The thermogelling PLGA–PEG–PLGA block copolymer as a sustained release matrix of doxorubicin
Lin
Yu
ab,
Tianyuan
Ci
a,
Shuchun
Zhou
a,
Wenjiao
Zeng
c and
Jiandong
Ding
*ab
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Advanced Materials Laboratory, Fudan University, Shanghai 200433, China. E-mail: jdding1@fudan.edu.cn; Fax: +086-021-65640293; Tel: +086-021-65643506
bKey Laboratory of Smart Drug Delivery of Ministry of Education and PLA, School of Pharmacy, Fudan University, Shanghai 201203, China
cDepartment of Pathology, Shanghai Medical College, Fudan University, Shanghai 200032, China
First published on 3rd January 2013
Abstract
This study is aimed at extending a thermo-induced physical hydrogel as a localized delivery system of the antitumor drug doxorubicin (DOX). An amphiphilic triblock copolymer consisting of poly(lactic acid-co-glycolic acid) (PLGA) and poly(ethylene glycol) (PEG) was synthesized. The PLGA–PEG–PLGA triblock copolymer/water system exhibited a reversible sol–gel transition with increasing temperature, and the gel window covered the physiological temperature (37 °C). After a subcutaneous injection of the aqueous polymer solution into Sprague–Dawley rats, in situ gelling occurred, and the gel persisted over 20 days in the body. The addition of DOX did not alter the basic thermogelling property, yet our rheological measurements revealed that the increased viscosity of the sol state influenced the available drug concentration, which should be taken into consideration with respect to the injectability. Despite being a small molecule and a water-soluble drug, DOX with an appropriate drug concentration was released from the physical hydrogel in a sustained manner following an initial burst. To evaluate antitumor efficacy in vivo, the formulation was injected subcutaneously into mice bearing sarcoma-180 solid tumors. A single injection of the DOX-loading gel presented higher therapeutic efficacy and lower toxic effects compared to two injections of free DOX under the same total dose.
Introduction
Among the various antitumor drugs used clinically, doxorubicin (DOX), an anthracycline drug, is one of the most effective antineoplastic agents developed for the treatment of solid tumors.1–3 Yet its side effects such as cardiotoxicity partially limit its clinical use.1–3 Another crucial problem of DOX in the traditionally systemic administration comes from the instability of DOX in the blood stream due to rapid metabolization resulting in the formation of inactive derivatives.4 In order to overcome these problems and meanwhile to improve the drug efficacy, many delivery systems of DOX have been explored.5–12Injectable in situ-forming gels have demonstrated numerous advantages such as improved patient compliance,13–17 and afford a type of biomaterial potentially used in localized drug delivery.18–24 Especially, a polymeric aqueous system possessing a reversible sol–gel transition as a function of temperature is an ideal injectable hydrogel system. Drugs can be mixed with polymer solutions at ambient temperature, and the mixture forms a gel depot at the target site simply due to the physiological heat at 37 °C after a syringe injection; the loaded drugs are then slowly released. Typical thermogelling polymers are Pluronic (block copolymer of poly(ethylene glycol) (PEG) and poly(propylene glycol)) and its derivatives,24–26 amphiphilic block copolymers composed of PEG and biodegradable polyesters including poly(lactide-co-glycolide) (PLGA),27–30 poly(ε-caprolactone),31,32 poly(propylene fumarate),33 poly(ε-caprolactone-co-lactide),34–37 PEG/polypeptide,38,39 and poly(phosphazenes).20,40
The most popular biodegradable thermogelling polymers are PLGA–PEG–PLGA triblock copolymers due to their convenient synthesis and excellent safety profile.13,15,28–30,41–43 This thermogelling system has been used as the carrier of local anticancer drug delivery.44–46 For example, the paclitaxel-loading PLGA–PEG–PLGA hydrogel (ReGel) can sustain in vitro release of the drug over 50 days; compared to the commercial paclitaxel product Taxol, the higher efficacy against human breast tumor xenografts was achieved via the treatment of the ReGel/paclitaxel formulation.45 The clinical trial of this formulation (OncoGel) also demonstrated its safety and efficacy against the solid tumors.47
The present study tried to extend the PLGA–PEG–PLGA thermogel as a sustained release carrier of DOX. Although PLGA–PEG–PLGA is the most promising biodegradable thermogelling polymer and DOX is a very common antitumor drug, we cannot find any report of the PLGA–PEG–PLGA hydrogel system encapsulating DOX. The thermogelling system is sensitive to additives,27,28,48 so we would like to know whether or not the presence of DOX will destroy the sol–gel transition of the PLGA–PEG–PLGA system and whether or not the polymer solution containing DOX is still injectable (the injectability is dependent upon the viscosity of the sol state). Meanwhile, it is well known that hydrogels are usually appropriate for sustained release carriers of water-insoluble hydrophobic small molecular drugs or water-soluble macromolecular drugs, but less probably appropriate for carriers of water-soluble small molecular drugs. Since DOX is a water-soluble small molecular drug, it is meaningful for checking the feasibility of the sustained release of DOX from the thermogel. Due to a localized delivery system used in this study, the instability of DOX in the blood stream will be avoided. Yet the side effect especially myocardial damage remains a crucial candidate problem. Hence, besides the in vivo efficacy of released DOX against sarcoma-180 (S-180) solid tumors, we will examine the DOX-induced side effect from the thermogel formulation in a mice model.
Materials and methods
Materials
PEG (1500) (Aldrich-Sigma), DL-lactide (LA) (Purac), glycolide (GA) (Purac) and 95% of stannous octoate (Aldrich-Sigma) were used as received without further purification. DOX in the form of DOX·HCl was purchased from YuanCheng Technology Development Co., Ltd. All solvents were of reagent grade.Synthesis of PLGA–PEG–PLGA
PLGA–PEG–PLGA triblock copolymers were synthesized through the bulk ring-opening copolymerization of LA and GA in the presence of PEG with stannous octoate as the catalyst. Details of the synthetic procedure have been reported in our previous publication.49NMR measurements
The copolymer composition and structure were examined by 1H-NMR.49 The data were recorded with a Bruker DMX500 spectrometer operating at 500 MHz using CDCl3 as a solvent and tetramethylsilane (TMS) as an internal standard.Gel permeation chromatography characterization
Gel permeation chromatography (GPC, Agilent1100) measurements were carried out to evaluate the molecular weight (MW) and its distribution of the copolymer. The apparatus was equipped with a differential refractometer as the detector, and tetrahydrofuran (THF) was used as the mobile phase with a flow rate of 1.0 mL min−1 at 35 °C. The columns were calibrated with polystyrene (PS) standards.Determination of the sol–gel transition
The temperature-induced sol–gel transition of PLGA–PEG–PLGA triblock copolymers in phosphate buffer saline (PBS, pH 7.4) solution was determined using the vial inversion approach.49 The polymers were dissolved in PBS via continuous stirring and thus the samples with varied concentrations were obtained. The vials containing 0.5 mL polymer solutions were immersed into a water bath and kept for 15 min prior to measurements. Each step of the temperature increment was set as 1 °C. The criterion of a gel was defined when no visual flow was observed within 30 s after inversion of the vial.Rheological analysis
The sol–gel transitions of the polymer formulations with or without DOX were also studied by the rheological measurements (ARES Rheolometric Scientific). About 10 mL polymer solution was added into the Couette cell (Couette diameter, 34 mm; bob diameter, 32 mm; bob height, 33.3 mm; bob gap, 2 mm) and then a few drops of low-viscosity silicone oil were overlaid on the surface of the solution to avoid solvent evaporation. An environment controller (Neslab, RTε-130) was used to modulate the temperature with an accuracy of ±0.05 °C. The heating rate and angular frequency were set at 0.5 °C min−1 and 10 rad s−1, respectively.In vitro degradation
The test tubes containing 0.5 mL polymer solutions were placed in a shaking bath. The incubation temperature was set at 37 °C and the shaking rate was 50 strokes per minute. After incubation for 10 min, the polymer solutions were transformed into gel depots. Then, 4.5 mL of PBS solution was added. The buffer solution was exchanged every 4 days. At each examined degradation time, some specimens were withdrawn from the shaking bath and the top buffer was decanted to separate the gel matrix. The copolymers in the remaining thermogels were collected via freeze-drying and then analyzed via GPC.SEM measurements of in vitro gel morphology
The interior morphology of remaining gel depots was observed by field-emission scanning electron microscopy (FE-SEM S-4800, Hitachi, Japan). At specified sample collection times, the specimen was quickly withdrawn and the gel morphology was fixed by liquid nitrogen. A metal stub coating a layer of conductive adhesive was used to load the gel. The SEM observations were performed at 0.5 kV acceleration voltage on a deceleration mode at −140 °C under a nitrogen atmosphere.In vivo gel formation and degradation
Sprague–Dawley (SD) rats (male 180 ± 20 g) were anesthetized by diethyl ether. Then aqueous polymer solutions (25 wt%, 0.5 mL) were injected into the subcutaneous layer of rats via a 2 mL syringe equipped with a 23-gauge needle. The animal experiments were carried out in accordance with “Principles of Laboratory Animal Care” (NIH publication #85-23, revised 1985) and approved by the Ethics Committee of Fudan University. After injection for the predetermined time intervals, the rats were sacrificed, and the remaining gels were removed from the injection sites for analyses of MW of polymers.In vitro DOX release
Polymer solutions containing DOX were dispensed into 10 mL vials and incubated in a shaking bath for 10 min at 37 °C to form the gel depots. Then, 5 mL of PBS was added into each gel formulation, and the samples were shaken at 50 strokes per minute. At the designated time points, a 4 mL solution was taken out of the vial and the same amount of fresh PBS was added. The amount of DOX in the supernatant was analyzed via a UV-vis assay at 495 nm. Three independent samples were examined for each release group, and the release medium of drug-free hydrogels at the same time intervals was used as the control to eliminate the influence of degradation products.In vivo antitumor activity in mice
The tumor model was established by intradermally implanting S-180 cells (1–2 × 107 mL−1, 0.2 mL) into the armpits of Kunming strain mice (female, 20 ± 2 g) at day −1. After implantation for 24 h, the animals were divided randomly into seven groups (n = 10 for each group). The gel formulation (polymer concentration 25 wt%, and drug loading 1 mg mL−1) was subcutaneously injected in the vicinity of the tumors using a disposable (1 mL) syringe with a 26-gauge needle. The seven groups were (1) normal saline as the negative control; (2) 5-fluorouracil (5-Fu) as the positive control group (at day 0 and day 3, the drug was injected intravenously); (3) the drug-free polymer solution; (4–6) drug-loading polymer solutions (doses: 0.4, 0.2 and 0.1 mL, respectively); (7) DOX solution (at day 0 and day 3, the drug was injected intravenously). After treatment for 7 days, the animals were sacrificed and the tumors were withdrawn and weighed. The formulation effects against S-180 sarcomas were evaluated in accordance with the mean tumor weight as| Tumor inhibition ratio (%) = (A1 − A2)/A1 × 100% |
Here, A1 and A2 represent the mean tumor weight of the negative control group without drug and that of the underlying drug-treated group, respectively. If the tumor inhibition ratio is ≥40% and no significant weight loss was observed, the result was regarded as efficient. The antitumor test adheres to guidelines evaluated and approved by the Ethics Committee of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
In addition, the hearts and tumors of mice were taken out and histologically processed using haematoxylin–eosin (H&E) stains for examination of therapeutic and side effects of DOX released from the copolymer hydrogel matrix.
Results
Synthesis and thermogelling properties of PLGA–PEG–PLGA triblock copolymers
The LA and GA were copolymerized onto the hydroxyl-terminated PEG, resulting in the formation of PLGA–PEG–PLGA triblock copolymers. The obtained product was characterized via1H NMR and GPC measurements. The integration of peaks from 1H NMR at 1.55, 3.60 and 5.20 ppm was used to determine the average MW of 1675(PLGA)–1500(PEG)–1675(PLGA) and the LA/GA molar ratio of 10/1. A unimodal MW distribution of 1.23 was achieved through the GPC measurement, indicating that the sample was well synthesized.Fig. 1 shows the phase diagram of PLGA–PEG–PLGA triblock copolymers in PBS solutions. At low temperatures, the polymer/water system formed a free-flowing liquid. As the temperature increased, the system underwent a reversible sol–gel transition. Another transition of gel to turbid suspension of the polymer in water occurred at higher temperatures, and the upper transition is more dependent on polymer concentration than the lower transition. In addition, the critical gel concentration (CGC) was determined from the phase diagram, and the value was about 9 wt% (an 8 wt% solution did not exhibit the sol–gel transition at all temperatures and 10 wt% exhibited the indicated two transitions). At the physiological temperature (37 °C), the polymer/water system with concentration over CGC was in the hydrogel state, indicating that this sample is suitable as an injectable biomaterial.
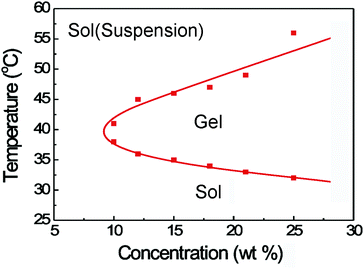 | ||
| Fig. 1 Phase diagram of PLGA–PEG–PLGA triblock copolymers in PBS solutions. | ||
In vitro degradation of the PLGA–PEG–PLGA hydrogel
We carried out in vitro degradation of the PLGA–PEG–PLGA hydrogel (25 wt%). The degradation was due to the hydrolysis of the PLGA chain and resulted in lactic acid, glycolic acid and PEG1500 as the final degradation products. The weight-average MW and polydispersity (PDI) of the remaining copolymers were obtained through GPC measurements, and the results are shown in Fig. 2. A steady decrease of MW over the period of one month was found. After incubation for 40 days, the loss of MW was more than 35% while the PDI increased from 1.20 to 1.34. The gel could not maintain its integrity at day 40, and became a free-flowing liquid.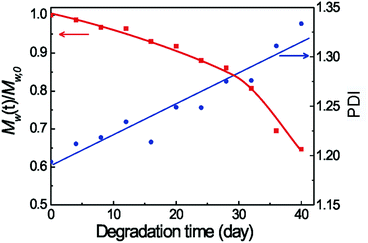 | ||
| Fig. 2 M w and PDI of copolymers in the remaining hydrogels during in vitro degradation in PBS at 37 °C. The initial polymer concentration was 25 wt%. | ||
The change of the interior morphology of the thermogel during in vitro degradation was observed by SEM, with the results shown in Fig. 3. Before incubation in PBS at 37 °C, no significant pore appeared in the thermogel. In contrast, striking porous morphology was generated as the erosion time increased.
 | ||
| Fig. 3 SEM images of the thermogel at the indicated days during degradation in PBS at 37 °C. The observations were made at freezing state. | ||
In vivo gelling and degradation of the PLGA–PEG–PLGA hydrogel
To confirm the in vivo gel formation and maintenance, the copolymer solutions were subcutaneously injected into SD rats through a conventional syringe at ambient temperature. The in vivo gelling occurred rapidly (about 30 s) following the injection due to the body heat at 37 °C. At the predetermined time points, the rats were sacrificed and dissected. The results showed that the gels under the skin were round or irregularly shaped with some protrusions. The integrity of the gel depot persisted over 3 weeks. A typical image taken at the 14th day is presented in Fig. 4.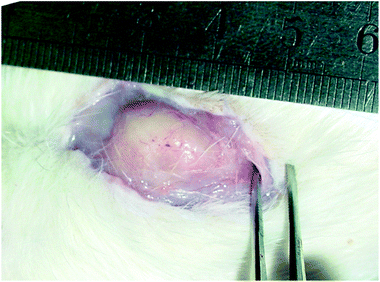 | ||
| Fig. 4 In situ gel formation of the PLGA–PEG–PLGA aqueous solution (25 wt%, 0.5 mL) in the neck of an SD rat. The photograph was taken after 2 weeks following the subcutaneous injection. | ||
The remaining samples in rats were further collected and freeze-dried. THF was used to extract the copolymers in the dried specimens, and the collected copolymers were analyzed by GPC. Fig. 5 displays the GPC traces of the remaining copolymers during in vitro and in vivo degradation tests. A single-peak profile is basically maintained; the increase of the retention time verified the decrease of MW in the examined periods of in vitro degradation. A multi-peak profile appeared in the in vivo degradation.
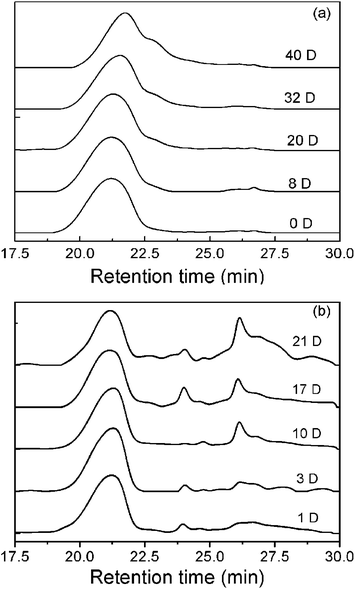 | ||
| Fig. 5 GPC traces of the PLGA–PEG–PLGA triblock copolymer collected during in vitro degradation (a) and in vivo degradation after being subcutaneously implanted in SD rats (b). | ||
Effects of DOX on the thermogelling properties of PLGA–PEG–PLGA copolymers
It is convenient to construct the DOX-loading hydrogel formulation simply by mixing a PLGA–PEG–PLGA aqueous solution with the drug at ambient or lower temperatures. The mixture was still a free-flowing solution at room temperature. With increasing temperature, the drug-loading system turned into a semisolid gel at the physiological temperature (37 °C) (Fig. 6). | ||
| Fig. 6 Photographs of the DOX-loading PLGA–PEG–PLGA formulation at indicated temperatures. The red color comes from DOX. | ||
The influence of drug concentration on the sol–gel transition was further assayed via dynamic rheological measurements, with the results shown in Fig. 7. When the drug-loading amount was 1 mg mL−1 or 2 mg mL−1, the viscosities of hydrogel formulations at the sol state were lower than 0.1 Pa s and the thermogelling behaviors were very similar to the drug-free system. However, on increasing the drug-loading to 4 mg mL−1, the viscosity at the sol state remarkably increased and thus the injectability suffered a certain difficulty. This phenomenon implies the nonlinear relation between the global rheological behaviors and the interaction of DOX/PLGA–PEG–PLGA triblock copolymers in water. The change of viscosity after addition of DOX into the aqueous polymer solution should be taken into consideration in design of the corresponding injectable formulation. Therefore, in the following in vitro and in vivo experiments, only the drug concentrations 1 and 2 mg mL−1 were adopted.
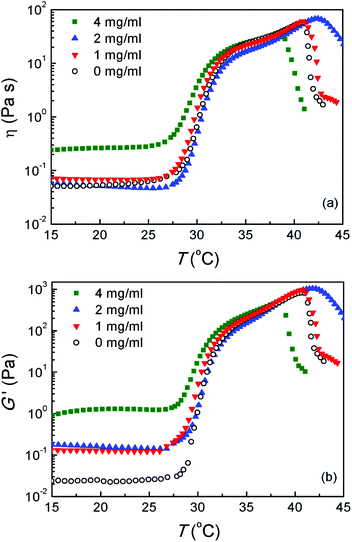 | ||
| Fig. 7 (a) Viscosity η and (b) storage modulus G′ of the PLGA–PEG–PLGA aqueous solutions (25 wt%) with and without DOX at varied temperatures. Heating rates: 0.5 °C min−1; oscillatory frequency: 10 rad s−1. | ||
In vitro release of DOX from the thermogel
The in vitro DOX release from the polymer formulations was examined up to 3 weeks. The burst release occurred at the first two days, then a slow release up to about 10 days. With increasing drug-loading from 1 to 2 mg mL−1, the amount of cumulative release decreased, as shown in Fig. 8a. With the increment of polymer concentration, both the burst release and the cumulative release slightly diminished, as illustrated in Fig. 8b.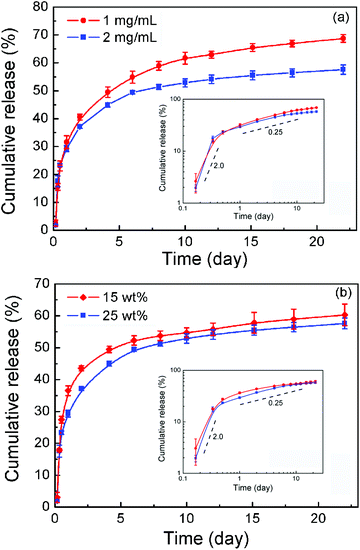 | ||
| Fig. 8 (a) Effect of drug-loading on in vitro drug release profiles in PBS (pH 7.4) at 37 °C. The polymer concentration was 25 wt%. (b) Effect of polymer concentration on in vitro drug release profiles. The drug-loading was 2 mg mL−1. Error bars represent the standard deviation (n = 3). Insets in both (a) and (b) are double logarithm plots; the values are the slopes of the indicated dashed lines. | ||
The release data were further fitted via the Higuchi equation written as 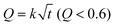 and the Peppas–Korsmeyer equation written as Q = ktm (Q < 0.6), where Q denotes the cumulative release, t is release time, k and m are constants. It is well-known that a good correlation coefficient R with m close to 0.5 suggests that drug release obeys a diffusion mechanism.16,44 However, the value of R2 was less than 0.90 outputted by the Higuchi equation and less than 0.93 with optimal m about 0.3 outputted via the Peppas–Korsmeyer equation. So, the mechanism governing the DOX release from the thermogel matrix deviated significantly from an ideal diffusion mechanism. We then presented the data in double logarithmic plots as shown in the insets of Fig. 8. It is clear that the release profile could be roughly divided into two stages, the early burst stage and the main diffusion stage. Yet the slopes in the log–log plots from both stages reflect a significant deviation from the typical Higuchi diffusion with slope 0.5. DOX has two forms, the hydrophilic form DOX·HCl under an acidic environment and the hydrophobic form under a neutral or basic environment.50 DOX·HCl was used initially, yet the drug was encapsulated at neutral PBS; hence the two forms might co-exist in the thermogel matrix. As is known, such a physical hydrogel has an internal structure of a percolated micelle network.49,51 The micellar cores and intermicellar region might interact with the two forms of DOX in different ways, which complicates the release profile. The details of the release process are open at the moment.
and the Peppas–Korsmeyer equation written as Q = ktm (Q < 0.6), where Q denotes the cumulative release, t is release time, k and m are constants. It is well-known that a good correlation coefficient R with m close to 0.5 suggests that drug release obeys a diffusion mechanism.16,44 However, the value of R2 was less than 0.90 outputted by the Higuchi equation and less than 0.93 with optimal m about 0.3 outputted via the Peppas–Korsmeyer equation. So, the mechanism governing the DOX release from the thermogel matrix deviated significantly from an ideal diffusion mechanism. We then presented the data in double logarithmic plots as shown in the insets of Fig. 8. It is clear that the release profile could be roughly divided into two stages, the early burst stage and the main diffusion stage. Yet the slopes in the log–log plots from both stages reflect a significant deviation from the typical Higuchi diffusion with slope 0.5. DOX has two forms, the hydrophilic form DOX·HCl under an acidic environment and the hydrophobic form under a neutral or basic environment.50 DOX·HCl was used initially, yet the drug was encapsulated at neutral PBS; hence the two forms might co-exist in the thermogel matrix. As is known, such a physical hydrogel has an internal structure of a percolated micelle network.49,51 The micellar cores and intermicellar region might interact with the two forms of DOX in different ways, which complicates the release profile. The details of the release process are open at the moment.
In vivo antitumor efficacy
We further assessed the in vivo antitumor efficacy of DOX released from the PLGA–PEG–PLGA hydrogel in S-180 bearing KM mice. Fig. 9 shows the S-180 sarcoma isolated from the mice at the seventh day after the indicated treatments. Compared with saline and gel-treated groups, a single injection of a DOX-loading gel formulation significantly inhibited the growth of tumors. The inhibition effect was dependent upon the dose of the drug.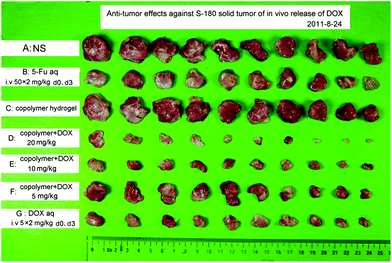 | ||
| Fig. 9 S-180 sarcomas captured at the seventh day after a subcutaneous injection of DOX-loading PLGA–PEG–PLGA solution (25 wt%). The scheduled dose and the route of administration are listed in Table 1. | ||
The quantitative measurements and associated statistics were also carried out, with the results summarized in Table 1. In the case of the whole dose of 10 mg kg−1, the group G of two intravenous injections of free DOX resulted in an 80.4% tumor inhibition ratio. The tumor inhibition increased to 87.2 wt% via a single subcutaneous injection of gel formulation at the same dose (group E). The results suggest that the gel formulation is more effective against tumor inhibition than free DOX.
| Groupa | Doseb (mg kg−1) | Route of medicationc | Animals | Mean weight of animal (g) | Mean weight of tumor (g) | Tumor inhibition ratio % | p | ||
|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | X ± SD | |||||
| a NS: the negative control with just the physiological saline solution injected; 5-FU: the positive control with a sufficient amount of 5-FU injected twice. b 20 mg kg−1, 10 mg kg−1, 5 mg kg−1 denote the whole drug dose relative to weight of mouse; 50 mg kg−1 × 2 denotes the 5-FU drug dose of every time, and at day 0 and day 3 the drug was intravenously injected; 5 mg kg−1 × 2 denotes the free DOX drug dose of every time, and at day 0 and day 3 the drug was intravenously injected. c i.v.: intravenous injection, s.c.: subcutaneous injection. d The p values resulting from Student t-tests of tumor weight versus the negative control (NS group). For each group, n = 10. | |||||||||
| A: NS | 0 | s.c. | 10 | 10 | 23.4 | 35.9 | 2.09 ± 0.79 | ||
| B: 5-Fu aq | 50 × 2 | i.v. | 10 | 10 | 22.5 | 29.4 | 1.02 ± 0.32 | 51.4 | <0.001 |
| C: hydrogel | 0 | s.c. | 10 | 10 | 23.4 | 34.4 | 2.07 ± 0.71 | 0.8 | >0.05 |
| D: DOX-loading gel | 20 | s.c. | 10 | 10 | 23.3 | 24.6 | 0.14 ± 0.13 | 93.4 | <0.001 |
| E: DOX-loading gel | 10 | s.c. | 10 | 10 | 20.4 | 28.7 | 0.27 ± 0.16 | 87.2 | <0.001 |
| F: DOX-loading gel | 5 | s.c. | 10 | 10 | 23.2 | 31.6 | 0.79 ± 0.47 | 62.1 | <0.001 |
| G: DOX aq | 5 × 2 | i.v. | 10 | 10 | 22.8 | 26.8 | 0.41 ± 0.18 | 80.4 | <0.001 |
The S-180 sarcomas captured from the mice were histologically processed using H&E staining. The slices were observed in order to assess the therapeutic effect. Some typical images are presented in Fig. 10, where the disappearance of cell nuclei reflects the necrosis of tissue. Hence, the tumor tissue exhibited a serious necrosis after receiving a single injection of the DOX-loading gel.
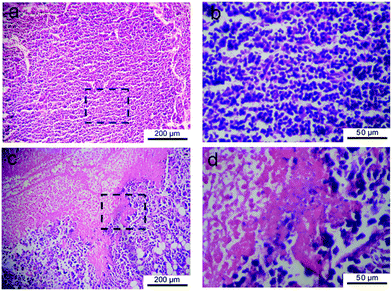 | ||
| Fig. 10 H&E-stained histological sections of tumors captured from S-180 bearing KM mice at the seventh day after subcutaneous injection of saline (a) and DOX-loading gel formulation (c). (b) and (d) are magnified images of the indicated regions in (a) and (c), respectively. Serious necrosis of tumor tissue was observed in the upper left of (c) and (d). | ||
Adverse effects
After the drug administration, the growth of mice was influenced due to the toxicity of the anticancer drug. We measured the change of body weight of mice as a function of treatment time, with the results shown in Fig. 11. In the case of the high dose of 20 mg kg−1, a significant inhibition of growth of body weight and a phenomenon of hair slip at the injection site were observed, indicating the high toxic effect. On decreasing the drug dose, the body weight of mice increased and no hair slip occurred. It is interesting that the animals in group E treated with the gel formulation presented higher tolerance compared to group G by free DOX under the same drug dose, evidenced by relatively low weight loss. This feature also suggests that the local drug delivery diminishes the systemic toxicity compared to the traditional intravenous administration.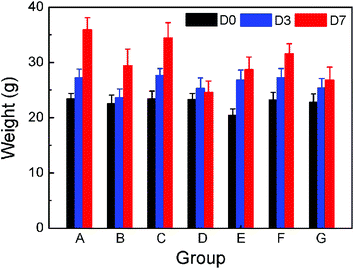 | ||
| Fig. 11 Body weight of KM mice as a function of time after the treatment of the groups as indicated in Table 1. | ||
The cardiotoxicity of DOX was also estimated. In the cases of gel formulation with different drug doses, tissue edema and inflammatory cells around blood vessels were observed in the heart tissue (Fig. 12). The tissue edema presented the increment of interval between myocardial cells and the inflammatory cells were mainly attributed to macrophages. Fig. 12b displays the most serious response in our experiments of this group, which is, however, much weaker than that reported in the literature about DOX formulations.1–3
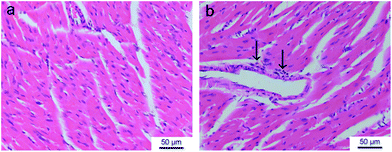 | ||
| Fig. 12 H&E-stained histological sections of heart tissues isolated at the seventh day after subcutaneous injection of saline (a) and DOX-loading gel formulation (b) in S180-bearing KM mice. The increment of interval between myocardial cells reflects the edema of tissue. The arrows denote the inflammatory cells (macrophage). | ||
Discussion
In the clinical treatment of solid tumors, the first choice is usually the surgical removal. However, it is not easy to entirely remove tumor tissues through surgery and a recurrence of cancer may take place near the site of the previous surgical excision after a few years. One supplementary treatment option to overcome this problem is the postsurgery injection of anticancer drugs at the dissection site. This local chemotherapy is capable of maximizing efficacy of antitumor drugs along with minimum side effects. Novel drug delivery systems provide localized but prolonged release in local chemotherapy. The thermogelling polymer PLGA–PEG–PLGA, which is biocompatible and biodegradable, has been tried to locally deliver anticancer drugs including paclitaxel, topotecan, PEGylated camptothecin etc.44–46 In this study, a DOX-loading PLGA–PEG–PLGA formulation was prepared and the feasibility as the local DOX delivery system was examined.Amphiphilic PLGA–PEG–PLGA triblock copolymers in water form core–corona micelles with hydrophilic PEG blocks as the coronas and hydrophobic PLGA blocks as the cores.49,51 The physical gelation occurs at high concentrations (>CGC) and temperatures (>Tgel) (Fig. 1), which might be due to the hydrophobic aggregation between micelles into a percolated micelle network.49,51 Different from Pluronic F-127, the intra- and/or intermolecular interactions between hydrophobic moieties of the PLGA–PEG–PLGA system resulted in the long-lasting maintenance in vivo after subcutaneous injection (Fig. 4). Meanwhile, following the in vivo degradation, a multi-peak profile for the remaining copolymers was observed (Fig. 5). The peaks with long retention time were attributable to the low MW fractions. This feature reflected that the degraded fragments of low MW were not rapidly dissolved away in the subcutaneous layer of the rats during in vivo degradation. In contrast, due to the frequent exchange of the release medium during the in vitro degradation tests, a single-peak profile remained and no low MW fractions were detected. In addition, we also found that in vivo degradation of this thermogel was faster than that in vitro (Fig. 5), which should be attributed to the cellular and enzymatic involvement. PLGA copolymers52,53 and thermogelling PEG/polyester copolymers42,54 presented a similar phenomenon after subcutaneous implantation.
The gelation process of the aqueous polymer solution with or without DOX was detected via the dynamic rheological measurements (Fig. 7). When the drug concentration was 1 or 2 mg mL−1, the addition of DOX had no significant influence on the polymeric thermogelling behaviors. However, the thermogelation properties of the poly(phosphazenes) such as sol–gel transition temperature and maximum viscosity were obviously changed after adding a small amount of DOX (0.1% or 0.2%).50 This finding reflects that our PLGA–PEG–PLGA system can adjust the drug-loading to a large extent compared with poly(phosphazenes).
Drug release from hydrogel depots can be influenced by many factors including pore size, degradable rate of hydrogel, size and morphology of matrix, concentration of drug and the specific interactions between the hydrogel and the loaded drug. Generally speaking, the release profile from a biodegradable hydrogel is mainly controlled by the drug diffusion and/or the matrix degradation.16 DOX was a glycoside composed of a hydrophobic aglycon moiety and a hydrophilic glucosamine moiety. Therefore, it is essentially an amphiphilic molecule and might interact with PLGA–PEG–PLGA triblock copolymers; and DOX has two forms depending upon initial pH. The partitioning of the two forms of DOX in the hydrophobic domain of the copolymer micelles might be important for the sustained release of DOX at a later stage. As a result, the complex release profiles as shown in Fig. 8 might rely on the dissociation rate of the drug from the thermogel matrix. The formation of a more tightly packed micelle network at higher polymer concentrations led to the slower drug release rate.
The aqueous polymer solution containing 1 mg mL−1 DOX was easily injected into the target site of mice by a 26-gauge needle. The high efficacy of inhibition against S-180 was achieved via a single administration, and the dose-dependent effect was observed as well (Fig. 9). These in vivo results, combined with the in vitro release profile, implied that the initial burst of DOX from the gel depot inhibited the initial tumor growth, and the later release of DOX induced the long-lasting suppression.
Under the same total dose, the mice showed the lower weight loss using the DOX formulation than free DOX (Fig. 11). This feature suggests that the injection of the DOX formulation in the vicinity of the tumors is beneficial for accumulating DOX in the tumor tissue and decreasing its distribution in other healthy organs, which produces the low drug-induced side effect compared to traditional intravenous administration.
The most characteristic adverse effect of DOX is its strong myocardial damage including disorganization of myofibrillar arrays and cytoplasmic vacuolization.1–3 In the current study, only some tissue edema and inflammatory reaction around blood vessels in the heart tissue was observed, and thus the myocardial damage was very low. Two possible reasons seem to be responsible for this result. One is the high concentration of DOX in tumors and the low distribution in the heart due to the localized drug delivery. Secondly, the lower release of DOX via a gel matrix may reduce the DOX-induced cardiotoxicity. By combining all of our results, we believe that the DOX-loading gel formulation with a 10 mg kg−1 dose was the optimal treatment due to the high therapeutic efficacy and the relatively low adverse effect.
Our experiments on the system composed of DOX and linear copolymers of PLGA and PEG are consistent with the previous report by Lee et al. on the system of DOX and star-shaped PLGA–PEG block copolymers.19 It seems necessary to indicate that the case of star copolymers cannot predict that of linear copolymers and vice versa the results of linear copolymers cannot replace those of star ones, because the thermogelling properties are very sensitive to polymer composition, architecture, end group, and additives.27–30,41,48,51,55 While it is popular to form micelles for amphiphilic block copolymers in water in a wide window, their thermogellable windows are quite narrow. Compared to star-shaped PLGA–PEG block copolymers, the linear PLGA–PEG–PLGA thermogel can be easily synthesized via a simple one-step reaction and has good reproducibility. This feature suggests that the PLGA–PEG–PLGA system has more potential as a medical material. Different from the previous publication,19 the present study examined the influence of DOX on the thermogelling property and revealed, for the first time, that the presence of DOX enhanced the viscosity of the copolymer aqueous solution significantly, which should be taken into consideration as an injectable hydrogel. We also assayed the adverse effect, and illustrated the insignificant myocardial damage of the DOX formulation of thermogels of copolymers composed of PEG and PLGA, which is also absent in the pertinent literature.19
Conclusions
Biodegradable PLGA–PEG–PLGA triblock copolymers with a sol–gel transition upon temperature rise were synthesized and characterized. After a subcutaneous injection of the polymer aqueous solution into SD rats, a gel rapidly formed in situ and remained in the target site for up to 20 days. The presence of DOX did not damage the sol–gel transition of the aqueous polymer system, but increased the viscosity significantly. In spite of it, the sol was still injectable when the drug concentration was lower than 2 mg mL−1. The DOX-loading hydrogel formulation sustained drug release in vitro after an initial burst. Due to sustained delivery and efficient accumulation of DOX in the tumor site, S-180 bearing animal models displayed significant inhibition of tumor growth and strong apoptosis of tumor cells after a subcutaneous injection of the mixture of DOX and copolymer solution. Compared to two intravenous injections of free DOX, a single injection of the DOX-loading gel enhanced the chemotherapeutic effect and reduced its systemic toxicity. The myocardial damage was relatively weak. Hence, the thermogelling PLGA–PEG–PLGA copolymer is promising as an injectable matrix of antitumor drugs.Acknowledgements
The group was supported by the Chinese Ministry of Science and Technology (973 Programs no. 2009CB930000 and no. 2011CB606203), NSF of China (Grants no. 50903021, no. 21034002, and no. 91127028), and the Specialized Research Fund for the Doctoral Program of Higher Education of China (no. 20090071120014).Notes and references
- R. C. Young, R. F. Ozols and C. E. Myers, N. Engl. J. Med., 1981, 305, 139 CrossRef CAS.
- S. E. Sallan and L. A. Clavell, Semin. Oncol., 1984, 11, 19 Search PubMed.
- R. M. Lowenthal and K. Eaton, Hematol. Oncol. Clin. North Am., 1996, 10, 967 Search PubMed.
- R. S. Benjamin and M. Gee, J. Pharmacol. Exp. Ther., 1971, 177, 567 Search PubMed.
- T. Lammers, V. Subr, K. Ulbrich, P. Peschke, P. E. Huber, W. E. Hennink and G. Storm, Biomaterials, 2009, 30, 3466 CrossRef CAS.
- R. Tong and J. J. Cheng, J. Am. Chem. Soc., 2009, 131, 4744 CrossRef CAS.
- G. T. Chang, C. Li, W. Y. Lu and J. D. Ding, Macromol. Biosci., 2010, 10, 1248 CrossRef CAS.
- W. L. Zhang, Y. L. Li, L. X. Liu, Q. Q. Sun, X. T. Shuai, W. Zhu and Y. M. Chen, Biomacromolecules, 2010, 11, 1331 CrossRef CAS.
- L. L. Zhao, L. J. Zhu, F. Y. Liu, C. Y. Liu, Shan-Dan, Q. R. Wang, C. L. Zhang, J. L. Li, J. G. Liu, X. Z. Qu and Z. Z. Yang, Int. J. Pharm., 2011, 410, 83 CrossRef CAS.
- Q. Q. Sun, D. Cheng, X. S. Yu, Z. Q. Zhang, J. Dai, H. Li, B. L. Liang and X. T. Shuai, J. Mater. Chem., 2011, 21, 15316 RSC.
- F. X. Zhan, W. Chen, Z. J. Wang, W. T. Lu, R. Cheng, C. Deng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomacromolecules, 2011, 12, 3612 CrossRef CAS.
- X. J. Loh, J. del Barrio, P. P. C. Toh, T. C. Lee, D. Z. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84 CrossRef CAS.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473 RSC.
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Macromolecules, 2011, 44, 6629 CrossRef CAS.
- M. H. Park, M. K. Joo, B. G. Choi and B. Jeong, Acc. Chem. Res., 2012, 45, 424 CrossRef CAS.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853 CrossRef CAS.
- W. X. Wang, H. Liang, R. C. Al Ghanami, L. Hamilton, M. Fraylich, K. M. Shakesheff, B. Saunders and C. Alexander, Adv. Mater., 2009, 21, 1809 CrossRef CAS.
- W. S. Shim, J. H. Kim, K. Kim, Y. S. Kim, R. W. Park, I. S. Kim, I. C. Kwon and D. S. Lee, Int. J. Pharm., 2007, 331, 11 CrossRef CAS.
- S. J. Lee, Y. Bae, K. Kataoka, D. Kim, D. S. Lee and S. C. Kim, Polym. J., 2008, 40, 171 CrossRef CAS.
- C. Chun, S. M. Lee, C. W. Kim, K. Y. Hong, S. Y. Kim, H. K. Yang and S. C. Song, Biomaterials, 2009, 30, 4752 CrossRef CAS.
- Y. M. Kang, G. H. Kim, J. Il Kim, D. Y. Kim, B. N. Lee, S. M. Yoon, J. H. Kim and M. S. Kim, Biomaterials, 2011, 32, 4556 CrossRef CAS.
- T. Ozeki, D. Kaneko, K. Hashizawa, Y. Imai, T. Tagami and H. Okada, Int. J. Pharm., 2012, 427, 299 Search PubMed.
- A. D. Baldwin, K. G. Robinson, J. L. Militar, C. D. Derby, K. L. Kiick and R. E. Akins, J. Biomed. Mater. Res., Part A, 2012, 100, 2106 Search PubMed.
- X. J. Loh, W. Guerin and S. M. Guillaume, J. Mater. Chem., 2012, 22, 21249 RSC.
- X. J. Loh, Y. X. Tan, Z. Y. Li, L. S. Teo, S. H. Goh and J. Li, Biomaterials, 2008, 29, 2164 CrossRef CAS.
- H. J. Chung, Y. H. Lee and T. G. Park, J. Controlled Release, 2008, 127, 22 CrossRef CAS.
- B. Jeong, Y. H. Bae and S. W. Kim, Macromolecules, 1999, 32, 7064 CrossRef CAS.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Y. Chang, Macromol. Rapid Commun., 2001, 22, 587 CrossRef.
- L. Yu, H. Zhang and J. D. Ding, Colloid Polym. Sci., 2010, 288, 1151 CrossRef CAS.
- L. Yu, Z. Zhang and J. D. Ding, Biomacromolecules, 2011, 12, 1290 CAS.
- S. J. Bae, J. M. Suh, Y. S. Sohn, Y. H. Bae, S. W. Kim and B. Jeong, Macromolecules, 2005, 38, 5260 CrossRef CAS.
- J. I. Kim, S. H. Lee, H. J. Kang, D. Y. Kwon, D. Y. Kim, W. S. Kang, J. H. Kim and M. S. Kim, Soft Matter, 2011, 7, 8650 RSC.
- E. Behravesh, A. K. Shung, S. Jo and A. G. Mikos, Biomacromolecules, 2002, 3, 153 CrossRef CAS.
- Z. Q. Jiang, X. M. Deng and J. Y. Hao, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4091 CrossRef CAS.
- Z. Zhang, Y. X. Lai, L. Yu and J. D. Ding, Biomaterials, 2010, 31, 7873 CrossRef CAS.
- Z. Zhang, J. Ni, L. Chen, L. Yu, J. W. Xu and J. D. Ding, Biomaterials, 2011, 32, 4725 CrossRef CAS.
- Z. Zhang, J. Ni, L. Chen, L. Yu, J. W. Xu and J. D. Ding, J. Biomed. Mater. Res., Part B, 2012, 100, 1599 Search PubMed.
- C. T. Huynh, M. K. Nguyen, J. H. Kim, S. W. Kang, B. S. Kim and D. S. Lee, Soft Matter, 2011, 7, 4974 RSC.
- S. H. Park, B. G. Choi, H. J. Moon, S. H. Cho and B. Jeong, Soft Matter, 2011, 7, 6515 RSC.
- B. H. Lee and S. C. Song, Macromolecules, 2004, 37, 4533 CrossRef CAS.
- L. Yu, H. Zhang and J. D. Ding, Angew. Chem., Int. Ed., 2006, 45, 2232 CrossRef CAS.
- L. Yu, Z. Zhang, H. Zhang and J. D. Ding, Biomacromolecules, 2010, 11, 2169 CrossRef CAS.
- L. Yu, Z. Zhang and J. D. Ding, Macromol. Res., 2012, 20, 234 Search PubMed.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, Int. J. Pharm., 2008, 348, 95 CrossRef CAS.
- G. M. Zentner, R. Rathi, C. Shih, J. C. McRea, M. H. Seo, H. Oh, B. G. Rhee, J. Mestecky, Z. Moldoveanu, M. Morgan and S. Weitman, J. Controlled Release, 2001, 72, 203 CrossRef CAS.
- G. T. Chang, T. Y. Ci, L. Yu and J. D. Ding, J. Controlled Release, 2011, 156, 21 CrossRef CAS.
- N. L. Elstad and K. D. Fowers, Adv. Drug Delivery Rev., 2009, 61, 785 CrossRef CAS.
- H. Zhang, L. Yu and J. D. Ding, Macromolecules, 2008, 41, 6493 CrossRef CAS.
- L. Yu, Z. Zhang, H. Zhang and J. D. Ding, Biomacromolecules, 2009, 10, 1547 CrossRef CAS.
- G. D. Kang, S. H. Cheon and S. C. Song, Int. J. Pharm., 2006, 319, 29 CrossRef CAS.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1122 CrossRef CAS.
- M. Sandor, J. Harris and E. Mathiowitz, Biomaterials, 2002, 23, 4413 CrossRef CAS.
- Q. Cai, G. X. Shi, J. Z. Bei and S. G. Wang, Biomaterials, 2003, 24, 629 CrossRef CAS.
- S. H. Park, B. G. Choi, M. K. Joo, D. K. Han, Y. S. Sohn and B. Jeong, Macromolecules, 2008, 41, 6486 CrossRef CAS.
- S. J. Lee, B. R. Han, S. Y. Park, D. K. Han and S. C. Kim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 888 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |