesicles filled with various degradative enzymes), nuclear localization and DNA unloading.21–24 However, the exact mechanisms of how these polycationic chains overcome each intracellular obstacle, as well as their particular physical–chemical properties that contribute to efficient intracellular trafficking remain largely unexplained, making it rather difficult to further improve the efficacy of non-viral polymeric vectors in vitro and in vivo.
In this context, deciphering the intracellular trafficking mechanism and establishing the structure–function relationship of non-viral polymeric vectors is of great importance because they will rationally guide our design and construction of multi-potent polymeric gene-delivery vehicles with well-defined structures and superior efficacy. There have been a number of review articles and chapters that summarize the recent developments of novel polymer materials for plasmid DNA10,16,25–33 and oligonucleotide delivery.34–39 In this perspective, we will not cover all the aspects in the field of non-viral polymeric gene delivery. Instead, we will first give a brief overview of several important polymeric vectors and, in particular highlight some promising candidates for clinical applications. Our main focus is on summarizing and discussing some new insights in understanding the intracellular trafficking of DNA–polymer complexes (“polyplexes”). Particularly, we will identify and discuss four critical, but previously overlooked, issues for successful DNA–polymer intracellular processing; namely, (1) the effect of free, uncomplexed polycationic chains in the DNA–polymer solution mixture on the gene transfection; (2) the effect of the endocytosis pathway on the intracellular fate of polyplexes; (3) the effect of the so-called and well-accepted “proton sponge” concept; and (4) the effect of the nuclear localization and unloading of DNA inside the nucleus.
2. Non-viral vectors made of commercial and specifically designed polymers
For understandable reasons, many earlier gene-delivery studies used commercially available polymers, such as poly(L-lysine) (PLL),17,40 polyethylenimine (PEI)18 and polyamidoamine dendrimers (PAMAM).41–44 These off-the-shelf polymers have been extensively studied and formed a literature basis for the development of non-viral gene delivery. In recent years, a broad diversity of polymer materials have been specifically designed and synthesized for gene delivery. In most cases, they were designed to pass one or more particular extra-/intra-cellular obstacles (“slits”), such as the avoidance of aggregation in blood circulation, the release from endolysosomes and translocation into cell nucleus. Some of these polymeric vectors perform better than the best off-the-shelf polymers. However, none of them are able to rival viral vectors in clinical trials because of their lower efficacy and potential toxicity, especially when long polycationic chains are used for in-vivo gene therapy.45,46In general, polymers designed and explored for gene delivery include: (1) polyethylenimine (PEI) and its derivatives; (2) polymethacrylate; (3) carbohydrate-based polymers, generally with β-cyclodextrin, chitosan, dextran, poly(glycoamidoamine) and schizophyllan as their carbohydrate functionalities; (4) poly(L-lysine) (PLL); (5) linear poly(amidoamine) (PAA); (6) dendrimer-based vectors, such as PAMAM and poly(propylenimine) (PPI) dendrimers; (7) biodegradable polymers, primarily involving phosphorus-containing polymers, poly(amino-ester), poly(4-hydroxy-L-proline ester) and poly[α-(4-aminobutyl)-L-glycolic acid] (PAGA); (8) polypeptide vectors,47–49 such as Tat-based peptide,50–52 antennapedia homeodomain peptide,53 MPG peptide54 and transportan peptide;55 (9) polycationic “clusters” assembled by several small molecules or oligomers with different desired functions;56 and many other examples reviewed in ref. 10. To illustrate their specific advantages and disadvantages, we choose to review several important classes of cationic polymers and emphasize their promising biomedical applications as follows.
Polyethylenimine (PEI)
The introduction of PEI as a non-viral vector represented a big leap because of its much higher gene transfection efficiency compared to other early polymeric vectors (e.g., PLL).18,19 PEI mainly has two different topologies: linear and branched structures. Branched PEI (bPEI) is synthesized via the acid-catalyzed polymerization of aziridine,57 whereas linear PEI (lPEI) is normally made by the ring opening polymerization of 2-ethyl-2-oxazoline followed by hydrolysis (Scheme 1).58 Several lPEIs have been made as commercial transfection agents, including ExGen500 and jetPEI. Both of them are derivatives of lPEI with a molar mass of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.
000 g mol−1.
 | ||
| Scheme 1 Schematic of synthesis of PEI by (A) branched: acid-polymerization of aziridine; and (B) linear: ring-opening polymerization of 2-ethyl-2-oxazoline followed by hydrolysis. | ||
Scheme 1 shows that PEI contains nitrogen at every third atom, leading to a high charge density on the chain, especially in acidic conditions. Theoretical calculation shows that bPEI contains primary, secondary and tertiary amino groups with a 1:2:1 ratio.59 These amines have pKa values spanning the physiological pH range, acted as a buffer. The degree of protonation of these amines increases from ∼20% to ∼45% as the pH decreases from ∼7.4 to ∼5.0.60 Previous studies have attributed its high transfection efficiency to the so-called “proton sponge” effect. Namely, further protonation of PEI chains inside the endolysosomes would lead to an influx of counter (chloride) ions and increase the osmotic pressure inside, which could burst the endocytic vesicle and release the polyplexes.18,61 Many people, especially those who joined the research field later, have taken such an explanation as granted. However, a number of researchers in the field have always questioned whether such a “proton sponge” effect plays a dominant role in promoting gene transfer because of some realistic estimations of the additional osmotic pressure and some contradictive results,16 which will be discussed later.
It is well-known in the field that both the gene transfection efficacy and toxicity of PEI are strongly related to its chain length and topology (branched or linear).62–66 Long PEI chains are highly effective but more cytotoxic. It has been shown that free cationic PEI chains, in particular long ones (bPEI with a molar mass of 25000 g mol−1, denoted as bPEI-25K), can induce the membrane damage (necrotic-like alteration) in the early stages (at 0.5-h post-treatment), assessed by a considerable release of cytosolic lactate dehydrogenase (LDH) and a concomitant increase in phosphatidylserine exposure from the inner plasma membrane to the outer cell surface (without the activation of apoptotic factors);45,46,67 while in the later stage (at 24-h post-treatment), they initiate mitochondrial-mediated apoptosis, reflected by an enhanced apoptotic gene expression (Bcl2l1, Bax, Atm, Ercc4 and Anxa5),68 the release of proapoptotic cytochrome c, a subsequent activation of executioner caspase-3, and a significant loss of mitochondrial membrane potential (MMP).46,67 Notably, such PEI-induced MMP loss (mitochondrial depolarization) mainly results from the direct interaction and permeabilization of mitochondria with those cationic PEI chains and/or PEI-mediated polyplexes,69 rather than the activation of mitochondrial permeability transition pore (MTP)67,69 or the perturbation of mitochondrial membrane pump.69 In addition to necrosis and apoptosis, it was recently reported that autophagy is also associated with PEI-mediated cytotoxicity and gives rise to aggravated cell damage.70 Particularly, in the early stage (at 3-h post-addition), autophagy is mainly correlated to the lysosomal damage, while in the later stages (after a 24-h recovery), autophagy is primarily associated with mitochondrial injury.70 Note that PEI is just one example. Many other polycationic chains also suffer from this “malignant” correlation of efficacy and toxicity. How to solve such a catch-22 problem has puzzled researchers for years. The search for a high efficient and low cytotoxic polymeric vector is an endless endeavor.
One of the approaches to circumvent such a catch-22 problem is to link short PEI chains into a long one by some degradable coupling agents, i.e., ester, β-aminoester and disulfide, to reduce the inherent toxicity of long polycationic chains inside the cell.71–75 The former two linkers have a hydrolysis half-life time ranging from hours to days, which might not be sufficiently fast. In contrast, the reductive degradation of a disulfide is quicker in the presence of glutathione (GSH) in the cytosol.76,77 Goepferich et al.73 clearly confirmed the intracellular degradation of such disulfide cross-linked PEI chains and the release of nearly non-toxic short PEI fragments (cell survival 98.69 ± 4.79%) (Fig. 1A). In comparison with seven commercial transfection agents in seven different cell lines, these disulfide cross-linked lPEI vectors exhibited a superior efficiency and a substantially lower toxicity, clearing manifesting that reductive degradation is promising in designing novel non-viral vectors (Fig. 1B).
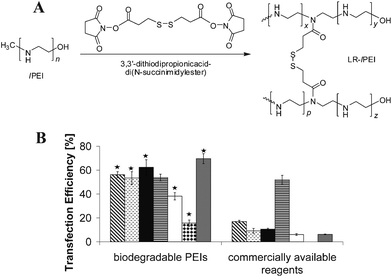 | ||
| Fig. 1 (A) Synthesis of disulfide cross-linked low-molecular-weight lPEI, where LR refers to Lomant's reagent, a cross-linking reagent. (B) Comparison of the best gene transfection efficiency of seven disulfide cross-linked lPEI vectors with seven commercially available transfection agents under conditions where the cell viability is >90%, in (from left to right) CHO-K1, COS-7, NIH/3T3, HepG2, HCT116, HeLa and HEK-293 cells, respectively. Statistically significant differences of biodegradable PEIs compared with those commercial transfection agents are denoted by ★ (p < 0.01). (Reprinted from ref. 73 with permission of National Academy of Sciences, USA). | ||
Recently, Deng et al.78 developed a laser light scattering (LLS) method to monitor the disulfide-coupling reaction in situ under a programmable mixing of short bPEIs (Mw = 2 × 103 g mol−1) and the cross-linking agent, dithiobis(succinimidyl propionate) (DSP). In this way, a series of linked PEI chains with different molar masses can be obtained from one reaction mixture. A comparative study of the transfection activity and cytotoxicity of two such linked PEI samples (PEI-7K-L and PEI-400K-L, respectively with Mw = 6.5 × 103 and 3.8 × 105 g mol−1) reveals that PEI-7K-L with an extended chain structure is less cytotoxic and 2–10 times more efficient than both the “golden standard” bPEI-25K and the widely-used commercial Lipofectamine 2000.78 On the other hand, PEI-400K-L with a microgel structure is ineffective in spite of the fact that it is much less cytotoxic. This study clearly demonstrates that a proper control of the chain structure is more important than that of the overall molar mass.
Previous studies have shown that the “naked” PEI-based polyplexes, although they possess positive surface charges, tend to aggregate in a time-dependent manner in physiological buffers (ionic strength equals to that of 150 mM NaCl).79 When administered in vivo, they are prone to absorb to the serum albumin and other negatively charged proteins in the bloodstream, giving rise to further aggregation and a rapid clearance by phagocytic cells and the reticuloendothelial system (RES).80 To unravel such problems, the surface of polyplexes was usually modified with a layer of hydrophilic polymers. Specifically, when polyethylene glycol (PEG) is used, it is often called PEGylation. The steric and hydrophilic shell stabilizes the resultant polyplexes in physiological condition, reduces their undesirable interaction with anionic proteins, and increases their intravenous circulation time as well.19,81 It should be noted that increasing the length and grafting density of PEG chains impedes the DNA complexation. Short PEG chains (Mw ≤ 500 g mol−1) fail to provide the shielding effect, while a molar mass of at least 2000–5000 g mol−1 seems to be sufficient to achieve such an effect.19 Unfortunately, there is a dilemma about PEGylation because it makes the polyplexes more “stealthy” in the body but reduces the cellular internalization, hinders the intracellular unpacking, and hampers the following release of DNA in the nucleus.82,83
The attachment of properly chosen cell-targeting ligands at the end of each PEG chain can enhance the cellular uptake.19,84 In clinical applications, it is often beneficial and sometimes critical to target the polyplexes to a specific cell type or tissue. Over the past few decades, much effort has been made to conjugate targeting moieties to PEI chains to enhance their cellular uptake and cell specificity.84–89 Many receptor proteins on cell membranes are chosen for targeting via receptor-mediated endocytosis. For instance, galactose was attached to PEI chains to target the asialoglycoprotein receptors on hepatocytes,85 while iron-transport protein transferrin (Tf),84 epidermal growth factor (EGF)86 and folic acid87,88 were conjugated to PEI chains to target their corresponding receptors that are typically up-regulated on cancer cells. The efficient cell-specific targeting requires careful optimization of various parameters, including the length of a spacer between ligand and polyplex, the number of ligands per polyplexes, and the ligand-receptor binding strength.2 Notably, the attachment of proper targeting ligands to the periphery of polyplexes not only improves their cellular internalization, but also alters their subsequent intracellular trafficking pathways (as will be discussed later in this article).16,88
Poly[2-(dimethylamino) ethyl methacrylate] (PDMAEMA)
PDMAEMA bearing a tertiary amino group in the side chain was utilized as a gene transfer agent in the early studies.90–92 Linear93 and star-shaped94,95 PDMAEMAs with precise, discrete molar mass and well-defined architectures can be synthesized via atom transfer radical polymerization (ATRP) (Scheme 2).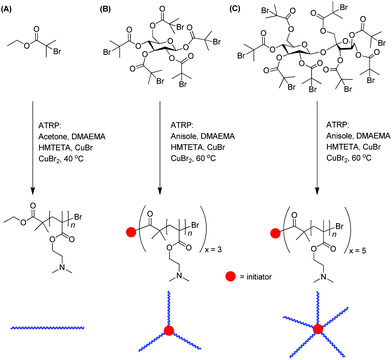 | ||
| Scheme 2 Synthesis of (A) linear, (B) 3-arm and (C) 5-arm star-shaped PDMAEMA with different desired chain lengths via atom transfer radical polymerization (ATRP).95. | ||
Note that PDMAEMA is generally less effective than PEI in nucleic acid delivery. The choice of PDMAEMA in the study is mainly due to its well-documented synthesis and characterization so that it becomes an excellent model for the evaluation of relationships between the chain structures and functions.90,93,95–98 van de Wetering et al.90 and Long et al.96 found that the transfection efficacy of linear PDMAEMA was dramatically enhanced with the increase of chain length in the molar mass range of 0.4–92 × 104 g mol−1, highlighting how significant the polycationic chain length is in the gene transfection. On the other hand, polyplexes made by different PDMAEMAs [Mw = (4.3–92) × 104 g mol−1] showed a comparably high level of cellular uptake, clearly indicating that intracellular trafficking, rather than cellular internalization, is the rate-limiting step in PDMAEMA-mediated gene transfection.96 In addition to chain length, the influence of chain architecture on the transfection efficacy and toxicity was recently investigated by using a family of linear, 3-arm and 5-arm star-shaped PDMAEMAs prepared via ATRP [Mw = (1.9–28) × 104 g mol−1].95 Unlike linear PDMAEMAs, an increase in the molar mass of star-shaped polymers does not necessarily lead to an improved transfection activity. It is also interesting to note that the cytotoxicity at a given molar mass is generally reduced with the increasing arm-number, indicating that PDMAEMA with a branched architecture (lower toxicity) and an intermediate molar mass (Mw ∼ 12 × 104 g mol−1 for 5-arm polymer) shows promise for efficient gene delivery.95
In terms of cytotoxicity, Cai et al.93 found that in the concentration range normally used for in-vitro gene transfection (10–110 μg mL−1), linear PDMAEMA chains with different lengths are cytotoxic to HepG2 cells by different mechanisms. Namely, (1) for short PDMAEMA chains [Mw = (1.1–1.7) × 104 g mol−1], their cytotoxicity, membrane disruption, and apoptosis are very low, independent of the chain length; (2) in the medium range (1.7 × 104 < Mw < 3.9 × 104 g mol−1), their cytotoxicity increases with the chain length and polymer concentration, mainly due to the cooperative effect of membrane disruption and apoptosis; and (3) long chains [Mw = (3.9–4.8) × 104 g mol−1] become more disruptive to cellular membranes and pro-apoptotic so that they are able to pass through the cytoplasm and enter the nucleus much faster than short ones but their high cytotoxicity is less dependent on the chain length.
Cyclodextrin-based polymers
Cyclodextrins (CDs), cyclic oligosaccharides made of 6, 7 or 8 glucose units (called α-, β- and γ-CD, respectively) are pharmaceutically attractive; namely, (1) they can form water-soluble inclusion complexes with small, hydrophobic “guest” molecules, e.g., adamantine (AD); and (2) they elicit no immune responses and have very low in-vivo toxicities and are approved by the FDA as solubilizing agents in pharmaceutical formulations.99,100 In 1999, Davis et al.101 first incorporated β-CD into the backbone of linear polycationic chains to introduce a new class of CD-based gene-delivery vehicles (Fig. 2A). The initial study showed that these CD-containing polymers are not only as effective as PEI and Lipofectamine, but also have minimal toxicity in both BHK-21 and CHO-K1 cell lines.101 The effect of CD size, charge centre and charge density on the gene-delivery efficacy and polymer toxicity were explored and summarized in later publications, as reviewed in ref. 102. Furthermore, a host of cationic polymers, such as PEI and PAMAM dentrimers, were modified by grafting CD moieties onto the polymers, and exploited as therapeutic gene carriers in various tumor cells and cultured neutrons.103–109 In particular, a folate grafted PEI600-CD polymer developed by Tang et al. has shown the capability to mediate a comparable level of transgene expression to that of adenovirus-mediated transduction in B16 melanoma-bearing mice, without eliciting any obvious toxicity at the administered dose.108 More recently, a series of polycationic star-shaped CD conjugates were developed by incorporating multiple oligoethylenimine (OEI)110 or PDMAEMA111 arms onto a CD-core for nucleic acid delivery. In particular, Reineke et al.112 and Fernandez et al.113–115 independently generated a small library of monodisperse polycationic β-CD “click clusters” by linking different functional building blocks to a per-azido-β-CD core via click coupling chemistry. This strategy not only aids the creation of polycationic CD-based delivery vehicles with well-defined structures and superior efficacy, but also provides feedback for the investigation of the structure–function relationship.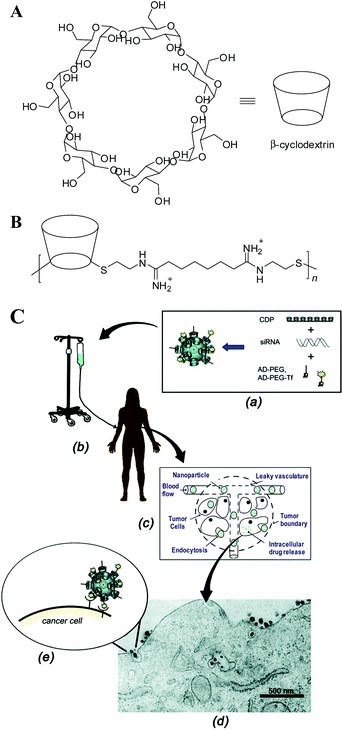 | ||
| Fig. 2 (A) Chemical structure of β-cyclodextrin (CD). β-CD has a hydrophobic interior and hydrophilic exterior surface. (B) Chemical structure of a β-CD-based polymer (CDP) designed for gene delivery, where n = 5. (C) Schematic of a targeted CDP-based nanoparticle delivery system made of a water-soluble CDP, an adamantine(AD)-PEG conjugate, a human transferrin conjugated at one end of PEG-AD for targeting, and siRNA, where an aqueous solution of nanoparticles is infused into patients, circulates in the blood, leaks via the effect of EPR into tumor tissues, penetrates though the tumor, and finally enters into the cancer cell via receptor-mediated endocytosis, as shown by a transmission electron micrograph. (Reprinted from ref. 102 with ACS permission). | ||
The CD-containing polyplexes readily form inclusion complexes with some hydrophobic compounds. For instance, they can be decorated with short AD-terminated PEG chains to improve their stability in biological fluids,116 or with AD-terminated galactose116 and transferrin (Tf)117 to target hepatocytes and cancer cells, respectively. This strategy has been used for a variety of CD-based polymers,102 yielding a targeted delivery system made of a linear, CD-based polycation (CDP), a folate or a human Tf protein ligand because these two receptors are typically up-regulated in cancer cells, a PEG steric stabilization agent, and plasmid DNA (pDNA) or small interfering RNA (siRNA) (Fig. 2B). Quickly, people realized that this kind of nanoparticle can be made by adding all the components together at one time. Namely, all the delivery components are placed in one vial; and DNA or siRNA, in another vial. Simply mixing them can lead to stable nanoparticles with a diameter of 60–80 nm, even at a very high nucleic acid concentrations.118
Such a targeted CDP-based delivery system first met success in delivering pDNA119 and DNAzyme (short catalytic single-strand DNA)120 to subcutaneous tumors via intravenous (i.v.) injection into mice. Later, it was shown that the Tf-targeted CDP-based nanoparticles with anti-cancer siRNA can effectively limit the tumor proliferation in a disseminated murine model of Ewing's sarcoma.121 A combination of bioluminescence imaging (BLI) and positron emission tomography (PET) further revealed the effect of tumor-specific targeting on their in-vivo biodistribution and efficacy.122 It should be noted that similar to pDNA and DNAzyme deliveries,119,120 the non-targeted siRNA nanoparticles are able to accumulate in the tumor region through the effect of enhanced permeability and retention (EPR), but their internalization into tumor cells is much less efficient, leading to poor expression of their carried genes inside the cell.122 Therefore, it should be reminded that the primary objective of targeting is to enhance the uptake of the nanoparticles by tumor cells, rather than their accumulation around tumor cells, in spite of that, the accumulation is a necessary and important step for cancer cell uptake, endocytosis.
The early in-vivo success of CDP-based gene delivery systems motivated people to entail its translation from laboratory to clinic. It has been shown that a targeted CDP-based delivery system, clinical version denoted as CALAA-01, is well-tolerated in multi-dosing experiments in a variety of non-human primates.123 To our knowledge, Davis et al.124 are now conducting the first in-human phase I clinical trial, which involves systemic administration of siRNA therapeutics to patients with solid tumors via this delivery system. Post-treatment tumor biopsies from melanoma patients showed that the amount of nanoparticles localized inside tumor cell was correlated to the administrated dose level. Moreover, a reduction of both the targeted messenger RNA (mRNA) and protein levels was detected in the post-dosing tumor tissue.124
pH-Sensitive, membrane-disruptive polymers
Intracellular trafficking is critical to deliver a therapeutic gene because of its degradation susceptibility in lysosomes by various enzymes. In nature, many viruses have evolved some specific acidic peptides in their protein coat that can be protonated at an acidic environment and thus become fusogenic with the endosomal membrane, allowing the release of the therapeutic genes directly into the cytoplasm.125 It motivated people to design and prepare a myriad of acid-responsive membrane-disruptive polymers in the hope that they can facilitate endosomal release in a similar manner to that of viruses.2,126–129 Such endosomolytic polymers include both polyanions and polycations. Typically, a polyanionic endosomolytic system comprises (1) acid-responsive functionalities, especially –COOH and anhydride groups with a pKa in the endosomal range of 5.5–6.5; (2) hydrophobic groups to interact with and disrupt the endosomal membrane; (3) cationic pendant groups to complex/conjugate with a therapeutic gene; and (4) a tumor cell-targeting ligand. One of the early examples is poly(2-ethylacrylic acid) (PEAA). It undergoes a hydrophilic-to-hydrophobic transition at pH < 6 so that it can partition into and disrupt the membranes of phospholipid vesicles130,131 and red blood cells.132 Following this study, two related polymers, poly(2-propylacrylic acid) (PPAA) and poly(2-butylacrylic acid) (PBAA), were synthesized to examine whether making the pendant alkyl group more hydrophobic would increase the hemolytic activity, a reflection of the ability of agents to disrupt membranes.132,133 It was found that PPAA could disrupt the red blood cells 15-fold more efficiently than PEAA at pH ∼ 6, yet showed no hemolytic activity at pH ∼ 7.4. In contrast, PBAA led to a severe hemolysis even at physiological pH, making it undesirable for the development of non-viral vectors.133 Inspired by its acid-responsive hemolytic activity, PPAA was incorporated into some cationic DNA/(1,2-dioleoyl-3-trimethylammonium-propane) (DOTAP) lipoplexes134,135 and DNA–chitosan polyplexes,136 respectively; and remarkably improved their intracellular gene delivery in vitro and in a murine excisional wound healing model.The early success of PPAA and its derivatives motivated recent developments in making a family of modular diblock copolymers that are composed of a cationic block, PDMAEMA, to condense therapeutic genes, and a second endosomolytic block comprising DMAEMA, 2-propylacrylic acid and butyl methacrylate (BMA), using controlled reversible addition fragmentation chain transfer (RAFT) polymerization.137 These diblock copolymers become sharply hemolytic at the endosomal pH regime and their hemolytic activity increases with the BMA content in the second block. When used for siRNA delivery, their transfection efficacy, reflected by the reduction of targeted mRNA and protein, was steadily enhanced with their pH-dependent hemolytic activity.137 Further, a protein antigen, ovalbumin, is successfully delivered to a mouse tumor model when it is conjugated with the PPAA-based carriers via reducible disulfide bonds, showing the great potential of PPAA-based polymers for therapeutic vaccine delivery.138
In the polycation category, an N-substituted poly(aspartamide) bearing 1,2-diaminoethane side chains [PAsp(DET)] shows minimal toxicity and great efficacy in mediating the release of polyplexes from endosomes due to its acid-stimulated membrane destabilization (Fig. 3A).139,140 Similar to other endosomolytic agents, PAsp(DET) manifests neglectable membrane perturbation at the physiological pH but becomes membrane-disruptive at the endosomal pH regime (pKa ∼ 6.3). Such a property is attributed to the protonation alteration in the flanking diamine unit, i.e., the monoprotonated gauche form at the neutral pH and the diprotonated anti form at the acidic pH, as shown in the inset of Fig. 3A. In other words, how 1,2-diaminoethane is protonated plays a pivotal role in triggering endosomal disruption.139 It was also shown that the facile degradation of PAsp(DET), which is induced by a rapid self-catalytic reaction between the PAsp backbone and the side-chain amide nitrogen, minimizes the cumulative toxicity caused by polycationic chains.140 Most recently, Kataoka et al.141 utilized a PEG-b-PAsp(DET) derivative together with the intravital real-time confocal laser scanning microscopy (IVRTCLSM), for the first time, to in situ quantify the dynamic states of polyplexes in the bloodstream. The efficacy of PEGylation in stabilizing polyplexes against platelet-induced agglomeration was visually demonstrated, as shown in Fig. 3B.
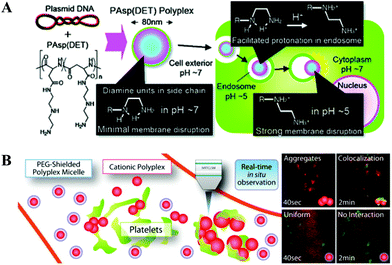 | ||
| Fig. 3 (A) Schematic of PAsp(DET)-mediated gene transfection, where the inset shows protonation of 1,2-diaminoethane moiety (Reprinted from ref. 139 with ACS permission); and (B) Schematic of interaction of polyplexes (red) with/without PEG coating with the platelets (green) in the bloodstream of a mouse earlobe. (Reprinted from ref. 141 with Elsevier permission). | ||
Alternatively, Duncan et al.142,143 introduced a family of linear poly(amidoamine)s (PAA), which have a pH-dependent conformation and membrane perturbation ability, to deliver genes and protein drugs. The protonation of PAA reduces the freedom of chain conformation and leads to a more rigid chain structure. Such a conformational change in lower pH values enhances its hemolytic activity so that it can function as an endosomolytic agent.144 Very recently, Richardson et al.145 provided a direct evidence of how a PAA derivative (ISA1) in-vivo permeabilizes the endocytic vesicular membranes, as shown in Fig. 4. In this study, radioactive-labeled ISA1 was combined with a liver sub-cellular fractionation to monitor the dose- and time-dependant passage of ISA1 along the endocytic pathway after its i.v. administration to rats, wherein the vesicular permeabilization (a reflection of perturbation of late endosomes/lysosomes) is quantified by the release of N-acetyl-D-glucosaminidase (NAG) from the vesicular fraction to the cytosolic fraction. The escalation of either the ISA1 dose or its incubation time enhances the release of both the radioactive polymer and NAG into the cytosol. Moreover, it was suggested that the endosomolytic activity of PAA chains might be due to their physical interactions with endocytic vesicular membranes rather than the popular “proton sponge” effect.145 Of note, this study provides a general methodology to acquire “quantitative” information on the intracellular localization of polymeric vectors and their therapeutic cargos in vivo.
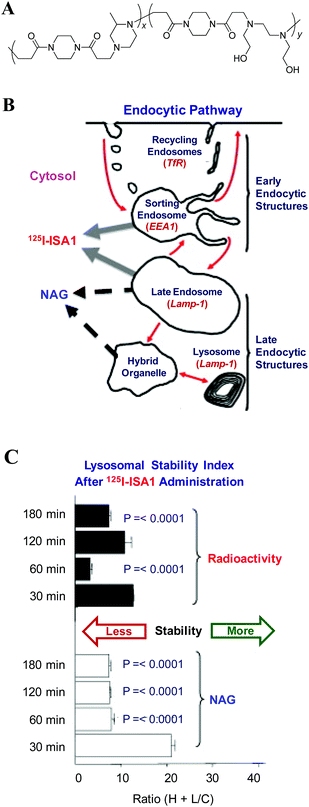 | ||
| Fig. 4 (A) Chemical structure of a poly(amidoamine) derivative, ISA1; (B) Schematic of endocytic system and markers used in sub-cellular fractionation studies; and (C) Time dependence of lysosomal stability index after the administration of 125I-labelled ISA1 at 10 mg kg−1, where the lower the index, the greater the vesicle permeability. (Reprinted from ref. 145 with Elsevier permission). | ||
3. Important remaining issues
In order to deliver genes from a solution mixture of anionic DNA and cationic polymer all the way from extracellular to intracellular space, crossing through the cellular membrane, the cytoplasm and the nuclear membrane before releasing DNA inside the cell nucleus, the complexes made of polymeric vectors and DNA therapeutics have to pass through a number of narrow gaps (“slits”), not “barriers” as widely described before in the literature because one can pass a barrier as long as one can jump higher. These “slits” mainly include endocytosis, escape from endolysosomal entrapment, transport through the cytoplasm, localization on and passing through the nuclear membrane, and the eventual release of DNA from the polyplexes (ideally inside the nucleus).2,16 The DNA–polymer complexes could be blocked by any of these “slits”. Currently, one of the most difficult issues is how to create a multi-functional delivery system so that the polyplexes are able to waltz through these “slits”. Although more than 100000 papers have been published over the last three decades, we still have not gained a clear and thorough understanding of the intracellular trafficking pathway(s) of polyplexes; one of the first important questions in the development of efficient non-viral vectors. In the following, we will mainly discuss four critical, but previously over-looked, issues for efficient DNA–polymer intracellular trafficking.
Role of cationic chains free in solution mixtures of DNA and polymer
The complexation and condensation of long anionic DNA with cationic polymer chains into small aggregates (∼102 nm) is the first and necessary step in the non-viral polymer-mediated gene transfection.146–153 It is worth noting that in the literature, the driving force of such complexation is often mistaken as electrostatic attraction; namely, an enthalpy driven process. Actually, it is driven by the gain of entropy, i.e., the release of small counter ions from both anionic DNA and polycationic chains during the complexation.146 Due to the huge gain of translational entropy, the formation of DNA–polymer complexes is normally instantaneous and spontaneous upon the mixing of two aqueous solutions (DNA and polymer). Great efforts have been made to correlate the size, density and surface charge (zeta-potential) of the polyplexes to their final transfection efficiency,62–66,154–158 but a coherent picture remains lacking. Previous studies revealed and confirmed two facts; namely that in order to achieve a reasonable transfection efficiency, (1) the periphery of the polyplexes in the solution mixture should be slightly positively charged; and (2) the molar ratio of nitrogen from the polymer to phosphate from the DNA (N:P) should be around 10. It is easy to understand that a positively charged periphery can facilitate the attachment of polyplexes to the negatively charged cell membrane and thus improve the endocytosis. For a long time, few people have asked why the N:P ratio has to be much higher than that required for charge neutrality (N:P ∼ 1).159–163 In the early 2000's, Mely et al.160,161 and Wagner et al.162 independently found that a large amount of PEI chains are unbound to DNA and exist as individual chains free in the solution mixture. It was also found that these free cationic PEI chains are more toxic than those bound to DNA inside the polyplexes.162,164 Moreover, the removal of free polycationic chains by size exclusion chromatography significantly reduced the gene transfection efficiency.162 However, they did not follow up such a finding; namely, why and how do those free polycationic chains help gene transfection?
Most recently, complexation between DNA and PEI in both water and phosphate buffered saline (PBS) has been revisited using a combination of laser light scattering and gel electrophoresis.165–167 The results clearly confirmed that nearly all the DNA chains are complexed with PEI to form small polyplexes (∼102 nm) when N:P ∼ 3, irrespective of the chain length of PEI and solvent used. However, a high in-vitro gene transfection efficiency is only achieved when N:P ≥ 10. Putting these two facts together, it has been concluded that (1) each solution mixture with a higher N:P ratio actually contains two kinds of cationic chains: bound to DNA and free in the solution (∼70%), as schematically shown in Fig. 5A; and (2) it is those free PEI chains that actually promote gene transfection no matter whether they exist (are added) many hours before or after the addition of polyplexes (N:P = 3).166 These findings were further confirmed by different polycations (PLL, PDMAEMA, chitosan 168) and cell lines (293T, HepG2, HeLa and CHO).169
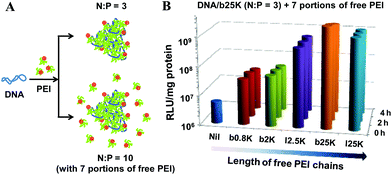 | ||
| Fig. 5 (A) Schematic of complexation between anionic DNA and cationic bPEI chains in solution mixtures; and (B) Effect of length and topology of free PEI chains on the gene transfection efficiency in 293T cells, where 7 portions of different free PEI chains were applied at 0, 2 or 4 h after adding the DNA–bPEI-25K polyplexes (N:P = 3).167 The overall and final N:P remains 10, identical for all the tests. “Nil” means that no free PEI chains were added, i.e., N:P = 3. | ||
Furthermore, the effects of length and topology of both the bound and free polycationic chains on gene transfection were studied.167 Notably, both short (∼2 K) and long (∼25 K) PEI chains are capable of condensing DNA completely at N:P ∼ 3 but long free chains are ∼102-fold more effective in enhancing gene transfection (Fig. 5B), indicating that the length of the free chains plays a vital role in gene transfection. It is also interesting to note that for long free PEIs, the chain topology has nearly no effect on the transfection efficiency; but for short PEI chains, linear free chains are ∼10-fold more effective than their branched counterparts (Fig. 5B). These results illustrate that the bound chains mainly provide cationic charges to neutralize the long anionic pDNA chains so that they become insoluble in water and collapse into small 102-nm particles, no more and no less. It is those polycationic chains free in the solution mixture that should get our attention.
Currently, it remains a challenge to elucidate how those free polycationic chains with a proper length/topology facilitate the intracellular trafficking of polyplexes since direct observation of their trafficking between different organelles is rather difficult. To visualize them, either DNA or polymer or both are often labeled with different fluorescence probes. The kinetic study of cellular uptake of labeled polyplexes by flow cytometry reveals that long free PEI chains boost the uptake rate, presumably due to their disruptive nature to the anionic cell membrane.167 However, the major contribution of free PEI chains is in the intracellular space.166 In the endolysosomal pathway, the shut-down of proton pump on endolysosomes using an specific inhibitor (bafilomycin A1) reduces the gene transfection efficiency by a factor of ∼15 for the DNA–bPEI-25K polyplexes at N:P = 10, but such a reduced transfection efficiency is still ∼20 times higher than that without free chains (N:P = 3).167 This clearly indicates that even after the complete removal of the so-called possible “proton sponge” effect, long free cationic PEI chains are still able to prevent the polyplexes from entrapment into the acidic lysosomes,167 presumably via (1) blocking the signal proteins on the inner cell membrane (i.e., on the periphery of the initial endocytic vesicles formed after endocytosis) so that the endolysosomes development is prevented or slowed down or (2) promoting the escape of polyplexes from the initial endocytic vesicles and/or early endosomes.
Quantitatively, cellular uptake and the subsequent intracellular distribution of the Cy3-DNA–bPEI-25K polyplexes without/with free PEI chains were compared using a confocal image-assisted three-dimensionally integrated method (Fig. 6A). It is found that 6 h after polyplex addition, the ultimate uptake amount is ∼1.0 × 105 DNA copies/cell, almost independent of the addition of free PEI chains. On the other hand, the transgene expression at N:P = 10 is ∼103-fold higher than that at N:P = 3, further indicating that those free cationic PEI chains mainly facilitate the intracellular processing of the DNA payload. In the endolysosomal pathway, Fig. 6B shows that with the aid of long free bPEI-25K chains, the fraction of Cy3-DNA entrapped into lysosomes (Flyso) slowly increases to ∼20% after 3 h but slightly decreases to ∼15% after 6 h. In contrast, without free PEI chains, the fraction of Cy3-DNA inside lysosomes keeps escalating and reaches ∼40% in the first 6 h. These results are in line with the study of intracellular pH variation around polyplexes. Namely, without free PEI chains (N:P = 3), the intracellular pH around the polyplexes decreases from ∼7.4 to ∼5.7 in the first 6 h, whereas in the presence of long free bPEI-25K chains (N:P = 10), the pH value only slightly decreases to ∼6.8. A combination of these results quantitatively suggest that long free cationic PEI chains are able to prevent the development of later endolysosomes and facilitate the release of polyplexes from the endosomes or even from the original endocytic vesicles.
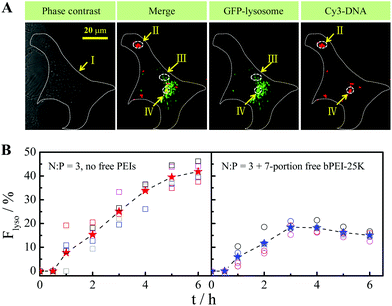 | ||
| Fig. 6 Effect of long free bPEI-25K chains on the fraction of DNA–PEI polyplexes (N:P = 3) entrapped into lysosomes per HepG2 cell (Flyso) monitored by using a confocal three-dimensionally integrated method (detailed in ref. 170 and 171), where DNA and lysosomes were labeled with Cy3 (red) and Lamp-1 GFP (green, indicated by arrow III), respectively. At each indicated time, 10 Z-scan images with a 1-μm step were captured from each cell (cell membrane is indicated by arrow I). Sum of Cy3-fluorescence intensity of yellow clusters (overlay of red and green, indicated by arrow IV) in ten images (Ilyso) indexes pDNA content inside lysosomes; whereas sum of Cy3-intensity of both red and yellow clusters (Itot, indicated by arrow II and IV) indexes total pDNA inside each cell. It has been demonstrated by us that the average Cy3-fluorescence intensity per cell (Iavg) linearly increases with the DNA concentration (CDNA) so that the average amounts of Cy3-DNA inside each cell and lysosomes are estimated from mDNA = CDNA × Vcell and Flyso = Ilyso/Itot × 100%, respectively, where Vcell is the cell volume, and at least 5 cells were analyzed under each experimental condition. | ||
Endocytosis pathway on intracellular fate of polyplexes
Recently, different possible modes of polyplex internalization have been correlated to their subsequent intracellular trafficking routes as well as the ultimate gene transfection efficiency.16,172,173 It is generally known that small polyplexes can be internalized by cells via multiple mechanisms,174 including clathrin-mediated endocytosis (CME, for endocytic vesicles with a size of ∼100–150 nm), caveolae-mediated endocytosis (∼50–80 nm), micropinocytosis (∼90 nm) and macropinocytosis (∼500–2000 nm).175–178In CME pathway, polyplexes are taken up by clathrin-coated pits, transferred to early/late endosomes and ultimately destined to lysosomes (Fig. 7A).179 Alternatively, small polyplexes can be internalized by caveolae, flask-shaped invaginations on the cell surface that bud from microdomains rich in cholesterol and caveolin, and subsequently delivered to caveosomes, pre-exsiting organelles with a stable neutral pH (Fig. 7B).176,180 The caveolae-mediated pathway might be more favorable for gene transfection because there is a relatively less chance for caveolar vesicles to fuse with the late endosomes or lysosomes,16,181 presumably due to the lack of proper signal molecules required for inter-vesicular fusion.182 Micropinocytosis initiates at the non-coated vesicles on the plasma membrane, which bud into the cytosol to form micropinosomes. Such non-coated vesicles become acidified and merge with early endosomes in common with the CME pathway.175,177 Macropinocytosis accompanies the actin-driven membrane ruffling which is regulated by growth factors or other signals. Such membrane protrusions collapse onto and fuse with the plasma membrane to generate large endocytic vesicles, called macropinosomes, that could engulf large polyplexes aggregates (>∼1 μm), as schematically shown in Fig. 7B.177,183–185 Currently, the last three pinocytosis pathways remain poorly understood in comparison with the well-studied and documented CME pathway.
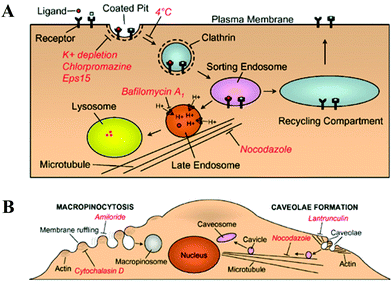 | ||
| Fig. 7 (A) Schematic of clathrin-mediated endocytosis, where internalized ligands are delivered either through a degradative pathway (leading to lysosomes) or a recycling pathway (leading to recycling back to the cell surface). (B) Schematic of macropinocytosis and caveolae-mediated endocytosis, where red italics delineate the inhibitors for indicated functions. (Reprinted from ref. 179 with Nature permission). | ||
Previous studies showed that the internalization of polyplexes made of “off-the-shelf” polymer, such as PEI and PDMAEMA, mainly follows the clathrin- and caveolae-mediated pathways.88,186,187 Blocking either one of them with a specific inhibitor only led to a partial, and sometimes, marginal decrease (<10%) in the cellular uptake of polyplexes, indicating that these two uptake routes might be interchangeable.187 On the other hand, gene transfection was almost completely abolished when the caveolae-mediated pathway was blocked; whereas the gene transfection efficiency remained unchanged or even increased up to 2-fold after the CME pathway was inhibited in A549, HeLa88,186 and COS-7 cell lines.187 Note that in these experiments, the N:P ratio used was in the range 4–6; namely, free polycationic chains are limited in the solution mixture used. If the polyplexes were internalized merely via the CME pathway, such a small amount of free PEI chains might not be sufficient to prevent the entrapment of polyplexes into the late endosomes/lysosomes. Therefore, at lower N:P ratios, the caveolae-dependent route is more likely to lead to an effective gene transfection.
Further, Pack and Gabrielson88 investigated the effects of two cell-targeting ligands, Tf and folic acid, on the intracellular trafficking of the DNA–PEI polyplexes (N:P ∼ 4). It is known that Tf and folic acid are typically internalized via the clathrin- and caveolae-mediated pathways, respectively. The attachments of the two ligands to PEI chains via a covalent bond enables the delivery of polyplexes through the respective pathways. Similar to the previous results, the gene transfection efficiency was not adversely affected after the CME pathway was inhibited, but was entirely abolished after the caveolae-mediated pathway was blocked by small molecular drugs or RNA interference.88 It is further shown that targeting the polyplexes through the caveolae-mediated pathway prevents the rapid and direct fusion of small endocytic vesicles with more acidic late endosomes or lysosomes. These recent results reveal that an optimized targeting ligand for gene therapy should (1) be able to associate with receptors that are typically up-regulated in tumor cells to improve the cellular uptake; and (2) favor the caveolae-mediated endocytosis over other pathways to avoid the delivery of polyplexes into the acidic lysosomes so that the enzymatic degradation of DNA could be prevented.16 Meanwhile, internalization of polyplexes via macro- and micro-pinocytosis should be further elucidated in order to precisely control the intracellular trafficking of polyplexes.
The “proton sponge” concept
For polycationic chains with a proton buffer capacity, such as PEI and other pH-responsive polymers, the so-called “proton sponge” effect on the intracellular trafficking is often taken as granted in the explanation of their high transfection efficiency. The heart of the “proton sponge” effect is that different amines on polymer chains can be further protonated inside endolysosomes, leading to an influx of counter ions (Cl−) and an increase of osmotic pressure inside so that the endolysosomes are finally burst.18,61 Despite its popularity in the field of non-viral gene delivery,188,189 this model has not yet been rigorously validated.16 Two fundamental issues relating to this well-accepted model have to be considered. Firstly, the buffer capacity of a polymeric vector sometimes does not or even reversely correlate(s) to the gene transfection efficiency. For instance, for a given topology, long PEI chains are generally more effective than short ones in gene transfection.167 On the other hand, if only considering their colligative properties, we know from thermodynamics that for a given weight concentration (g mL−1), short chains should generate a higher osmotic pressure inside the endocytic vesicles. The previous results also showed that a simple coupling of 3–4 short bPEI-2K chains via a disulfide linker slightly decreases their buffer capacity but hugely enhances the gene transfection by a factor of ∼104–105 times,78 depending on the N:P ratio, which could not be simply explained by the “proton-sponge” effect.
To investigate the structure–efficacy relationship, Thomas and Klibanov190 performed a set of modifications on the primary, secondary and tertiary amines of bPEI-25K and bPEI-2K chains, which decrease the number of protonable amines and thus lower their buffer capacities. Surprisingly, N-acylation of bPEI-25K with alanine nearly doubled its gene transfection efficacy in the presence of serum. Moreover, dodecylation and hexadecylation of primary amines on the short bPEI-2K enhanced its transfection efficiency by ∼400 times in the presence of serum, even ∼5-fold higher than that of bPEI-25K. Further, Pack et al.191 synthesized a series of modified PEI chains by acetylating different amounts of primary and secondary amines. Their results showed that partial acetylation reduces the buffer capacity, but increases the in-vitro transfection efficiency of those modified PEIs. Particularly, the acetylation of ∼43% primary amines made PEI ∼20-fold more efficient than its parent at N:P ∼ 15–20 no matter whether serum was added. On the other hand, Hennink et al.192 attempted to improve the endosomolytic ability of PDMAEMA by introducing an additional tertiary amino group to each monomeric unit in the hope ofboosting the “proton sponge” effect. Unexpectedly, such a modified PDMAEMA analogue exhibited much lower transfection efficiency even though it was less cytotoxic. However, adding an endosomolytic peptide, INF-7, restored the gene transfection efficiency, clearly indicating that the higher buffer capacity of modified PDMAEMAs are not able to mediate the polyplex release from endolysosomes via the “proton sponge” effect. In a similar way, Schacht et al.193 showed that the imidazole-modified PDMAEMA derivatives had a similar buffer capacity as PEI but were still not able to transfect COS-1 cells, much worse than PDMAEMA with only tertiary amines. Further study revealed that these modified PDMAEMA chains were actually less effective in preventing the entrapment of polyplexes into the acidic late endosomes or lysosomes.194 These studies do not necessarily deny a possible effect of buffer capacity on the non-viral gene transfection. Instead, they indicate that the buffer capacity might not play a dominant and decisive role in promoting the intracellular trafficking of polyplexes.
The second issue is whether the osmotic pressure generated by the “proton sponge” effect is sufficiently high to rupture the endocytic vesicles by itself; or other mechanisms, such as the polycation-membrane physical interaction at a lower pH, are simultaneously involved. Assuming that one clathrin-coated vesicle contains one DNA–PEI complexes of N:P = 7, Won et al.16 estimated that the maximum osmotic pressure produced inside this vesicle was ∼8.3 × 104 Pa when the pH was decreased from 7.4 to 5.0. Such a change in osmotic pressure would expand the membrane area only by 2.3%, whereas lipid vesicles can generally withstand an area expansion up to ∼5% before they start to lose their integrity.195 Note that in reality the proton-absorbing capacity of the polycationic chains must be greatly attenuated because of their complexation with anionic DNA chains as well as their absorption to other anionic membranes and proteins. Therefore, the increase of osmotic pressure inside endocytic vesicles during the acidification alone is theoretically insufficient to rupture them, although it might be a cooperative factor to mediate the eventual release of polyplexes from various endocytic vesicles.
Besides a possible increase of osmotic pressure via the “proton sponge” effect, PEI can also destabilize the anionic membrane via charge neutralization,22,167,196,197 thereby facilitating the release of polyplexes from different kinds of endocytic vesicles. Note that long bPEI-25K chains are much more disruptive to the cellular membrane than short bPEI-2K chains for a given polymer weight concentration, especially when CbPEI ≥ 2.7 μg mL−1, corresponding to N:P ≥ 10 in a typical gene transfection experiment.167 It was also found that long cationic bPEI-25K chains can reverse the charge of the synthetic phospholipid vesicles at a much lower concentration (∼2 μg mL−1) than their short counterparts.167 It seems that the destabilization/disruption of the phospholipid membrane by long free polycationic chains is correlated to the lesser entrapment of polyplexes into the late endolysosomes, and to some extent, to the enhanced uptake of polyplexes from the extracellular space into the cell.
In summary, the aforementioned results have indicated that the escape of polyplexes from endolysosomes is not necessarily mediated by the proposed osmotic-pressure-induced membrane rupture; and the buffer capacity of polycationic chains is only partially responsible for the safe trafficking of polyplexes in the intracellular space. Recently, Wu et al.167 proposed a hypothesis to account for why free long polycationic chains in the solution mixture are able to promote the intracellular trafficking of polyplexes. Namely, it is proposed that long free polycationic chains embedded in or on the membranes might actually block the signal proteins for inter-vesicular fusion so that most of the endocytic vesicles with the polyplexes inside do not fuse with the later endolysosomes in CME pathway. As a result, most of the polyplexes will not be trapped into the acidic lysosomes and suffer from degradation. Such a hypothesis is based on a large amount of experimental evidences as follows.
After being added into the cell culture medium, long polycationic chains are able to quickly penetrate different membranes of a cell and cross the cytosol all the way into the cell nucleus within one hour.166,167 Some of them are inevitably embedded in the membranes.161,198 Typical phospholipid bi-layer membranes with two anionic surfaces have a thickness of 5–6 nm. On one hand, those embedded polycationic chains can destabilize/disrupt the membranes by interacting with anionic phospholipids and thus facilitate the escape of the polyplexes trapped inside (here the “proton sponge” effect might be helpful). On the other hand, those embedded chains with a sufficient length (∼15–20 nm) can interact with the membrane proteins. It is generally known that lysosomes differentiate the endocytic vesicles with entrapped foreign subjects from those vesicles generated from different organelles inside the cell by the signal proteins attached at the inner surface of the cell membrane. Shielding or malfunctioning these signal proteins will attenuate or hinder the inter-vesicular fusion and block the development of the later endolysosomes.
Using this hypothesis, one is able to explain many of the currently observed differences and contradictions in gene transfection, e.g., (1) why long linear and branched PEI-25K chains have a similar transfection efficiency but short free PEI chains are less effective? (e.g. bPEI-0.8K ∼ 4 nm and bPEI-2K ∼ 6 nm, too short to shield the signal proteins);167 (2) why lPEI-2.5K is more effective than bPEI-2K? (lPEI-2.5K chain is ∼18 nm, much longer than its branched counterpart);167 (3) why coupling 3–4 short bPEI-2K chains into a long one (∼20 nm) can enhance the transfection efficiency by a factor of ∼104 times;78 and (4) why attaching a short hydrophobic chain190,199 or cholesterol200,201 to the less effective bPEI-2K can significantly promote the gene transfection? (The short hydrophobic chain and cholesterol can insert into the membrane so that bPEI-2K can stick out to shield the signal proteins).
It should be noted that the longer the polycationic chains, the more cytotoxic they become, because long polycationic chains disrupt the membranes, cause the leakage of cytoplasm into extracellular space, and induce necrosis and apoptosis.46 Therefore, there is a dedicate balance between the cytotoxicity and the transfection efficiency. The hypothesis leads to a better and practical strategy in the development of non-viral polymeric vectors. Namely, one might use a small amount of long and more toxic polycationic chains (e.g., bPEI-25K) to effectively condense DNA into small polyplexes with N:P = 3; and use short and less toxic chains (e.g., bPEI-2K, ∼5 nm) modified with a proper hydrophobic molecule (sticker, ∼2–5 nm in size) as the 7 portions of free chains so that their hydrophobic part can insert into the cell membrane to expose their short cationic part to shield the signal proteins. In this way, such a catch-22 “transfection efficiency”-versus-“cytotoxicity” problem could be solved. Previously, such a strategy was exploited but not established on the above hypothesis. For example, Mahato et al.200 have shown that attaching one cholesterol to each short bPEI-1.8K chain greatly boosted its transfection efficiency, while the modification on bPEI-10K had no such enhancement in the gene transfection, presumably because bPEI-10K is long enough to insert into the membranes. Alternatively, Uludag et al.199 modified bPEI-2K with a set of aliphatic lipids with different lengths and found that attaching an aliphatic lipid to bPEI-2K can turn the ineffective bPEI-2K into an effective gene-delivery vehicle. Notably, linoleic acid (LA, C17H31CO–) and palmitic acid (PA, C15H31CO–) substituted PEI derivatives led to a much higher gene transfection efficiency than caprylic acid (CA, C7H15CO–) substituted ones (lipid: PEI molar ratio ∼ 1, N:P ∼ 35), comparable to the potent bPEI-25K but much less cytotoxic.
Nuclear localization and unloading of DNA
Once internalized into the cell and avoiding lysosomal entrapment, polyplexes have to move towards and enter the cell nucleus, and unload/release the DNA inside for transcription. In the cytosolic transport step, some of the polyplexes might first escape from the endocytic vesicles and then travel along microtubules to the perinuclear region, similar to adenoviruses.202 Alternatively, they are more likely to reach the nucleus periphery within the endocytic vesicles (which are transported on microtubules) and then release from them before entering the nucleus, resembling adeno-associated viruses.203 Either way, experimental evidence showed that the cytosolic delivery of polyplexes to the nucleus periphery is an active (not passive diffusive) process,204–207 with a linear speed of v ∼ 10−1 μm s−1 in both COS-7204 and HUH-7 cells,206 measured by a real-time multiple particle tracking (MPT) technique. It is worth noting that such transportation is generally not a rate-limiting step in the intracellular trafficking. However, less attention has been paid to the subsequent nuclear localization, which does impose a great hurdle in gene transfection.208 The cell nucleus is separated from the cytoplasm by a double-layer membrane with tightly regulated pores that govern the import and export of a specific set of biomacromolecules (RNAs and proteins). The nuclear pore complexes (NPCs) allow passive diffusion of small molecules (diameter < 3–5 nm), while larger proteins have to be actively transported via specific nuclear proteins, such as importins.2 Viruses have evolved functions to utilize this nuclear import machinery, but unmodified polymers or pDNA clearly have no such an ability. Early studies showed that the polyplexes (or pDNA) mainly entered the nucleus during cell mitosis when the nuclear membrane was temporarily dismantled.2,209 This partially explains why the gene transfection efficiency is extremely low when non-dividing or growth-arrested cells are used.209Many proteins are naturally targeted to the nucleus via some nuclear localization signals (NLS), short cationic peptides whose sequences are recognized by importins.2 Using such a nuclear import machinery, one can attach a synthetic peptide with a NLS peptide to DNA so that the hybrid DNA-NLS can be identified as a nuclear import substrate. Initial studies showed that the conjugation of a NLS peptide to a circular210 or linear DNA211 enhanced the importin-induced nuclear translocation in the gene transfection. Recent studies also revealed that the nuclear factor kappa B (NFκB), a family of transcription factors that shuttle between the cytoplasm and cell nucleus under specific conditions, is a desirable intracellular target to increase the nuclear import of pDNA.212 The NFκB binding sequences were optimized and constructed into pDNA, leading to an effective nuclear import and a prolonged in-vivo transgene expression.213 Note that in such a strategy, the unloading of pDNA from the polyplexes in the cytosol, preferably near the nuclear membrane, is a prerequisite. Jeong et al.214 developed poly(amido ethylenimine), whose backbone is degradable in the cytoplasm by reduction, to facilitate the release of pDNA from the polyplexes in the cytosol. They showed that upon the activation of NFκB by interleukin-1β, most of the pDNA released due to the poly(amido ethylenimine) degradation were translocated into the cell nucleus, leading to a much higher transfection efficiency in comparison with the PEI-mediated transfection. In another study, Choi et al.215 improved the nuclear import of polyplexes by attaching a glucocorticoid steroid molecule, dexamethasone, to bPEI-2K because dexamethasone can dilate the NPCs upon binding to its glucocorticoid receptor and thereby create a “giant pore” for impermeable macromolecules.216 In this way, the dexamethasone-conjugated bPEI-2K and large bPEI-25K exhibited a similar gene transfection efficiency for higher N:P ratios but the bPEI-2K derivatives were much less cytotoxic.
Incorporating a viral component into a non-viral gene delivery system is another approach to enhance the nuclear translocation. Very recently, Pack et al.217 constructed a hybrid polymer-virus vector by coating the small non-infectious retroviral-like particles without a viral protein envelope with cationic PLL or PEI chains. The cationic polymer coatings are used to mediate the cellular uptake and release of the hybrid particles from endosomes. Such hybrid vectors are efficient in gene transfection, retaining some important viral-like functions, including nuclear import, genomic integration, and infection of non-dividing cells; and at the same time, avoid some disadvantages inherent to the native viruses, such as the notorious fragility to physical forces in common processing conditions.
In the extracellular space, we like to compact anionic DNA by cationic polymer chains as much as possible so that polyplexes can be brought to cross the cell membrane and be protected inside the cytoplasm before they hit the nuclear membrane, but then we wish the DNA–polymer complexation to be weak so that DNA could be easily released for transcription inside the nucleus. Again, this is a narrow “slit” between these two requirements, another catch-22 problem.218–220 A quantitative comparison of the intracellular trafficking between adenovirus and non-viral vectors (cationic lipids and PEI) revealed that in addition to the nuclear import, another rate-limiting step for non-viral gene delivery is the transcription and translation of the exogenous DNA,221,222 which might be related to the slow release of DNA from the polyplexes, i.e., the replacement of DNA molecules by other polyanionic chains near or inside the nucleus, presumably other proteins or DNA/RNA chains. This leads to another question: should DNA be released before or after its nuclear entry? Early studies observed that pDNA entered the cell nucleus together with its cationic vector,198 but later, it was found that pDNA was (at least partially) dissociated from the polyplexes upon their release from endosomes.16,223 Using real-time CLSM, our recent studies revealed the existence of the released DNA in the cytosol as well as the polymer-bound DNA inside the nucleus. Nevertheless, most DNA chains are still inside the polyplexes in the cytosol. It is also found that for DNA–bPEI-25K of N:P = 10, the transgene expression was detectable as early as 6 h after addition of the polyplex to the HepG2 cells, while the corresponding transgene expression is ∼10% of the maximum value at 36 h.
Currently, it is rather difficult, if not impossible, to elucidate whether this early transgene expression is prevailingly mediated by the pDNA released inside the cytosol or the nucleus, or both. We still question whether and how those free polycationic chains in the solution mixture play a vital role in this process. It is only generally known that for an efficient polymeric gene delivery, the polyplexes should be properly “programmed” to release their DNA payloads after they reach the nuclear membrane or enter the nucleus.16 The advancements of modern analytic methods, such as live cell imaging with high spatio-temporal resolution, real-time particle tracking and intravital real-time CLSM, start to enable us to “see” the cytosolic and nuclear delivery of the therapeutic genes, and more importantly, to elucidate how those free polycationic chains help the polyplexes to navigate through each of many “slits” in the intracellular space. Therefore, in addition to synthesizing more polymeric vectors, we should also pay great attention to the detailed mechanism of the intracellular pDNA unpacking via well-designed comparing/differentiating experiments so that the future developments of non-viral polymer vectors can be better guided.
4. Future research and development of non-viral polymeric vectors
Over the past few decades, polycationic chains with different sizes and topologies, sometimes exotic, have been designed and synthesized in vitro and in vivo to deliver genes into a variety of cells and tissues. However, their transfection efficacy remains disappointing, orders of magnitude lower than their viral counterparts.16 It is our opinion that this is, at least partially, due to the lack of some fundamental understanding on how DNA is delivered into the cell nucleus; namely, a detailed pathway(s) for the intracellular trafficking of DNA–polymer complexes. In the last 15 years, the astonishing advancement of molecular cell biology and its related commercially available analytic tools/kits have now enabled us to study this complicated problem. In our opinion, it might be improper nowadays to keep fishing potential non-viral polymer vectors in a lottery fashion. Our suggestions are as follows.(1) Much attention has already been paid to endocytosis in the past, as well as the DNA complexation and condensation in the extracellular space. More and more evidence has been accumulated to show that the complexation is mainly due to charge neutrality, a counterion-related entropically driven process.146 Therefore, we should shift our attention away from it. Note that ingesting part of the plasma membrane (endocytosis) is a constant cell activity. It normally takes 1–2 h for a typical cell to replace its entire plasma membrane via the endo-and-exo-cytosis circle. Thus, endocytosis of polyplexes with appropriate cell-targeting ligands should not impose an immense hurdle as long as we are able to bring them sufficiently close to the cell membrane. This is why a slightly positively charged periphery of polyplexes is important and the attachment of proper ligands to target receptors on the cell membrane is helpful. Meanwhile, internalization of polyplexes via multiple clathrin-independent pathways should be further examined and elucidated in order to precisely control the intracellular trafficking of polyplexes.
(2) Much attention has also been paid to the release of the polyplexes from endolysosomes by using polycationic chains that have a buffer capacity or are pH-sensitive. Recent experimental results have revealed that it is the free polycationic chains in the solution mixture (N:P ≥ 6) that actually promote gene transfection, especially in the intracellular trafficking pathway(s).165–167 Therefore, in addition to stimulating the escape of polyplexes from late endosomes/lysosomes, we should also consider how to block inter-vesicular fusion between the initial polyplex-containing endocytic vesicles and early endosomes so that they will not be developed into endolysosomes. In this way, the escape of trapped polyplexes from lysosomes will not be an issue. The important two related issues here will be the detailed molecular mechanisms of (a) how free polycationic chains possibly prevent inter-vesicular fusion; and (b) how the polyplexes escape from the small initial endocytic vesicles into the cytosol.
(3) More attention should be given to the transport of polyplexes through the cytosol. Note that the cytosol is a fairly concentrated protein solution (∼30% by weight) with a high viscosity. It is hard to imagine that polyplexes are able to passively move towards the cell nucleus by thermal diffusion. Some past experiments showed that the polyplex-filled endocytic vesicles can be actively transported to the nuclear periphery via microtubules.204–206 More studies and convincing evidence are required to confirm such a pathway; even though the migration of polyplexes towards cell nucleus might not be a rate-determining step it is certainly important.
(4) More efforts should be directed to a better understanding of how the polyplexes or large DNA chains actually pass through the nuclear membrane, especially when the cells are not in their mitosis state. A few subsequent questions are as follows: (a) Are DNA chains released from the polyplexes before or after passing through the nuclear membrane? (b) How are the polyplexes or even the released DNA chains able to pass through the nuclear pores much smaller than them? (c) How can we artificially induce the temporal formation of large pores on the nuclear membrane, or the temporal dismantlement of nuclear membrane to allow the released DNA or polyplexes into the nucleus?
(5) Chemists should learn from molecular cell biologists to understand and recognize some subtle differences between micro-environments in the cytosol and cell nucleus so that one can use them to design and prepare a new generation of superior non-viral polymeric vectors for gene delivery; namely, these novel vectors can release their captured DNA cargos in a more controllable manner inside the cell. This problem is extremely complicated and multidimensional and its researchers need to be similarly multidimensional. Polymer researchers who are interested in the development of useful, efficient non-viral vectors have no choice but to sit down and learn sufficient molecular cell biology and pharmacology because our future is multidisciplinary.
In summary, a huge amount of literature (∼105 publications) has been accumulated over the past 3–4 decades in the search of non-viral polymeric vectors. We have made much important progress, but our success is still limited with respect to clinical applications. For simple questions, we can sometimes rely on our intuition to solve them, but for complicated biological problems, such as the development of superior non-viral vectors, a hypothesis-driven strategy might be more favorable. It should be emphasized that there is still a hope to design and construct a multi-functional polymeric delivery system that can navigate (waltz) through various intracellular “slits” (obstacles) if we can properly address and elucidate the above questions. To do so, a combination of chemistry, molecular cell biology and polymer physics is not only a necessary but also a sufficient approach. We will also have to learn and use many innovative biophysical characterization methods, especially those single molecule techniques specifically developed for in situ cell studies. Collaboration and communication between viral (biologists) and non-viral (chemists) fields should be enhanced. Finally, it should be noted that the in vivo animal or clinic tests of non-viral polymeric vectors involve very different kind of problems,31,224–227 such as the particle stability in the blood circulation, immune responses and cell-targeting property, which beyond our discussion in this review.
Acknowledgements
The financial support of the National Natural Scientific Foundation of China (NNSFC) Projects (20934005 and 51173177), the Strategic Investment Fund—Scheme B, The Chinese University of Hong Kong (1903020), and Hong Kong Special Administration Region Earmarked (RGC) Projects (CUHK4042/09P, 2160396; and CUHK4042/10P, 2130241; 2060405) is gratefully acknowledged.Notes and references
- R. Mulligan, Science, 1993, 260, 926 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581 CrossRef CAS.
- I. M. Verma and N. Somia, Nature, 1997, 389, 239 CrossRef CAS.
- R. G. Vile, A. Tuszynski and S. Castleden, Mol. Biotechnol., 1996, 5, 139 Search PubMed.
- M. J. During, Adv. Drug Delivery Rev., 1997, 27, 83 Search PubMed.
- A. Vasileva and R. Jessberger, Nat. Rev. Microbiol., 2005, 3, 837 Search PubMed.
- E. Check, Nature, 2002, 420, 116 CrossRef CAS.
- S. Hacien-Bey-Abina, Science, 2003, 302, 568 Search PubMed.
- S. E. Raper, N. Chirmule, F. S. Lee, N. A. Wivel, A. Bagg, G. P. Gao, J. M. Wilson and M. L. Batshaw, Mol. Genet. Metab., 2003, 80, 148 CrossRef CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259 CrossRef CAS.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413.
- J. Zabner, Adv. Drug Delivery Rev., 1997, 27, 17 CrossRef CAS.
- I. Koltover, T. Salditt, J. O. Radler and C. R. Safinya, Science, 1998, 281, 78 CrossRef CAS.
- W. J. Li and F. C. Szoka, Pharm. Res., 2007, 24, 438 CrossRef CAS.
- S. D. Li and L. Huang, J. Controlled Release, 2007, 123, 181 CrossRef CAS.
- Y.-Y. Won, R. Sharma and S. F. Konieczny, J. Controlled Release, 2009, 139, 88 CrossRef CAS.
- G. Y. Wu and C. H. Wu, J. Biol. Chem., 1987, 262, 4429 CAS.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297 CAS.
- M. Neu, D. Fischer and T. Kissel, J. Gene Med., 2005, 7, 992 CrossRef CAS.
- U. Lungwitz, M. Breunig, T. Blunk and A. Gopferich, Eur. J. Pharm. Biopharm., 2005, 60, 247 CrossRef CAS.
- H. Pollard, J. S. Remy, G. Loussouarn, S. Demolombe, J. P. Behr and D. Escande, J. Biol. Chem., 1998, 273, 7507 CrossRef CAS.
- T. Bieber, W. Meissner, S. Kostin, A. Niemann and H. P. Elsasser, J. Controlled Release, 2002, 82, 441 CrossRef CAS.
- Y. K. Oh, D. Suh, J. M. Kim, H. G. Choi, K. Shin and J. J. Ko, Gene Ther., 2002, 9, 1627 CrossRef CAS.
- K. G. de Bruin, C. Fella, M. Ogris, E. Wagner, N. Ruthardt and C. Brauchle, J. Controlled Release, 2008, 130, 175 CrossRef CAS.
- T. Niidome and L. Huang, Gene Ther., 2002, 9, 1647 CrossRef CAS.
- T. G. Park, J. H. Jeong and S. W. Kim, Adv. Drug Delivery Rev., 2006, 58, 467 CrossRef CAS.
- S. Y. Wong, J. M. Pelet and D. Putnam, Prog. Polym. Sci., 2007, 32, 799 CrossRef CAS.
- J. Luten, C. F. van Nostruin, S. C. De Smedt and W. E. Hennink, J. Controlled Release, 2008, 126, 97 CrossRef CAS.
- J. A. Wolff and D. B. Rozema, Mol. Ther., 2008, 16, 8 CrossRef CAS.
- D. Schaffert and E. Wagner, Gene Ther., 2008, 15, 1131 CrossRef CAS.
- M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J. P. Benoit, Biomaterials, 2008, 29, 3477 CrossRef CAS.
- B. F. Canine and A. Hatefi, Adv. Drug Delivery Rev., 2010, 62, 1524 CrossRef CAS.
- Y. W. Won, K. S. Lim and Y. H. Kim, J. Controlled Release, 2011, 152, 99 CrossRef CAS.
- S. Akhtar, M. D. Hughes, A. Khan, M. Bibby, M. Hussain, Q. Nawaz, J. Double and P. Sayyed, Adv. Drug Delivery Rev., 2000, 44, 3 CrossRef CAS.
- S. Akhtar and I. F. Benter, J. Clin. Invest., 2007, 117, 3623 CrossRef CAS.
- S. B. Zhang, B. Zhao, H. M. Jiang, B. Wang and B. C. Ma, J. Controlled Release, 2007, 123, 1 CrossRef CAS.
- K. A. Howard, Adv. Drug Delivery Rev., 2009, 61, 710 CrossRef CAS.
- J. H. Jeong, H. Mok, Y. K. Oh and T. G. Park, Bioconjugate Chem., 2009, 20, 5 CrossRef CAS.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129 CrossRef CAS.
- G. Y. Wu and C. H. Wu, J. Biol. Chem., 1988, 263, 14621 CAS.
- J. Haensler and F. C. Szoka, Bioconjugate Chem., 1993, 4, 372 CrossRef CAS.
- J. F. KukowskaLatallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897 CrossRef CAS.
- X. Q. Zhang, X. L. Wang, S. W. Huang, R. X. Zhuo, Z. L. Liu, H. Q. Mao and K. W. Leong, Biomacromolecules, 2005, 6, 341 CrossRef CAS.
- Y. Y. Cheng, L. B. Zhao, Y. W. Li and T. W. Xu, Chem. Soc. Rev., 2011, 40, 2673 RSC.
- A. C. Hunter, Adv. Drug Delivery Rev., 2006, 58, 1523 CrossRef CAS.
- S. M. Moghimi, L. Parhamifar, A. K. Larsen, A. C. Hunter and T. L. Andresen, Soft Matter, 2010, 6, 4001 RSC.
- B. Gupta, T. S. Levchenko and V. P. Torchilin, Adv. Drug Delivery Rev., 2005, 57, 637 CrossRef CAS.
- A. Mann, G. Thakur, V. Shukla and M. Ganguli, Drug Discovery Today, 2008, 13, 152 Search PubMed.
- T. Endoh and T. Ohtsuki, Adv. Drug Delivery Rev., 2009, 61, 704 CrossRef CAS.
- J. S. Suk, J. Suh, K. Choy, S. K. Lai, J. Fu and J. Hanes, Biomaterials, 2006, 27, 5143 CrossRef CAS.
- S. L. Lo and S. Wang, Biomaterials, 2008, 29, 2408 CrossRef CAS.
- H. Y. Nam, J. Kim, S. Kim, J. W. Yockman, S. W. Kim and D. A. Bull, Biomaterials, 2011, 32, 5213 CrossRef CAS.
- A. Astriab-Fisher, D. Sergueev, M. Fisher, B. R. Shaw and R. L. Juliano, Pharm. Res., 2002, 19, 744 Search PubMed.
- F. Simeoni, M. C. Morris, F. Heitz and G. Divita, Nucleic Acids Res., 2003, 31, 2717 CrossRef CAS.
- A. Muratovska and M. R. Eccles, FEBS Lett., 2004, 558, 63 CrossRef CAS.
- N. P. Gabrielson and J. J. Cheng, Biomaterials, 2010, 31, 9117 CrossRef CAS.
- G. D. Jones, Dc Macwilli and N. A. Braxtor, J. Org. Chem., 1965, 30, 1994 Search PubMed.
- H. Cheradame, B. Brissault, A. Kichler, C. Guis, C. Leborgne and O. Danos, Bioconjugate Chem., 2003, 14, 581 CrossRef CAS.
- I. M. Klotz and Ar. Sloniews, Biochem. Biophys. Res. Commun., 1968, 31, 421 Search PubMed.
- J. Suh, H. J. Paik and B. K. Hwang, Bioorg. Chem., 1994, 22, 318 CrossRef CAS.
- J. P. Behr, Chimia, 1997, 51, 34 CAS.
- D. Fischer, T. Bieber, Y. X. Li, H. P. Elsasser and T. Kissel, Pharm. Res., 1999, 16, 1273 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 1999, 45, 268 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, Biomaterials, 2001, 22, 471 CrossRef.
- T. Reschel, C. Konak, D. Oupicky, L. W. Seymour and K. Ulbrich, J. Controlled Release, 2002, 81, 201 CrossRef CAS.
- T. Kissel, K. Kunath, A. von Harpe, D. Fischer, H. Peterson, U. Bickel and K. Voigt, J. Controlled Release, 2003, 89, 113 CrossRef CAS.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990 CrossRef CAS.
- A. Beyerle, M. Irmler, J. Beckers, T. Kissel and T. Stoeger, Mol. Pharmaceutics, 2010, 7, 727 CAS.
- G. Grandinetti, N. P. Ingle and T. M. Reineke, Mol. Pharmaceutics, 2011, 8, 1709 CAS.
- X. L. Gao, L. Yao, Q. X. Song, L. Zhu, Z. Xia, H. M. Xia, X. G. Jiang, J. Chen and H. Z. Chen, Biomaterials, 2011, 32, 8613 Search PubMed.
- M. A. Gosselin, W. J. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989 CrossRef CAS.
- M. Thomas, Q. Ge, J. J. Lu, J. Z. Chen and A. M. Klibanov, Pharm. Res., 2005, 22, 373 CrossRef CAS.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454 CrossRef CAS.
- Q. Peng, Z. L. Zhong and R. X. Zhuo, Bioconjugate Chem., 2008, 19, 499 CrossRef CAS.
- H. C. Kang, H. J. Kang and Y. H. Bae, Biomaterials, 2011, 32, 1193 CrossRef CAS.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS.
- Y. Lee, H. Mo, H. Koo, J. Y. Park, M. Y. Cho, G. W. Jin and J. S. Park, Bioconjugate Chem., 2007, 18, 13 CrossRef CAS.
- R. Deng, Y. Yue, F. Jin, Y. C. Chen, H. F. Kung, M. C. M. Lin and C. Wu, J. Controlled Release, 2009, 140, 40 CrossRef CAS.
- L. Wightman, R. Kircheis, V. Rossler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362 CrossRef CAS.
- P. R. Dash, M. L. Read, L. B. Barrett, M. Wolfert and L. W. Seymour, Gene Ther., 1999, 6, 643 CrossRef CAS.
- M. Ogris, S. Brunner, S. Schuller, R. Kircheis and E. Wagner, Gene Ther., 1999, 6, 595 CrossRef CAS.
- H. Petersen, P. M. Fechner, A. L. Martin, K. Kunath, S. Stolnik, C. J. Roberts, D. Fischer, M. C. Davies and T. Kissel, Bioconjugate Chem., 2002, 13, 845 CrossRef CAS.
- V. Knorr, L. Allmendinger, G. F. Walker, F. F. Paintner and E. Wagner, Bioconjugate Chem., 2007, 18, 1218 CrossRef CAS.
- M. Kursa, G. F. Walker, V. Roessler, M. Ogris, W. Roedl, R. Kircheis and E. Wagner, Bioconjugate Chem., 2003, 14, 222 CrossRef CAS.
- M. A. Zanta, O. Boussif, A. Adib and J. P. Behr, Bioconjugate Chem., 1997, 8, 839 CrossRef CAS.
- K. de Bruin, N. Ruthardt, K. von Gersdorff, R. Bausinger, E. Wagner, M. Ogris and C. Brauchle, Mol. Ther., 2007, 15, 1297 CrossRef CAS.
- H. Cheng, J. L. Zhu, X. Zeng, Y. Jing, X. Z. Zhang and R. X. Zhuo, Bioconjugate Chem., 2009, 20, 481 CrossRef CAS.
- N. P. Gabrielson and D. W. Pack, J. Controlled Release, 2009, 136, 54 CrossRef CAS.
- H. Y. Tian, L. Lin, J. Chen, X. S. Chen, T. G. Park and A. Maruyama, J. Controlled Release, 2011, 155, 47 Search PubMed.
- P. van de Wetering, J. Y. Cherng, H. Talsma and W. E. Hennink, J. Controlled Release, 1997, 49, 59 CrossRef CAS.
- P. van de Wetering, E. E. Moret, N. M. E. Schuurmans-Nieuwenbroek, M. J. van Steenbergen and W. E. Hennink, Bioconjugate Chem., 1999, 10, 589 CrossRef CAS.
- P. van de Wetering, N. M. E. Schuurmans-Nieuwenbroek, W. E. Hennink and G. Storm, J. Gene Med., 1999, 1, 156 CrossRef CAS.
- J. G. Cai, Y. A. Yue, D. Rui, Y. F. Zhang, S. Y. Liu and C. Wu, Macromolecules, 2011, 44, 2050 CrossRef CAS.
- F. A. Plamper, A. Schmalz, E. Penott-Chang, M. Drechsler, A. Jusufi, M. Ballauff and A. H. E. Muller, Macromolecules, 2007, 40, 5689 CrossRef CAS.
- C. V. Synatschke, A. Schallon, V. Jerome, R. Freitag and A. H. E. Muller, Biomacromolecules, 2011, 12, 4247 CrossRef CAS.
- J. M. Layman, S. M. Ramirez, M. D. Green and T. E. Long, Biomacromolecules, 2009, 10, 1244 CrossRef CAS.
- T. K. Georgiou, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2006, 7, 3505 CrossRef CAS.
- S. Uzgun, O. Akdemir, G. Hasenpusch, C. Maucksch, M. M. Golas, B. Sander, H. Stark, R. Imker, J. F. Lutz and C. Rudolph, Biomacromolecules, 2010, 11, 39 CrossRef CAS.
- J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023 CrossRef CAS.
- H. Gonzalez, S. J. Hwang and M. E. Davis, Bioconjugate Chem., 1999, 10, 1068 CrossRef CAS.
- M. E. Davis, Mol. Pharmaceutics, 2009, 6, 659 CrossRef CAS.
- F. Kihara, H. Arima, T. Tsutsumi, F. Hirayama and K. Uekama, Bioconjugate Chem., 2002, 13, 1211 CrossRef CAS.
- S. H. Pun, N. C. Bellocq, A. J. Liu, G. Jensen, T. Machemer, E. Quijano, T. Schluep, S. F. Wen, H. Engler, J. Heidel and M. E. Davis, Bioconjugate Chem., 2004, 15, 831 CrossRef CAS.
- M. L. Forrest, N. Gabrielson and D. W. Pack, Biotechnol. Bioeng., 2005, 89, 416 CrossRef CAS.
- G. P. Tang, H. Y. Guo, F. Alexis, X. Wang, S. Zeng, T. M. Lim, J. Ding, Y. Y. Yang and S. Wang, J. Gene Med., 2006, 8, 736 CrossRef CAS.
- J. Li and X. J. Loh, Adv. Drug Delivery Rev., 2008, 60, 1000 CrossRef CAS.
- H. Yao, S. S. Ng, W. O. Tucker, Y. K. T. Tsang, K. Man, X. M. Wang, B. K. C. Chow, H. F. Kung, G. P. Tang and M. C. Lin, Biomaterials, 2009, 30, 5793 CrossRef CAS.
- Q. L. Hu, J. L. Wang, J. Shen, M. Liu, X. Jin, G. P. Tang and P. K. Chu, Biomaterials, 2012, 33, 1135 CrossRef CAS.
- C. A. Yang, H. Z. Li, S. H. Goh and J. Li, Biomaterials, 2007, 28, 3245 CrossRef CAS.
- F. J. Xu, Z. X. Zhang, Y. Ping, J. Li, E. T. Kang and K. G. Neoh, Biomacromolecules, 2009, 10, 285 CrossRef CAS.
- S. Srinivasachari, K. M. Fichter and T. M. Reineke, J. Am. Chem. Soc., 2008, 130, 4618 CrossRef.
- A. Diaz-Moscoso, L. Le Gourrierec, M. Gomez-Garcia, J. M. Benito, P. Balbuena, F. Ortega-Caballero, N. Guilloteau, C. Di Giorgio, P. Vierling, J. Defaye, C. O. Mellet and J. M. G. Fernandez, Chem.–Eur. J., 2009, 15, 12871 CrossRef CAS.
- A. Diaz-Moscoso, D. Vercauteren, J. Rejman, J. M. Benito, C. O. Mellet, S. C. De Smedt and J. M. G. Fernandez, J. Controlled Release, 2010, 143, 318 CrossRef CAS.
- A. Mendez-Ardoy, N. Guilloteau, C. Di Giorgio, P. Vierling, F. Santoyo-Gonzalez, C. O. Mellet and J. M. G. Fernandez, J. Org. Chem., 2011, 76, 5882 CrossRef CAS.
- S. H. Pun and M. E. Davis, Bioconjugate Chem., 2002, 13, 630 CrossRef CAS.
- N. C. Bellocq, S. H. Pun, G. S. Jensen and M. E. Davis, Bioconjugate Chem., 2003, 14, 1122 CrossRef CAS.
- D. W. Bartlett and M. E. Davis, Bioconjugate Chem., 2007, 18, 456 CrossRef.
- N. C. Bellocq, M. E. Davis, H. Engler, G. S. Jensen, A. J. Liu, T. Machemer, D. C. Maneval, E. Quijano, S. Z. H. Pun, T. Schluep and S. F. Wen, Mol. Ther., 2003, 7, S290 Search PubMed.
- S. H. Pun, F. Tack, N. C. Bellocq, J. J. Cheng, B. H. Grubbs, G. S. Jensen, M. E. Davis, M. Brewster, M. Janicot, B. Janssens, W. Floren and A. Bakker, Cancer Biol. Ther., 2004, 3, 641 Search PubMed.
- S. Hu-Lieskovan, J. D. Heidel, D. W. Bartlett, M. E. Davis and T. J. Triche, Cancer Res., 2005, 65, 8984 CrossRef CAS.
- D. W. Bartlett, H. Su, I. J. Hildebrandt, W. A. Weber and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15549 CrossRef CAS.
- J. D. Heidel, Z. P. Yu, J. Y. C. Liu, S. M. Rele, Y. C. Liang, R. K. Zeidan, D. J. Kornbrust and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5715 CrossRef CAS.
- M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067 CrossRef CAS.
- D. Grimm and M. A. Kay, Curr. Gene Ther., 2003, 3, 281 Search PubMed.
- P. Midoux and M. Monsigny, Bioconjugate Chem., 1999, 10, 406 CrossRef CAS.
- J. M. Benns, J. S. Choi, R. I. Mahato, J. S. Park and S. W. Kim, Bioconjugate Chem., 2000, 11, 637 CrossRef CAS.
- M. E. H. El-Sayed, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2005, 101, 47 CrossRef CAS.
- I. K. Park, K. Singha, R. B. Arote, Y. J. Choi, W. J. Kim and C. S. Cho, Macromol. Rapid Commun., 2010, 31, 1122 CrossRef CAS.
- J. L. Thomas and D. A. Tirrell, Acc. Chem. Res., 1992, 25, 336 CrossRef CAS.
- J. L. Thomas, S. W. Barton and D. A. Tirrell, Biophys. J., 1994, 67, 1101 CrossRef CAS.
- N. Murthy, J. R. Robichaud, D. A. Tirrell, P. S. Stayton and A. S. Hoffman, J. Controlled Release, 1999, 61, 137 CrossRef CAS.
- N. Murthy, I. Chang, P. Stayton and A. Hoffman, Macromol. Symp., 2001, 172, 49 CrossRef CAS.
- C. Y. Cheung, N. Murthy, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 906 CrossRef CAS.
- T. R. Kyriakides, C. Y. Cheung, N. Murthy, P. Bornstein, P. S. Stayton and A. S. Hoffman, J. Controlled Release, 2002, 78, 295 CrossRef CAS.
- T. Kiang, C. Bright, C. Y. Cheung, P. S. Stayton, A. S. Hoffman and K. W. Leong, J. Biomater. Sci., Polym. Ed., 2004, 15, 1405 Search PubMed.
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221 CrossRef CAS.
- S. Foster, C. L. Duvall, E. F. Crownover, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2010, 21, 2205 CrossRef CAS.
- K. Miyata, M. Oba, M. Nakanishi, S. Fukushima, Y. Yamasaki, H. Koyama, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 16287 CrossRef CAS.
- K. Itaka, T. Ishii, Y. Hasegawa and K. Kataoka, Biomaterials, 2010, 31, 3707 CrossRef CAS.
- T. Nomoto, Y. Matsumoto, K. Miyata, M. Oba, S. Fukushima, N. Nishiyama, T. Yamasoba and K. Kataoka, J. Controlled Release, 2011, 151, 104 CrossRef CAS.
- S. C. W. Richardson, N. G. Pattrick, Y. K. S. Man, P. Ferruti and R. Duncan, Biomacromolecules, 2001, 2, 1023 CrossRef CAS.
- N. G. Pattrick, S. C. W. Richardson, M. Casolaro, P. Ferruti and R. Duncan, J. Controlled Release, 2001, 77, 225 Search PubMed.
- K. W. Wan, B. Malgesini, L. Verpilio, P. Ferruti, P. C. Griffiths, A. Paul, A. C. Hann and R. Duncan, Biomacromolecules, 2004, 5, 1102 Search PubMed.
- S. C. W. Richardson, N. G. Pattrick, N. Lavignac, P. Ferruti and R. Duncan, J. Controlled Release, 2010, 142, 78 Search PubMed.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269 CrossRef CAS.
- H. G. Hansma, R. Golan, W. Hsieh, C. P. Lollo, P. Mullen-Ley and D. Kwoh, Nucleic Acids Res., 1998, 26, 2481 CrossRef CAS.
- E. Lai and J. H. van Zanten, Biophys. J., 2001, 80, 864 CrossRef CAS.
- E. Vuorimaa, A. Urtti, R. Seppanen, H. Lemmetyinen and M. Yliperttula, J. Am. Chem. Soc., 2008, 130, 11695 CrossRef CAS.
- G. M. Pavan, A. Danani, S. Pricl and D. K. Smith, J. Am. Chem. Soc., 2009, 131, 9686 CrossRef CAS.
- J. Ziebarth and Y. M. Wang, Biophys. J., 2009, 97, 1971 Search PubMed.
- R. Deng, S. Diao, Y. N. Yue, T. Ngai, C. Wu and F. Jin, Biopolymers, 2010, 93, 571 Search PubMed.
- C. B. Sun, T. Tang and H. Uludag, J. Phys. Chem. B, 2012, 116, 2405 Search PubMed.
- E. Wagner, M. Cotten, R. Foisner and M. L. Birnstiel, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4255 CrossRef CAS.
- K. Morimoto, M. Nishikawa, S. Kawakami, T. Nakano, Y. Hattori, S. Fumoto, F. Yamashita and M. Hashida, Mol. Ther., 2003, 7, 254 CrossRef CAS.
- M. Koping-Hoggard, K. M. Varum, M. Issa, S. Danielsen, B. E. Christensen, B. T. Stokke and P. Artursson, Gene Ther., 2004, 11, 1441 CrossRef CAS.
- J. J. Green, R. Langer and D. G. Anderson, Acc. Chem. Res., 2008, 41, 749 CrossRef CAS.
- A. V. Ulasov, Y. V. Khramtsov, G. A. Trusov, A. A. Rosenkranz, E. D. Sverdlov and A. S. Sobolev, Mol. Ther., 2011, 19, 103 CrossRef CAS.
- D. Finsinger, J. S. Remy, P. Erbacher, C. Koch and C. Plank, Gene Ther., 2000, 7, 1183 Search PubMed.
- J. P. Clamme, J. Azoulay and Y. Mely, Biophys. J., 2003, 84, 1960 CrossRef CAS.
- J. P. Clamme, G. Krishnamoorthy and Y. Mely, Biochim. Biophys. Acta, Biomembr., 2003, 1617, 52 CrossRef CAS.
- S. Boeckle, K. von Gersdorff, S. van der Piepen, C. Culmsee, E. Wagner and M. Ogris, J. Gene Med., 2004, 6, 1102 CrossRef CAS.
- J. M. Saul, C. H. K. Wang, C. P. Ng and S. H. Pun, Adv. Mater., 2008, 20, 19 Search PubMed.
- J. Fahrmeir, M. Gunther, N. Tietze, E. Wagner and M. Ogris, J. Controlled Release, 2007, 122, 236 CrossRef CAS.
- C. Wu, Abstr. Pap. Am. Chem. S, 2009, 237.
- Y. A. Yue, F. Jin, R. Deng, J. G. Cai, Y. C. Chen, M. C. M. Lin, H. F. Kung and C. Wu, J. Controlled Release, 2011, 155, 67 CrossRef CAS.
- Y. A. Yue, F. Jin, R. Deng, J. G. Cai, Z. J. Dai, M. C. M. Lin, H. F. Kung, M. A. Mattebjerg, T. L. Andresen and C. Wu, J. Controlled Release, 2011, 152, 143 CrossRef CAS.
- M. Thibault, M. Astolfi, N. Tran-Khanh, M. Lavertu, V. Darras, A. Merzouki and M. D. Buschmann, Biomaterials, 2011, 32, 4639 CrossRef CAS.
- M. Hanzlikova, M. Ruponen, E. Galli, A. Raasmaja, V. Aseyev, H. Tenhu, A. Urtti and M. Yliperttula, J. Gene Med., 2011, 13, 402 Search PubMed.
- H. Akita, R. Ito, I. A. Khalil, S. Futaki and H. Harashima, Mol. Ther., 2004, 9, 443 CrossRef CAS.
- S. Hama, H. Akita, R. Ito, H. Mizuguchi, T. Hayakawa and H. Harashima, Mol. Ther., 2006, 13, 786 Search PubMed.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32 CrossRef CAS.
- P. Midoux, G. Breuzard, J. P. Gomez and C. Pichon, Curr. Gene Ther., 2008, 8, 335 Search PubMed.
- S. Grosse, Y. Aron, G. Thevenot, D. Francois, M. Monsigny and I. Fajac, J. Gene Med., 2005, 7, 1275 Search PubMed.
- N. E. Bishop, Rev. Med. Virol., 1997, 7, 199 CrossRef.
- L. Pelkmans and A. Helenius, Traffic, 2002, 3, 311 CrossRef CAS.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37 CrossRef CAS.
- C. G. Hansen and B. J. Nichols, J. Cell Sci., 2009, 122, 1713 CrossRef CAS.
- L. K. Medina-Kauwe, J. Xie and S. Hamm-Alvarez, Gene Ther., 2005, 12, 1734 CrossRef CAS.
- R. G. Parton and K. Simons, Nat. Rev. Mol. Cell Biol., 2007, 8, 185 CrossRef CAS.
- L. Pelkmans, T. Burli, M. Zerial and A. Helenius, Cell, 2004, 118, 767 CrossRef CAS.
- K. A. Joiner, S. A. Fuhrman, H. M. Miettinen, L. H. Kasper and I. Mellman, Science, 1990, 249, 641 Search PubMed.
- C. Goncalves, E. Mennesson, R. Fuchs, J. P. Gorvel, P. Midoux and C. Pichon, Mol. Ther., 2004, 10, 373 CrossRef CAS.
- H. Hufnagel, P. Hakim, A. Lima and F. Hollfelder, Mol. Ther., 2009, 17, 1411 Search PubMed.
- P. M. McLendon, K. M. Fichter and T. M. Reineke, Mol. Pharmaceutics, 2010, 7, 738 Search PubMed.
- J. Rejman, A. Bragonzi and M. Conese, Mol. Ther., 2005, 12, 468 CrossRef CAS.
- M. A. E. M. van der Aa, U. S. Huth, S. Y. Hafele, R. Schubert, R. S. Oosting, E. Mastrobattista, W. E. Hennink, R. Peschka-Suss, G. A. Koning and D. J. A. Crommelin, Pharm. Res., 2007, 24, 1590 CrossRef CAS.
- N. D. Sonawane, F. C. Szoka and A. S. Verkman, J. Biol. Chem., 2003, 278, 44826 CrossRef CAS.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657 CrossRef CAS.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14640 CrossRef CAS.
- M. L. Forrest, G. E. Meister, J. T. Koerber and D. W. Pack, Pharm. Res., 2004, 21, 365 CrossRef CAS.
- A. M. Funhoff, C. F. van Nostrum, G. A. Koning, N. M. E. Schuurmans-Nieuwenbroek, D. J. A. Crommelin and W. E. Hennink, Biomacromolecules, 2004, 5, 32 CrossRef CAS.
- P. Dubruel, B. Christiaens, B. Vanloo, K. Bracke, M. Rosseneu, J. Vandekerckhove and E. Schacht, Eur. J. Pharm. Sci., 2003, 18, 211 CrossRef CAS.
- P. Dubruel, B. Christiaens, M. Rosseneu, J. Vandekerckhove, J. Grooten, V. Goossens and E. Schacht, Biomacromolecules, 2004, 5, 379 CrossRef CAS.
- D. Needham and R. S. Nunn, Biophys. J., 1990, 58, 997 CrossRef CAS.
- P. R. Leroueil, S. Y. Hong, A. Mecke, J. R. Baker, B. G. Orr and M. M. B. Holl, Acc. Chem. Res., 2007, 40, 335 CrossRef CAS.
- J. M. Chen, J. A. Hessler, K. Putchakayala, B. K. Panama, D. P. Khan, S. Hong, D. G. Mullen, S. C. DiMaggio, A. Som, G. N. Tew, A. N. Lopatin, J. R. Baker, M. M. B. Holl and B. G. Orr, J. Phys. Chem. B, 2009, 113, 11179 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5177 CrossRef CAS.
- A. Neamnark, O. Suwantong, K. C. R. Bahadur, C. Y. M. Hsu, P. Supaphol and H. Uludag, Mol. Pharmaceutics, 2009, 6, 1798 CrossRef.
- D. A. Wang, A. S. Narang, M. Kotb, A. O. Gaber, D. D. Miller, S. W. Kim and R. I. Mahato, Biomacromolecules, 2002, 3, 1197 CrossRef CAS.
- A. Bajaj, P. Kondaiah and S. Bhattacharya, Bioconjugate Chem., 2008, 19, 1640 CrossRef CAS.
- P. L. Leopold, G. Kreitzer, N. Miyazawa, S. Rempel, K. K. Pfister, E. Rodriguez-Boulan and R. G. Crystal, Hum. Gene Ther., 2000, 11, 151 Search PubMed.
- G. Seisenberger, M. U. Ried, T. Endress, H. Buning, M. Hallek and C. Brauchle, Science, 2001, 294, 1929 CrossRef CAS.
- J. Suh, D. Wirtz and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3878 CrossRef CAS.
- R. P. Kulkarni, D. D. Wu, M. E. Davis and S. E. Fraser, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7523 Search PubMed.
- R. Bausinger, K. von Gersdorff, K. Braeckmans, M. Ogris, E. Wagner, C. Brauchle and A. Zumbusch, Angew. Chem., Int. Ed., 2006, 45, 1568 CrossRef CAS.
- R. P. Kulkarni, K. Castelino, A. Majumdar and S. E. Fraser, Biophys. J., 2006, 90, L42 Search PubMed.
- D. A. Dean, D. D. Strong and W. E. Zimmer, Gene Ther., 2005, 12, 881 CrossRef CAS.
- S. Grosse, G. Thevenot, M. Monsigny and I. Fajac, J. Gene Med., 2006, 8, 845 Search PubMed.
- L. J. Branden, A. J. Mohamed and C. I. E. Smith, Nat. Biotechnol., 1999, 17, 784 CrossRef CAS.
- M. A. Zanta, P. Belguise-Valladier and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 91 CrossRef CAS.
- S. Choi, S. Oh, M. Lee and S. W. Kim, Mol. Ther., 2005, 12, 885 Search PubMed.
- C. Goncalves, M. Y. Ardourel, M. Decoville, G. Breuzard, P. Midoux, B. Hartmann and C. Pichon, J. Gene Med., 2009, 11, 401 Search PubMed.
- J. H. Jeong, S. H. Kim, L. V. Christensen, J. Feijen and S. W. Kim, Bioconjugate Chem., 2010, 21, 296 Search PubMed.
- Y. M. Bae, H. Choi, S. Lee, S. H. Kang, Y. T. Kim, K. Nam, J. S. Park, M. Lee and J. S. Choi, Bioconjugate Chem., 2007, 18, 2029 Search PubMed.
- L. Kastrup, H. Oberleithner, Y. Ludwig, C. Schafer and V. Shahin, J. Cell. Physiol., 2006, 206, 428 Search PubMed.
- J. D. Ramsey, H. N. Vu and D. W. Pack, J. Controlled Release, 2010, 144, 39 CrossRef CAS.
- I. Honore, S. Grosse, N. Frison, F. Favatier, M. Monsigny and I. Fajac, J. Controlled Release, 2005, 107, 537 CrossRef CAS.
- R. N. Cohen, M. A. E. M. van der Aa, N. Macaraeg, A. P. Lee and F. C. Szoka, J. Controlled Release, 2009, 138, 185 Search PubMed.
- D. V. Schaffer, N. A. Fidelman, N. Dan and D. A. Lauffenburger, Biotechnol. Bioeng., 2000, 67, 598 CrossRef CAS.
- C. M. Varga, N. C. Tedford, M. Thomas, A. M. Klibanov, L. G. Griffith and D. A. Lauffenburger, Gene Ther., 2005, 12, 1023 CrossRef CAS.
- S. Hama, H. Akita, S. Iida, H. Mizuguchi and H. Harashima, Nucleic Acids Res., 2007, 35, 1533 Search PubMed.
- K. Itaka, A. Harada, Y. Yamasaki, K. Nakamura, H. Kawaguchi and K. Kataoka, J. Gene Med., 2004, 6, 76 Search PubMed.
- T. Merdan, J. Kopecek and T. Kissel, Adv. Drug Delivery Rev., 2002, 54, 715 CrossRef CAS.
- E. Wagner, Pharm. Res., 2004, 21, 8 CrossRef CAS.
- D. Putnam, Nat. Mater., 2006, 5, 439 CrossRef CAS.
- W. F. Lai, Expert Rev. Med. Devices, 2011, 8, 173 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |