Reduced in vitro immune response on titania nanotube arrays compared to titanium surface
Barbara S.
Smith
a,
Patricia
Capellato
bc,
Sean
Kelley
c,
Mercedes
Gonzalez-Juarrero
d and
Ketul C.
Popat
*ce
aDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
bDepartment of Materials, Faculty of Engineering Guaratinguetá, Sao Paulo State University-UNESP, Av. Ariberto Pereira da Cunha 333, Pedregulho, CEP 12516-410, Guaratinguetá, SP, Brazil
cSchool of Biomedical Engineering, Colorado State University, Fort Collins, CO 80523, USA. E-mail: ketul.popat@colostate.edu; Fax: +1 970-491-3827; Tel: +1 970-491-1468
dDepartment of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO 80523, USA
eDepartment of Mechanical Engineering, Colorado State University, Fort Collins, CO 80523, USA
First published on 28th November 2012
Abstract
Material surfaces that provide biomimetic cues, such as nanoscale architectures, have been shown to alter cell/biomaterial interactions. Recent studies have identified titania nanotube arrays as strong candidates for use in interfaces on implantable devices due to their ability to elicit improved cellular functionality. However, limited information exists regarding the immune response of nanotube arrays. Thus, in this study, we have investigated the short- and long-term immune cell reaction of titania nanotube arrays. Whole blood lysate (containing leukocytes, thrombocytes and trace amounts of erythrocytes), isolated from human blood, were cultured on titania nanotube arrays and biomedical grade titanium (as a control) for 2 hours and 2 and 7 days. In order to determine the in vitro immune response on titania nanotube arrays, immune cell functionality was evaluated by cellular viability, adhesion, proliferation, morphology, cytokine/chemokine expression, with and without lipopolysaccharide (LPS), and nitric oxide release. The results presented in this study indicate a decrease in short- and long-term monocyte, macrophage and neutrophil functionality on titania nanotube arrays as compared to the control substrate. This work shows a reduced stimulation of the immune response on titania nanotube arrays, identifying this specific nanoarchitecture as a potentially optimal interface for implantable biomedical devices.
For the 8–10% of Americans (20–25 million people) that have implanted biomedical devices, biomaterial failure and the need for revision surgery is a critical concern.1,2 In fact, the biggest shortcoming has been the lack of complete biointegration of materials.3,4 Biocompatibility can be defined as the ability of a material to coexist with natural tissue or organs without initiating harm. The degree of biocompatibility may therefore be determined by characterizing the extent of inflammatory reactions acting to neutralize or sequester the implanted biomedical device.5 The physiological response to implanted biomaterials has been well documented, identifying a complex cascade of events, which often results in a foreign body reaction on the surface of the biomaterial and in the surrounding tissue.1 The successful integration of long-term implantable devices is highly dependent on the early events of the tissue/material interaction, either promoting fibrosis (foreign body reaction) or a wound healing response (extracellular matrix production and vasculature).6 Immediately following implantation, biomaterials initiate immunological events in the form of protein adsorption, followed by platelet adhesion/activation, which recruit immune cells (monocytes and neutrophils) to the site of injury through biochemical cues. This inflammatory cell infiltration subsequently leads to acute and chronic inflammation, promoting additional cellular activation (monocyte differentiation into macrophages), apoptosis and intercellular communication (cytokines/chemokines). This cascade of events perpetuates cellular infiltration and activation (lymphocytes and fibroblasts) initiating granulocyte recruitment. The resulting foreign body reaction (foreign body giant cells, FBGC) and fibrosis potentially leads to fibrous encapsulation and complete implant rejection.3,4 The cells required for healthy tissue regeneration are compromised through this adverse immune response, thus hindering the natural wound healing response and the long-term success of implantable biomedical devices, such as intraosseous implants7,8 and stent grafts.9,10 In order to better understand this critical stage in biomaterial integration, a great need exists for a more in-depth look at the mechanisms behind immune–biomaterial interactions.
Titanium and titanium-based alloys have been the most widely utilized materials for use in implantable biomedical devices since the mid-1900s, when titanium was found to possess tissue-compatible properties.11,12 Clinical applications for these materials include dental,13,14 craniofacial and prosthetic implants,15–18 cardiovascular stents19 and hard and soft tissue grafts.20 The two main factors contributing to the widespread use of titanium in biomedical implants include their impressive mechanical and biocompatible properties.12,21 In addition, titanium reacts naturally with atmospheric oxygen to produce a passivating oxide layer on its surface, termed titania. Protecting the non-toxic metal from environmental factors, titania creates a hard and scratch-resistant material surface, improves corrosion and wear resistance, enables a low coefficient of friction and provides a favorable biocompatible interface for tissue integration.17,22 Although titanium and titanium-based alloys are among the better choices for biomedical implants, to date, all long-term implanted biomaterials have the potential of initiating physiological events shown to promote poor tissue/material integration.3,4
The human body is composed of a hierarchy of biological structures from the smallest molecule to the largest tissue. Therefore, when microscale cells interact with their macroscale environment, they do so through countless nanoscale topographies, biochemical cues and protein interactions. In fact, cells are in constant interaction with their surroundings, comprised of nanoscale subcellular structures, including fibers, pits, pores and protrusions, thus, identifying a need for better understanding the effect of topographical (micro- and nano-scale)23–26 or biochemical cues (biofunctional)27,28 on cellular functionality. Recent technological advances have enabled the fabrication of nano-engineered biomaterial surfaces with the goal of providing relevant nanoscale cues.29 Studies have identified enhanced integration of various cell types on nanoparticles,30 nanofibers,31,32 nanopores,33,34 nanowires,35,36 nanostructured hydrogels37 and nanotube arrays.38,39 Of these nanotopographical fabrications, current research has correlated favorable cellular functionality with nanotube array interfaces. Previous studies on titania nanotube arrays have demonstrated enhanced osseointegration,40,41 improved endothelialization,42 altered hemocompatibility and thrombogenicity,43 preferential fibroblast orientation,44 increased dermal matrix deposition44,45 and selective behavioral responses of stem cells.46–48 In addition, the nanotube arrays can be filled with drugs, such as antibiotics or growth factors to thwart infection and encourage tissue integration, respectively, through the localized delivery of drugs.49,50 However, prior to the application of this promising nano-architecture in implantable devices, it is imperative to obtain a better understanding of the effect of the nanotube interface on the immune reaction.
The ability to modulate the immune reaction at the tissue/biomaterial interface is critical for the long-term success of implantable devices. An evaluation of the immune response in vitro can be accurately represented through whole blood lysate, present in less than 1% of whole human blood. This immune-rich portion of blood is mainly comprised of white blood cells (leukocytes) and platelets (thrombocytes), which play a fundamental role in the human immune response and the foreign body reaction (FBR), potentially leading to poor implant integration in vivo. However, limited studies exist on the interaction of immune cells with titania nanotube arrays. Thus, in this study we have evaluated the effect of titania nanotube arrays (diameter: 170 nm, length: 1 μm) on physiologically relevant whole human blood lysate, in order to better understand the mechanisms behind immune/nano-biomaterial interactions. Immune cell viability, adhesion, proliferation, morphology, cytokine/chemokine expression and nitric oxide release were investigated after 2 hours and 2 and 7 days of culture using a cell viability assay, fluorescence microscope imaging, scanning electron microscopy (SEM), cytokine bead assays (CBA) and a Griess reagent kit on titania nanotube arrays as compared to biomedical grade titanium (as a control). In order to promote the long-term health following biomaterial integration, a reduction in adverse immune reactions is imperative.
Experimental methods
Fabrication and characterization of titania nanotube arrays
Titania nanotube arrays were fabricated using a simple anodization process, as previously described.51,52 Titanium foil (0.25 mm thick, 99.7%) was cleaned with soap, acetone and isopropanol, prior to anodization. A two-electrode cell was assembled with titanium foil as the anode and platinum foil as the cathode. The anode–cathode system was introduced into an electrolyte solution, prepared with diethylene glycol (DEG, 99.7%), 2% hydrofluoric acid (HF, 48% solution) and 3% de-ionized water. All experiments were carried out at 60 V for 24 hours at room temperature (RT). Following anodization, the titania nanotube arrays were rinsed with isopropanol and dried with nitrogen gas. Crystallized substrates were obtained by annealing the as-anodized titania nanotube arrays at 530 °C in an oxygen ambient environment for 3 hours.The nanotube architecture was identified using a field emission scanning electron microscope (SEM, JEOL JSM-6300). The substrates were coated with a 10 nm layer of gold prior to imaging at 15 kV. A 3-dimensional topographical analysis of the nanotube surface, indicating surface roughness (uniformity and modification), was examined by atomic force microscopy (AFM) using a confocal micro-Raman microscope alpha300 R (WITec). The material surface wettability was evaluated using a static sessile water-drop method. Images were taken immediately following DI water/substrate contact. A contact angle goniometer was used to identify the degree of phase separation, formed between the liquid–solid interface and the liquid–vapor interface. The images were analyzed to evaluate the degree of surface hydrophobicity/hydrophilicity, where higher angles correlate with a low surface energy and an increased hydrophobicity of the substrate. Biomedical grade titanium (as received) substrates were used as controls.
Whole human blood lysate isolation
Whole human blood was acquired through venopuncture from healthy individuals. All experiments were performed in compliance with the relevant laws and institutional guidelines. Blood was drawn into standard 6 ml vacuum tubes coated with ethylenediaminetetraacetic acid (EDTA) for its anti-coagulant properties. The first tube was discarded to account for the skin plug and locally activated platelets, resulting from the needle insertion. Whole human blood was mixed with Gey's balanced salt solution (150 mM of NH4Cl and 10 mM of KHCO3) at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and placed into a 15 ml conical tube. The blood: Gey's mixture was placed on a rocker plate for 5–10 min at RT, to allow for the red blood cells (RBCs) to isotonically lyse. The mixed vials were centrifuged at 300g for 5 min at 4 °C and the supernatant was discarded. The cell pellet was resuspended in 5 ml of Gey's solution and immediately centrifuged at 300g for 5 min at 4 °C to lyse all remaining RBCs. The supernatant was discarded. This was followed by washing the cell pellet (2×) via resuspension in Hank’s buffered salt solution (without Ca and Mg, Sigma) and centrifuged at 300g for 5 min at 4 °C. The supernatant was discarded after each wash. The cell pellets from 3 vials were combined by resuspension in cold Roswell Park Memorial Institute (RPMI) medium (Sigma) supplemented with 10% FBS and 1% penicillin/streptomycin. The cell density was determined by trypan blue dye exclusion, using a hemocytometer. Cell suspensions were further diluted in RPMI medium to obtain a concentration of 2 × 106 cells per ml. Isolated cells were seeded within 4 hours of removal from the body.
:2 and placed into a 15 ml conical tube. The blood: Gey's mixture was placed on a rocker plate for 5–10 min at RT, to allow for the red blood cells (RBCs) to isotonically lyse. The mixed vials were centrifuged at 300g for 5 min at 4 °C and the supernatant was discarded. The cell pellet was resuspended in 5 ml of Gey's solution and immediately centrifuged at 300g for 5 min at 4 °C to lyse all remaining RBCs. The supernatant was discarded. This was followed by washing the cell pellet (2×) via resuspension in Hank’s buffered salt solution (without Ca and Mg, Sigma) and centrifuged at 300g for 5 min at 4 °C. The supernatant was discarded after each wash. The cell pellets from 3 vials were combined by resuspension in cold Roswell Park Memorial Institute (RPMI) medium (Sigma) supplemented with 10% FBS and 1% penicillin/streptomycin. The cell density was determined by trypan blue dye exclusion, using a hemocytometer. Cell suspensions were further diluted in RPMI medium to obtain a concentration of 2 × 106 cells per ml. Isolated cells were seeded within 4 hours of removal from the body.
Incubation of whole blood lysate on titania nanotube arrays
In order to evaluate the immune response in vitro, the cells were seeded on titania nanotube arrays and biomedical grade titanium (0.5 cm × 0.5 cm) in a 24-well plate. Prior to seeding, all substrates were sterilized by incubation in 70% ethanol for 30 min, washed (2×) with PBS, air dried and subjected to 30 min of UV exposure. Immune cells were seeded at a density of 2 × 106 cells per ml. The substrates were incubated at 37 °C and 5% CO2 in 500 μl of cell-rich media and investigated for viability, adhesion, proliferation, morphology, cytokine/chemokine expression and NO release after 2 hours and 2 and 7 days of culture. Fresh culture medium, with and without lipopolysaccharide (LPS), was added for the long-term study in the amount of 250 μl after 3 and 6 days of culture. Additional substrates were incubated with 2 μg ml−1 lipopolysaccharide (LPS from E. coli, Sigma) in cell-rich media, as a positive cellular control, in order to evaluate the effect of LPS on cell/nano-biomaterial interactions. The substrates incubated in LPS-enriched media were investigated for cytokine/chemokine production using CBA assays, performed by flow cytometry after 2 hours and 2 and 7 days of culture. Prior to all measurements, unless otherwise stated, non-adhered cells were removed by aspirating the cell-rich media from the substrates followed by two gentle rinses with PBS. The substrates were then transferred to a new 24-well plate and examined using the following protocols.Viability of whole blood lysate on titania nanotube arrays
Cellular viability was characterized using a commercially available methylthiazol tetrazolium (MTT) assay kit (Sigma) and the standard manufacturer protocol was followed. In brief, the substrates were incubated in 10% MTT solution in PBS at 37 °C and 5% CO2. The resulting formazan crystals were disrupted by adding a mixture of 10% Triton-X in MTT solvent to each substrate in the amount equal to the incubated MTT solution in PBS. The absorbance of the solution was measured at a wavelength of 570 nm using a plate reader (BMG Labtech). The net absorbance was calculated by subtracting the background absorbance at a wavelength of 690 nm to measure the amount of viable cells adhered on the substrates.Adhesion and proliferation of whole blood lysate on titania nanotube arrays
Cellular adhesion and proliferation was characterized by fluorescently staining adhered cells with 5-chloromethylfluorescein diacetate (CellTracker green CMFDA, Invitrogen) live stain and 4′,6-diamidino-2-phenylindole-dihydrochloride (DAPI, Invitrogen) nuclei stain. The substrates were incubated with 3 μM CMFDA in PBS for 40 min at 37 °C and 5% CO2. They were then placed into warm RPMI medium and incubated for 30 min at 37 °C and 5% CO2. The substrates were fixed in 3.7 wt% formaldehyde in PBS for 15 min at RT, followed by 3 washes (5 min per wash) with PBS at RT. Adhered cells were further permeabilized by incubation in 1% Triton-X in PBS for 3 min and rinsed with PBS. The substrates were incubated in DAPI nuclear stain (300 nM) for 5 min at RT, rinsed with PBS and imaged with a fluorescence microscope (Zeiss) using 62 HE BP 474/28 green filter and 49 DAPI BP 445/50 blue filter. The number of cells adhered on the substrates was determined by counting the number of stained nuclei from the DAPI fluorescence images.Cytoskeletal organization of whole blood lysate on titania nanotube arrays
Cytoskeletal organization was evaluated using fluorescence microscope imaging after 2 hours and 7 days of culture. The same samples that were stained for DAPI were concurrently stained for F-actin with rhodamine-conjugated phalloidin (70 nM, Cytoskeleton Inc.). Following permeabilization and prior to DAPI staining, substrates were incubated in rhodamine-conjugated phalloidin (70 nM) in PBS for 20 min at RT. The substrates were imaged with a fluorescence microscope using a 62 HE BP 585/35 red filter.Morphology of whole blood lysate on titania nanotube arrays
Cellular morphology was investigated using SEM imaging. Prior to imaging, the adhered cells were fixed and further dehydrated on the substrate surface. The cells were incubated in a solution of primary fixative (3% glutaraldehyde (Sigma), 0.1 M sodium cacodylate (Polysciences) and 0.1 M sucrose (Sigma)) for 45 min. The substrates were then incubated in a secondary fixative (primary fixative without glutaraldehyde) for 10 min. They were subsequently dehydrated by incubation in consecutive solutions of increasing ethanol concentrations (35%, 50%, 70% and 100%) for 10 min each. Further dehydration was accomplished by incubating the substrates in hexamethyldisilazane (HMDS, Sigma) for 10 min. They were then air dried and stored in a desiccator until SEM imaging. The substrates were coated with a 10 nm layer of gold prior to imaging at 15 kV.Cytokine and chemokine expression
Human inflammatory cytokines/chemokines were detected using commercially available cytometric bead array (CBA, BD Biosciences) human plex flex sets. Human inflammatory cytokine/chemokine expression was identified through the presence of TNF, IFN-γ, TGF-β1, MIP-1β, MCP-1, IL-1β, IL-6, IL-8, IL-10 and IL-12p70 on the substrates with and without LPS. All immunoassays were run together. The standard manufacturer protocol was followed. In brief, substrate-exposed supernatant was removed at the designated time points and stored at −80 °C until further testing by CBA analysis using flow cytometry. Human flex set standards (×10), human soluble protein flex set capture beads (×100) and PE detection reagents (×100) were prepared as directed, such that each analyte was added at 1/10th of the final calculation. The procedure was initiated by adding the standards and samples (100 μl) to capture beads (50 μl) in a 96-well plate. The mixture was gently vortexed for 15 s and incubated for 1 hour at RT. Following incubation, PE detection reagent (50 μl) was added to each well. The mixture was gently vortexed for 15 s and incubated in a dark environment for 2 hours at RT. The samples were washed by adding the manufacturer-provided wash buffer (100 μl). The well plates were centrifuged at 1040 rpm for 3 min to pelletize the cell/bead population in the supernatant. The supernatant (200 μl) was gently discarded by aspiration, making sure not to disrupt the cell/bead pellet on the bottom of the wells. Immediately following the resuspension of the cell/bead pellet in wash buffer (150 μl), the cell/bead-rich samples were transferred to flow tubes (VWR 83009–678) and further tested by flow cytometry. Data was acquired on a FACS Array bioanalyzer. The standard samples were used to establish gated regions for the identification of each cytokine/chemokine. The results were analyzed using FCAP Array Software and Microsoft Office Excel data analysis software.Nitric oxide release on titania nanotube arrays
Cell-released nitric oxide (NO) was detected using a commercially available Griess reagent kit (Biotium Inc.). The standard manufacturer protocol was followed. In brief, substrate-exposed supernatant was incubated in a mixture of Griess reagent and DI water for 30 min at RT. The samples were photometrically measured at a wavelength of 548 nm using a plate reader (BMG Labtech). The absorbance values quantified the effect of titania nanotube arrays on the degree of NO release.Statistical analysis
Each experiment was performed on at least three different substrates from at least three different blood populations (nmin = 9). Furthermore, all of the quantitative results were evaluated on at least five different substrates from at least three different blood populations (nmin = 15), using an analysis of variance (ANOVA). Statistical significance was considered at p < 0.05. During the analysis, variance among each group were not assumed to be equal and a two sample t-test approach was utilized to test the significance between the titania nanotube arrays and the control substrate. This analysis was conducted using Microsoft Office Excel data analysis software.Results and discussion
Limited tissue–biomaterial integration and biomaterial failure often result from adverse immune responses in the form of a fibrous encapsulation, which acts to “wall off” the implant from natural tissue. Recent studies have identified titania nanotube arrays as potential biomimetic, biocompatible and non-biodegradable interfaces for implantable devices, making this specific nano-architecture appealing for use in implants. However, limited studies have investigated one of the most critical events in cell/nano-biomaterial integration: the biomaterial-generated immune response. After only 2 hours following implantation, monocytes and neutrophils adhere to the biomaterial surface and begin to release factors of communication. By day 2, however, adverse immune reactions are visible through immune cell activation, differentiation (monocytes to macrophages) and cytokine and chemokine expression, promoting additional cellular infiltration. Between 7 and 10 days of incubation, the formation of fused cells (3+ nuclei) and FBGCs may be present on the biomaterial surface, leading to further fibrous encapsulation and potential implant rejection. Thus, in this study, we have evaluated the short- and long-term cellular immune response on titania nanotube arrays as compared to biomedical grade titanium. All studies were performed after 2 hours and 2 and 7 days of culture unless otherwise stated.Immobilized, vertically oriented and high aspect ratio nanotubes were fabricated after 24 hours of anodization at 60 V on titanium substrates, as described elsewhere.40,44,52 The nanotube architecture was examined for uniformity and repeatability using SEM imaging (see Fig. 1). The results indicate nanotube dimensions of approximately 170 nm in diameter and 1 μm in length. Previous studies have demonstrated altered cellular functionality, including cell adhesion, activation, proliferation and protein expression,38,42,45 modified hemocompatibility43,53 and sustained drug release50 on similar titania nanotube arrays. The nanotube fabrication can be characterized by a localized dissolution model, which takes into account the competition between electrochemical etching and chemical dissolution of titanium.54,55 Recent studies have identified a precise correlation between the anodization voltage and the pore size; thus, by altering basic anodization parameters, nanotube arrays with different diameters and lengths can be fabricated.56,57 Furthermore, as-anodized titania nanotube arrays were annealed in an ambient atmosphere to create a crystalline structure, as described elsewhere.43
 | ||
| Fig. 1 Representative SEM images (5000×, 20000× and 50000× magnification, represented scale bars of 1μm, 1μm and 100nm respectively) showing the nano-architecture of titania nanotube arrays fabricated using an anodization technique. Titania nanotube arrays were coated with a 10 nm layer of gold and imaged at 15 kV. | ||
A 3-dimensional analysis of surface topography was performed using atomic force microscopy (AFM). A precise mechanical probing of the material surface was utilized to detect changes in topography and identify surface roughness. The results indicate nanoscale variations within an overall uniform micro- and macro-scale architecture present on the surface of titania nanotube arrays as compared to the micrometer-sized variations on the surface of the control substrate (see Table 1). Nanoscale variations, indicative of the topography found in natural tissue, have repeatedly been shown to promote protein-specific adsorption, further directing cell–material and cell–cell interactions, and subsequently cell behavior through chemo-mechanic mechanisms. Thus, these traits allow for a promising interface for improved cellular interaction due to the increased nanoscale roughness of the material surface.58,59 In addition to the material’s surface characteristics, such as topography and roughness, physiochemical properties, such as hydrophobicity, may direct environmental influences following implantation.
| AFM | Titanium | Titania nanotube arrays |
|---|---|---|
| Ra | 1.3 μm | 667.33 nm |
| Rq | 1.67 μm | 866.01 nm |
| Rz | 11.34 μm | 9.23 μm |
| Rt | 12.28 μm | 10.45 μm |
| Magnification | 10.24× | |
| Sampling | 820.29 nm | |
| Array size | 736 × 480 | |
Within the living body, myriads of hydrophobic (non-polar, water-fearing) and hydrophilic (polar and ionic, water-loving) molecules direct cell function, proving imperative for survival. Biomaterial wettability has been found to be a considerable factor for tissue integration and the success of implanted devices. Recent studies have shown that biomaterials that possess a heightened degree of hydrophilicity promote improved cell-specific interactions.60,61 Material surface wettability was evaluated by contact angle measurement to determine the degree of phase separation immediately following DI water/substrate contact. Higher contact angles (>90°) correlate to a low surface energy and a reduced water–material interaction, indicating hydrophobic surfaces. The opposite is true for hydrophilic surfaces, which possess a high surface energy. The resulting images identified a hydrophilic 25.8° contact angle for titania nanotube arrays as compared to the significantly increased hydrophobicity of the control substrates, with a contact angle of 81.8° (see Table 2). These results show titania nanotube arrays have a hydrophilic interface, which naturally promotes cellular integration through an altered cell–material interface.
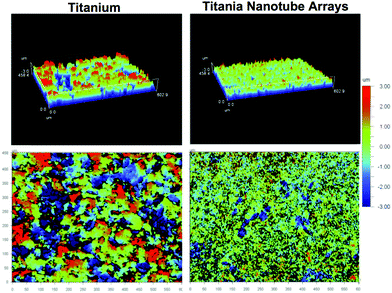 | ||
| Fig. 2 Representative atomic force microscopy (AFM) images of surface topographies for titania nanotube arrays and the control substrates. The size scale is indicated by the color bar (right side), from -3μm to 3μm. The resulting images identify a nano-architecture on titania nanotube arrays and a micro-architecture on biomedical grade titanium. | ||
| Titanium | Titania nanotube arrays | |
|---|---|---|
| Contact angle (°) | 81.8 ± 5.8 | 25.8 ± 4.8 |

|

|
The short- and long-term cell viability were characterized using a commercially available MTT assay kit (see Fig. 3). Spectrophotometric measurements identified a significant decrease notable in immune cells adhered on both substrates from 2 hours to 2 days of culture, without any further decrease after 2 days of culture. The short- to long-term reduction in cell viability on both substrates is likely due to the limited lifespan of monocytes. However, the results indicate a significantly reduced amount of metabolically-active immune cells present on titania nanotube arrays as compared to the control substrate at all time-points. The overall results clearly indicate a significant decrease in the viability of immune cells on titania nanotube arrays as compared to the control substrate. These results are likely due to the altered cellular interaction resulting from the nanotopography and the subsequent protein interactions. Further studies will be needed to determine the mechanisms of cellular response.
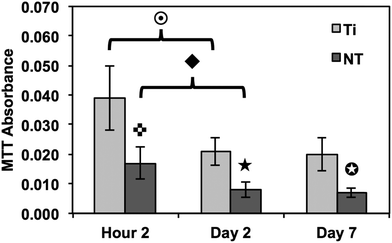 | ||
Fig. 3 Cellular viability measured using an MTT assay to identify adhered immune cells. The results indicate a significant decrease in the number of adhered immune cells on titania nanotube arrays as compared to biomedical grade titanium. ( , ★, , ★,  , ,  , ◆, , ◆, 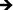 p ≤ 0.05). p ≤ 0.05). | ||
A complex cascade of events is initiated immediately upon implantation, where the body's natural response to the biomaterial interface directs immune cell interactions, thus regulating the level of biocompatibility. Cell adhesion and proliferation are therefore key factors in determining the fate of a successful implant. In this study, we have investigated the short- and long-term adhesion and proliferation of immune cells on titania nanotube arrays as compared to the control substrate. Fluorescence microscope imaging using CMFDA live staining and DAPI nuclei staining were performed in order to better understand the immune/nano-biomaterial interaction62 (see Fig. 4(a)). After only 2 hours of culture, fluorescence images showed small and unactivated monocytes and neutrophils adhered on both substrates, with significantly reduced amounts of adhered immune cells on titania nanotube arrays. Due to the lack of initial alteration in cell size and subsequent interaction with the material surface, it is immediately apparent that the adhered monocytes and neutrophils have not yet become activated or begun to differentiate. Thus, the short-term results show a decrease in adhered cells on titania nanotube arrays as compared to the control substrate. After 2 and 7 days of culture, the results indicate a reduction in the number of adhered cells on both substrates, with a greater reduction found on titania nanotube arrays. These findings are likely the result of either the short lifespan of monocytes or premature cell death due to ex vivo conditions. Cells adhered on both substrates showed an increase in size, identifying an increased long-term activation. However, when comparing the titania nanotube arrays with control substrates, a clear difference is apparent. The results indicate a substantial reduction in immune cell adhesion on titania nanotube arrays as compared to the control substrate. This decrease in cell adhesion continues on titania nanotube arrays between days 2 and 7, where limited cell–cell interactions and minimal cellular elongation become apparent. In contrast, images of the control substrates highlight a substantial increase in cell–cell interactions and cellular elongation after 2 and 7 days of culture. Therefore, the monocytes adhered on the control substrates after only 2 hours of culture clearly morph into their activated counterpart, macrophages, which exhibit an increased diameter, a more expansive material integration and an altered cellular structure. Images of the control substrates after 7 days of culture clearly show fusion and the production of foreign body giant cells (FBGCs), which are all but absent on titania nanotube arrays. The formation of FBGCs has been associated with the inability of macrophages to remove or destroy a biomaterial, which leads them to fuse in a gesture of “frustrated phagocytosis”. Multiple nuclei are readily apparent within the cell conglomerates of FBGCs, indicative of the first stages of fibrous encapsulation. Fluorescence images of titania nanotube arrays show a moderate initial adhesion followed by an extreme reduction in immune cell proliferation, while the control substrate promotes a strong immune cell adhesion early on, leading to FBGCs and an overall adverse immune response.63 The total number of adhered cells was further analyzed using the fluorescence images and ImageJ software (see Fig. 4(b)). The results show a significant reduction in cell count on titania nanotube arrays as compared to the control substrate after all time points, further indicating a vastly reduced short- and long-term immune response in terms of adhesion and proliferation on titania nanotube arrays as compared to the control substrate.
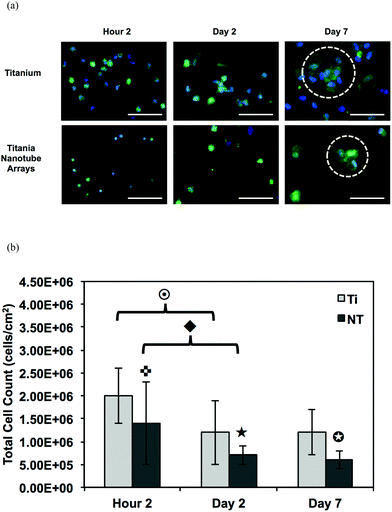 | ||
Fig. 4 (a) Representative fluorescence microscope images of adhered monocytes, macrophages, neutrophils and foreign body giant cells (FBGC, 3 or more nuclei) stained with DAPI (nuclei) and CMFDA (cytoplasm) on titania nanotube arrays and biomedical grade titanium after 2 hours and 2 and 7 days of incubation in human whole blood lysate. The images show a decrease in cellular adhesion, fusion and the formation of FBGCs (indicated by the circled regions) on titania nanotube arrays. The scale bar represents 50 μm. (b) Cell count calculated using the fluorescence images and ImageJ software. The results indicate a significant decrease in the number of adhered immune cells on titania nanotube arrays as compared to biomedical grade titanium. ( , ★, , ★,  , ,  , ◆, , ◆, 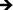 p ≤ 0.05). p ≤ 0.05). | ||
Composed of microfilaments, microtubules and intermediate filaments, the cytoskeleton plays a quintessential role as the structural support within the cell membrane and acts as the barrier between the cellular components and the extracellular world. Cellular activation often perpetuates cytoskeletal reorganization, facilitating fundamental events, such as cell division, motility, protein trafficking and secretion. In addition, this essential component enables cell–cell and cell–ECM communication. Thus, the effect of titania nanotube arrays was characterized by cytoskeletal organization after 2 hours and 7 days of culture using fluorescence microscope imaging by evaluating substrates stained with rhodamine-conjugated phalloidin (F-actin stain) (see Fig. 5). Short-term results clearly show limited cell activation and cytoskeletal reorganization on both substrates. After 7 days of culture, however, fluorescence images identify extremely reduced adhesion, activation and cell–cell interaction on titania nanotube arrays as compared to the control substrates. In addition, the cell–cell interaction occurring on the control substrate shows immune cell fusion, as seen by the interlinking of multiple cell cytoskeletons. The formation of fused cells with FBGCs on the control substrate indicates the potentially adverse immune response elicited by biomedical grade titanium, unlike that seen on the titania nanotube arrays.
 | ||
| Fig. 5 Representative fluorescence microscope images of adhered monocytes, macrophages, neutrophils and foreign body giant cells (3 or more nuclei) stained with DAPI (nuclei) and rhodamine-conjugated phalloidin (actin, cytoskeleton) on titania nanotube arrays and biomedical grade titanium after 2 hours and 2 and 7 day of incubation in human whole blood lysate. The images indicate a decrease in immune cell adhesion, activation and fusion on titania nanotube arrays. The scale bar represents 50 μm. | ||
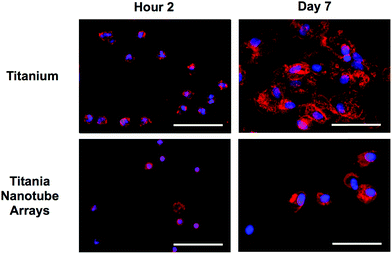
Cellular morphology is a strong indicator of cell activation, communication and cell–biomaterial integration. Thus, SEM imaging has been utilized to evaluate cell morphology in order to better understand the immune response elicited by titania nanotube arrays (see Fig. 6). Short-term results show reduced immune cell adhesion and morphology on titania nanotube arrays, clearly representing a limited initial immune reaction. However, the high levels of adhered immune cells on the control substrate identify preliminary morphological changes of monocytes to macrophages. This is exemplified by the changes in the cell structure from small round monocytes into the broader morphology of macrophages, and by their greater cytoplasmic to nuclear area ratio. The increased activation of immune cells on the control substrate can be attributed to the altered oxidation along the material surface, characteristics of nanotopography, which requires cells to spread out for attachment, and modified protein deposition for directed cellular functionality. The substrate-elicited immune cell activation is a strong factor towards promoting additional cell–material and cell–cell activation. The long-term results remain consistent where, even with the much reduced cell counts on both substrates, the trends of material interaction hold true. Titania nanotube arrays appear to facilitate a lack of immune cell adhesion, activation, morphological changes and overall integration, therefore reducing any possibility of cell–cell fusion. The control substrates, however, not only promote immune cell–biomaterial interactions, cellular activation (extreme morphological changes) and cell–cell fusion, but also result in the formation of FBGCs, showing pre-emptive signs of fibrous encapsulation. Thus, the specific nanotopography of the nanotube arrays directly correlates with a definitive reduction in initial immune cell adhesion, subsequent cell activation and further cell–material integration. These traits are extremely promising for promoting the success of long-term implantable devices with a reduced immune response. Cellular activation and differentiation were determined by the presence of cell extensions, heightened cellular size and structure and cell fusion.64
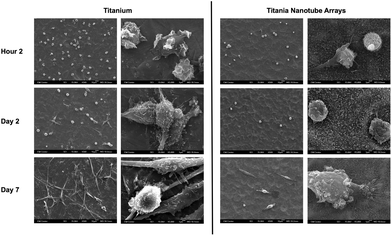 | ||
| Fig. 6 Representative SEM images of adhered immune cells on titania nanotube arrays and biomedical grade titanium after 2 hours and 2 and 7 days of incubation in human whole blood lysate. The substrates were coated with a 10 nm layer of gold and imaged at 15 kV. Representative images taken at 500× (left column of both substrate types, scale bar 10 μm) and 5000× (right column of both substrate types, scale bar 1 μm). The images indicate constrained cellular morphology on titania nanotube arrays, while showing increased cell–substrate integration and cell–cell interactions leading to the formation of foreign body giant cells on biomedical grade titanium. | ||
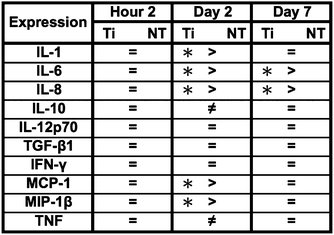
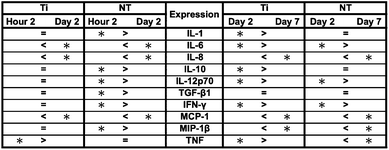
Inflammatory and wound healing cells, such as lymphocytes, monocytes, macrophages, neutrophils and fibroblasts release inflammatory cytokines (small proteins) and chemokines (low molecule cytokines) following cellular adhesion and activation.65 This complex network of small proteins acts to modulate cellular interaction, infiltration, communication and behavior.66 In time, additional activation and matrix formation perpetuates inflammatory or wound healing responses. Cytokines bind to surface receptors, either promoting (pro-inflammatory) or impeding (anti-inflammatory) further intracellular function, intercellular communication and extracellular matrix (ECM) development.67,68 Thus, cytokines and chemokines are key factors in evaluating cellular activation and communication. In this study, cellular expression was evaluated through the presence of cytokines and chemokines, which are known to affect specific functions within the inflammatory or wound healing processes, including TNF, IFN-γ, TGF-β1, MIP-1β, MCP-1, IL-1β, IL-6, IL-8, IL-10 and IL-12p70 (see Fig. 7). The results after 2 hours of culture indicate TNF, TGF-β1, MIP-1β and IL-8 cytokine activity. Equivalent cytokine expression is shown on both substrates, thus indicating no difference in the material cytotoxicity of titania nanotube arrays and the control substrate. The results after 2 days of culture show increased levels of TNF, TGF-β1, MIP-1β, MCP-1, IL-1β, IL-6, IL-8 and IL-10 cytokine expression. Adhered immune cells show significant differences in the up regulation of TNF and IL-10 expression, while significantly de-regulated expression of MIP-1β, MCP-1, IL-1β, IL-6 and IL-8 is present on titania nanotube arrays as compared to the control substrate. These findings identify the cellular attempt to reduce the immune response by the activation of the anti-inflammatory cytokine, IL-10, on both substrates. Furthermore, the results after 7 days of culture show TGF-β1, MIP-1β, MCP-1, IL-6 and IL-8 cytokine activity. TGF-β1, known for its pro-fibrotic properties and its ability to actively suppress inflammation, is consistently expressed at all time points. This is likely due to the fact that TGF does not differentiate between its latent and active states, but is activated only when associated with its receptor. Thus, our results only indicate the presence of TGF-β1 and do not determine its level of activation. In addition, TNF plays a large role in expanding the network of inflammatory cytokines. This cytokine shows a significantly decreased expression on both substrates between 2 hours and 2 days, indicating a reduced inflammatory response. The results show significant decreases in IL-6 and IL-8 expression on titania nanotube arrays as compared to the control substrate, further indicating a significant reduction in the long-term expression of pro-inflammatory cytokines and chemokines on titania nanotube arrays as compared to the control substrate. Thus, the results clearly confirm an overall reduction in adverse immune responses on titania nanotube arrays (see Tables 3–5). Located on the outer membrane of gram-negative bacteria, LPS has been widely attributed to activating the immune response.69,70 Thus, LPS was utilized as a positive control in determining its effect on immune cell activation in conjunction with titania nanotube arrays (see Fig. 8). As expected, the results show an unmistakable increase in cytokine expression on titania nanotube arrays in the presence of LPS and show that an increased immune cell behavior is possible and known to be a direct factor attributed to the presence of LPS.
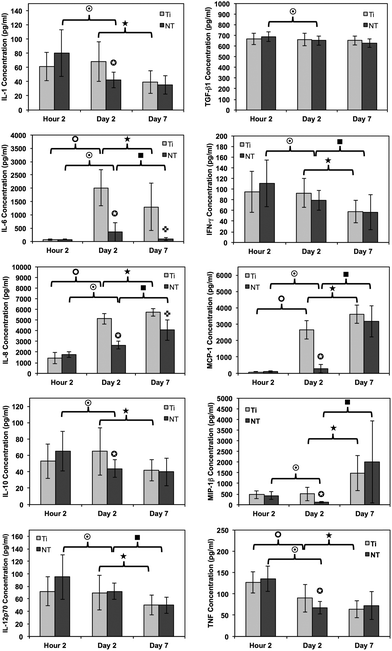 | ||
Fig. 7 Cytokine/chemokine expression was identified using a CBA assay and flow cytometry. The results identify the presence of 10 different cytokines/chemokines, including IL-1β, IL-6, IL-8, IL-10, IL-12p70, TNF, TGF-β1, IFN-γ, MCP-1 and MIP-1β. The long-term results after 2 and 7 days of incubation in human whole blood lysate show significant reductions in protein expression on titania nanotube arrays as compared to biomedical grade titanium ( , ,  , ,  , ,  , ★, , ★,   p ≤ 0.05). p ≤ 0.05). | ||
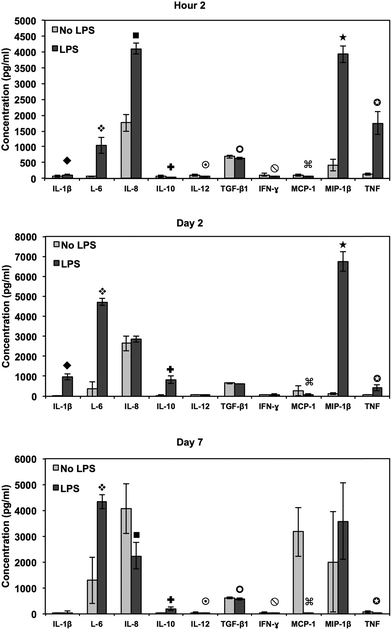 | ||
Fig. 8 Cytokine/chemokine expression was identified using a CBA assay and flow cytometry. Human whole blood lysate incubated in RPMI medium with and without LPS tested for cytokine/chemokine expression after 2 hours and 2 and 7 days of incubation in human whole blood lysate on titania nanotube arrays as compared to the biomedical grade titanium. The results indicate altered short- and long-term cytokine/chemokine expression with the addition of LPS positive medium (◆,  , ,  , ,  , ,  , ,  , ,  , ,  , ★, , ★,   p ≤ 0.05). p ≤ 0.05). | ||
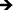 p ≤ 0.05).
p ≤ 0.05).
|
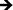 p ≤ 0.05).
p ≤ 0.05).
|
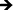 p ≤ 0.05).
p ≤ 0.05).
|
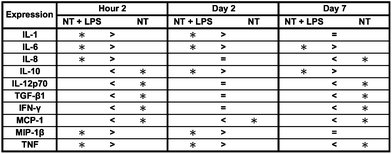
In an attempt to degrade or phagocytose the biomaterial, adherent macrophages and neutrophils release degradative enzymes, including nitric oxide synthase (iNOS) which plays a fundamental role in the immune response. This small molecule mediates a variety of physiological processes, including vasodilation, inflammation, thrombosis, immunity and neurotransmission, hypotension, intercellular communication and toxic defence against infectious organisms, as well as regulating the functional activity, growth and death of many immune and inflammatory cell types.71 Thus, the presence of this molecule determines the level of healthy integration of regions surrounding the implant. A commercially available Griess reagent kit was used in this study to detect the degree of nitrate present in the supernatant (see Fig. 9). The results showed a significant increase in NO between 2 hours and 7 days of culture on both substrates. In addition, the Griess reaction identifies a significant difference in NO released after 2 days of culture and a significant increase after 7 days of culture on titania nanotube arrays as compared to the control substrate. Studies have identified a connection between the activation of iNOS with IFN-γ and TNF.72 In addition, studies have shown that TGF-β, IL-4 and IL-10 provide inhibitory signals to iNOS.73,74 Due to the highly toxic effects of NO on cell functionality, the additional NO release seen on titania nanotube arrays may help to explain the reduced cellular proliferation and activation.
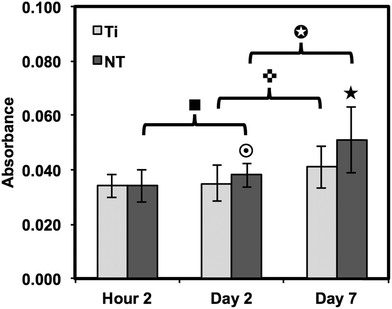 | ||
Fig. 9 Nitric oxide release was measured by a Griess reagent. The results indicate a significant increase in long-term NO presence in substrate-exposed supernatant on titania nanotube arrays as compared to biomedical grade titanium ( , ★, , ★,  , ■, , ■,  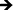 p ≤ 0.05). p ≤ 0.05). | ||
Conclusion
The ability to modulate the adverse immune response of implantable biomedical devices will prove to be implemental in the success of long-term implants. The results presented here indicate a reduced immune reaction on titania nanotube arrays as compared to biomedical grade titanium after 2 hours and 2 and 7 days of culture with whole human blood lysate. MTT assays and fluorescence microscope images show significantly decreased cell viability and count after all time points on titania nanotube arrays as compared to the control substrates. Short- and long-term monocyte, macrophage and neutrophil adhesion and proliferation evaluated by fluorescence microscope images indicated a reduction in cellular adhesion, proliferation and cytoskeletal reorganization on titania nanotube arrays. After 7 days of culture, fluorescence images identified extreme elongation and the formation of FBGCs on the control substrate, which were absent on titania nanotube arrays. In addition, SEM images showed a lack of cell–surface and cell–cell interactions on titania nanotube arrays, which were prevalent on the control substrate. Long-term cytokine and chemokine expression were shown to be significantly increased for MIP-1β, MCP-1, IL-6 and IL-8 on the control substrates, likely resulting from the cellular fusion and overall increased activation on the biomedical grade titanium substrates. A Griess reaction kit was used to show significantly increased amounts of NO present on the nanotube arrays. These results suggest the improved biocompatibility of titania nanotube arrays, identifying this specific nano-architecture as a potential interface for promoting the short- and long-term success of implantable biomedical devices. The results presented here identify a physiologically-relevant representation of the immune response on titania nanotube arrays as whole human blood lysate includes a cell population similar to that of the in vivo environment.Acknowledgements
Financial support for this work was provided by the National Science Foundation (CBET – 0827827) and the Colorado Office of Economic Development and International Trade. The authors would like to thank D. Bolchen and the staff at Garth Englund Blood Center for their professional assistance with the blood collection.References
- J. A. Jones, PhD 3263192, Case Western Reserve University, 2007.
- V. Mooney, P. K. Predecki, J. Renning and J. Gray, J. Biomed. Mater. Res., 1971, 5, 143–159 Search PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS.
- C. M. Jackson and Y. Nemerson, Annu. Rev. Biochem., 1980, 49, 765–811 CrossRef.
- D. T. Luttikhuizen, M. C. Harmsen and M. J. Van Luyn, Tissue Eng., 2006, 12, 1955–1970 CrossRef CAS.
- A. Remes and D. F. Williams, Biomaterials, 1992, 13, 731–743 CrossRef CAS.
- J. Cohen, J. Bone Joint Surg., 1959, 41, 152–166 Search PubMed.
- E. F. DiCarlo and P. G. Bullough, Clin. Mater., 1992, 9, 235–260 Search PubMed.
- G. Mani, M. D. Feldman, D. Patel and C. M. Agrawal, Biomaterials, 2007, 28, 1689–1710 CrossRef CAS.
- M. Peuster, C. Hesse, T. Schloo, C. Fink, P. Beerbaum and C. von Schnakenburg, Biomaterials, 2006, 27, 4955–4962 Search PubMed.
- D. John E, Biomaterials, 2007, 28, 5058–5067 CrossRef CAS.
- K. Wang, Mater. Sci. Eng., A, 1996, 213, 134–137 Search PubMed.
- F. H. Jones, Surf. Sci. Rep., 2001, 42, 75–205 CrossRef CAS.
- Y. Lin, G. O. Gallucci, D. Buser, D. Bosshardt, U. C. Belser and P. C. Yelick, J. Dent. Res., 2011, 90, 251–256 Search PubMed.
- D. Bhargava, P. Bartlett, J. Russell, M. Liddington, A. Tyagi and P. Chumas, Acta Neurochir., 2010, 152, 173–176 Search PubMed.
- H. Eufinger and M. Wehmöller, Plast. Reconstr. Surg., 1998, 102, 300–308 Search PubMed.
- M. Long and H. J. Rack, Biomaterials, 1998, 19, 1621–1639 CrossRef CAS.
- S. D. Messinger, Disability & Rehabilitation, 2009, 31, 2130–2134 Search PubMed.
- W. Nammas, Int. J. Cardiol., 2011, 146, 456–456 Search PubMed.
- B. F. Speroni S, P. Maridati, M. Beretta and C. Maiorana, Minerva Stomatol., 2011, 60, 123–131 Search PubMed.
- M. Niinomi, J. Mech. Behav. Biomed. Mater., 2008, 1, 30–42 Search PubMed.
- S. Young-Taeg, Biomaterials, 2003, 24, 3893–3907 Search PubMed.
- G. Mendonça, D. B. S. Mendonça, F. J. L. Aragão and L. F. Cooper, Biomaterials, 2008, 29, 3822–3835 CrossRef CAS.
- A. Curtis and C. Wilkinson, Biomaterials, 1997, 18, 1573–1583 CrossRef CAS.
- P. Decuzzi and M. Ferrari, Biomaterials, 2010, 31, 173–179 CrossRef CAS.
- G. E. W. Park and J. Thomas, J. Biomed. Nanotechnol., 2005, 1, 18–29 Search PubMed.
- V. Vogel and M. P. Sheetz, Curr. Opin. Cell Biol., 2009, 21, 38–46 CrossRef CAS.
- M. Dadsetan, J. A. Jones, A. Hiltner and J. M. Anderson, J. Biomed. Mater. Res., 2004, 71A, 439–448 Search PubMed.
- N. J. Sniadecki, R. A. Desai, S. A. Ruiz and C. S. Chen, Ann. Biomed. Eng., 2006, 34, 59–74 CrossRef.
- T. P. Kunzler, C. Huwiler, T. Drobek, J. Vörös and N. D. Spencer, Biomaterials, 2007, 28, 5000–5006 CrossRef CAS.
- D. Khang, M. Sato, R. L. Price, A. E. Ribbe and T. J. Webster, Int. J. Nanomed., 2006, 1, 65–72 Search PubMed.
- T. T. Ruckh, K. Kumar, M. J. Kipper and K. C. Popat, Acta Biomater., 2010, 6, 2949–2959 CrossRef CAS.
- K. E. La Flamme, K. C. Popat, L. Leoni, E. Markiewicz, T. J. La Tempa, B. B. Roman, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 2638–2645 CrossRef CAS.
- K. C. Popat, E. E. Leary Swan, V. Mukhatyar, K.-I. Chatvanichkul, G. K. Mor, C. A. Grimes and T. A. Desai, Biomaterials, 2005, 26, 4516–4522 CrossRef CAS.
- J. R. Porter, A. Henson and K. C. Popat, Biomaterials, 2009, 30, 780–788 Search PubMed.
- S. Bechara, L. Wadman and K. C. Popat, Acta Biomater., 2011, 7, 2892–2901 Search PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- K. S. Brammer, S. Oh, C. J. Cobb, L. M. Bjursten, H. v. d. Heyde and S. Jin, Acta Biomater., 2009, 5, 3215–3223 CrossRef CAS.
- J. Park, S. Bauer, K. von der Mark and P. Schmuki, Nano Lett., 2007, 7, 1686–1691 CrossRef CAS.
- K. C. Popat, L. Leoni, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 3188–3197 CrossRef CAS.
- C. von Wilmowsky, S. Bauer, S. Roedl, F. W. Neukam, P. Schmuki and K. A. Schlegel, Clin. Oral Implants Res., 2012, 23, 359–366 Search PubMed.
- L. Peng, M. L. Eltgroth, T. J. LaTempa, C. A. Grimes and T. A. Desai, Biomaterials, 2009, 30, 1268–1272 CrossRef CAS.
- B. S. Smith, S. Yoriya, L. Grissom, C. A. Grimes and K. C. Popat, J. Biomed. Mater. Res., Part A, 2010, 95A, 350–360 CrossRef CAS.
- B. S. Smith, S. Yoriya, T. Johnson and K. C. Popat, Acta Biomater., 2011, 7, 2686–2696 Search PubMed.
- S. D. Puckett, P. P. Lee, D. M. Ciombor, R. K. Aaron and T. J. Webster, Acta Biomater., 2010, 6, 2352–2362 CrossRef CAS.
- K. von der Mark, S. Bauer, J. Park and P. Schmuki, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, E60 CrossRef.
- S. Bauer, J. Park, J. Faltenbacher, S. Berger, K. von der Mark and P. Schmuki, Integr. Biol., 2009, 1, 525–532 RSC.
- K. M. Ainslie, S. L. Tao, K. C. Popat, H. Daniels, V. Hardev, C. A. Grimes and T. A. Desai, J. Biomed. Mater. Res., A, 2009, 91, 647–655 Search PubMed.
- A. Kodama, S. Bauer, A. Komatsu, H. Asoh, S. Ono and P. Schmuki, Acta Biomater., 2009, 5, 2322–2330 Search PubMed.
- K. Popat, M. Eltgroth, T. LaTempa, C. Grimes and T. Desai, Small, 2007, 3, 1878–1881 CrossRef CAS.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS.
- C. Ruan, M. Paulose, O. K. Varghese, G. K. Mor and C. A. Grimes, J. Phys. Chem. B, 2005, 109, 15754–15759 CrossRef CAS.
- S. C. Roy, M. Paulose and C. A. Grimes, Biomaterials, 2007, 28, 4667–4672 CrossRef CAS.
- J. M. Macák, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed., 2005, 44, 2100–2102 CrossRef CAS.
- M. Paulose, K. Shankar, S. Yoriya, H. E. Prakasam, O. K. Varghese, G. K. Mor, T. A. Latempa, A. Fitzgerald and C. A. Grimes, J. Phys. Chem. B, 2006, 110, 16179–16184 CrossRef CAS.
- G. K. Mor, O. K. Varghese, M. Paulose, K. Shankar and C. A. Grimes, Sol. Energy Mater. Sol. Cells, 2006, 90, 2011–2075 CrossRef CAS.
- D.-J. Yang, H.-G. Kim, S.-J. Cho and W.-Y. Choi, Mater. Lett., 2008, 62, 775–779 Search PubMed.
- S. Lavenus, J. C. Ricquier, G. Louarn and P. Layrolle, Nanomedicine, 2010, 5, 937–947 CrossRef CAS.
- M. Vandrovcova and L. Bacakova, Physiol. Res. Acad. Sci. Bohemosl., 2011, 60, 403–417 Search PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- P. Shokrollahi, H. Mirzadeh, O. A. Scherman and W. T. Huck, J. Biomed. Mater. Res., Part A, 2010, 95A, 209–221 Search PubMed.
- J. M. Anderson, Cardiovasc. Pathol., 1993, 2, 33–41 Search PubMed.
- H. J. Rhee, C. P. M. Burgh-de Winter and W. T. Daems, Cell Tissue Res., 1979, 197, 355–378 Search PubMed.
- D. M. Higgins, R. J. Basaraba, A. C. Hohnbaum, E. J. Lee, D. W. Grainger and M. Gonzalez-Juarrero, Am. J. Pathol., 2009, 175, 161–170 CrossRef CAS.
- A. Gschwind, E. Zwick, N. Prenzel, M. Leserer and A. Ullrich, Oncogene, 2001, 20, 1594–1600 CrossRef CAS.
- A. Mantovani and E. Dejana, Immunol. Today, 1989, 10, 370–375 Search PubMed.
- C. A. Dinarello, J. Biol. Regul. Homeost. Agents, 1997, 11, 91–103 Search PubMed.
- J. A. Hoffmann, Nature, 2003, 426, 33–38 Search PubMed.
- Y. Moret and P. Schmid-Hempel, Science, 2000, 290, 1166–1168 Search PubMed.
- C. Nathan, FASEB J., 1992, 6, 3051–3064 CAS.
- M. Munder, M. Mallo, K. Eichmann and M. Modolell, J. Exp. Med., 1998, 187, 2103–2108 Search PubMed.
- C. Brodie, N. Goldreich, T. Haiman and G. Kazimirsky, J. Neuroimmunol., 1998, 81, 20–30 Search PubMed.
- C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |