Bio-inspired silica–collagen materials: applications and perspectives in the medical field
Sascha Heinemann*a, Thibaud Coradin*b and Martin F. Desimone*c
aMax Bergmann Center of Biomaterials and Institute of Materials Science, Technische Universität Dresden, Budapester Str. 27, D-01069 Dresden, Germany. E-mail: sascha.heinemann@tu-dresden.de; Fax: +49-351-46339401; Tel: +49-351-463 39381
bUPMC Univ Paris 06, CNRS, Laboratoire de Chimie de la Matière Condensée de Paris, Collège de France, 11 place Marcelin Berthelot, F-75005 Paris, France. E-mail: thibaud.coradin@upmc.fr; Fax: +33-144271443; Tel: +33-144271528
cIQUIMEFA-CONICET and Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Junín 956 Piso 3°, (1113) Ciudad Autónoma de Buenos Aires, Argentina. E-mail: desimone@ffyb.uba.ar; Fax: +54-1149648254; Tel: +54-1149648254
First published on 25th April 2013
Abstract
Silica and collagen are two of the most abundant substances in the Earth's geosphere and biosphere, respectively. Yet, their close association in nature has never been clearly demonstrated despite increasing evidence for the key role of silicon in mammalians. Foreseeing the therapeutic benefits of their association within composites or hybrids, a wide diversity of bio-inspired silica–collagen materials have been prepared over nearly 15 years. These works not only generated materials with a large range of structures and properties, from soft mineralized hydrogels to hard compact xerogels, but also provided more fundamental information about the interplay between polymer self-assembly processes and inorganic condensation mechanisms. Biological in vitro and in vivo evaluations suggest their bioactivity, cyto- and biocompatibility as well as controlled drug delivery properties. Hence they can now fully integrate the family of materials with high potential for the development of innovative biomedical devices.
 Sascha Heinemann | Sascha Heinemann was born in Schmalkalden, Germany, in 1980. He studied Materials Science at the Technische Universität Dresden/Max-Bergmann-Center of Biomaterials and received his PhD in 2011. His research interests focus on the development of organic/inorganic composites for bone substitution including cell–biomaterial interactions. Furthermore, he manages transfer projects in this field. He co-authored over 50 journal publications, book chapters, and patent applications and is currently a postdoctoral fellow at the Collaborative Research Center TRR 79. |
 Thibaud Coradin | Thibaud Coradin, born in 1970, is Directeur de Recherche at the CNRS since 2007. He is currently leading the “Materials and Biology” group in the Laboratoire de Chimie de la Matière Condensée de Paris (UPMC-Paris 06). His research topics include biomineralization, bionanocomposites, biomaterials, bioencapsulation and green materials chemistry. He co-authored over 120 publications and 14 book chapters. He is a member of the Advisory Editorial Board of Current Medicinal Chemistry and Silicon. |
 Martin F. Desimone | Martin Federico Desimone was born in 1974 in Buenos Aires, Argentina. He graduated from Pharmacy and Biochemistry and received his PhD degree from the University of Buenos Aires. Currently, he holds an Adjunct Professor position in the Faculty of Pharmacy and Biochemistry at the University of Buenos Aires. His research topics include the development of organic, inorganic and biological hybrid materials and the study of the interactions in the nano- and microscale for biomedical and biotechnological applications. He co-authored over 40 journal publications, book chapters and one patent application. He is a CONICET researcher and the director of the biocompatible nanomaterials project. |
1. Introduction
Hydrated biopolymer networks are the main constituents of living tissues, in association with cells and, in some cases, mineral particles. Hence it is not surprising that (bio)-organic hydrogels are the favored class of materials for soft tissue replacement, repair or regeneration.1–3 Despite the versatility of their composition, structure, and reactivity, they suffer from mechanical properties that are unsuitable for several applications. Cross-linking strategies using organics4 or inorganic5 additives, or enzymatic reactions6,7 have been considered. Interpenetrating polymer networks constitute a promising approach to tailor hydrogel behavior.8 Finally, mineralization of hydrogels to form organic–inorganic hybrid or composite structures has been proposed.9Considering the selection of the most appropriate constituents of these hydrogels, a possible challenging approach is the ‘biomimetic’ one where the prepared biomaterial should resemble the tissue to be repaired as closely as possible, i.e. not only in terms of chemical composition but also taking into account the size, organization and, ultimately, in vivo conditions of formation.10 A typical illustration is provided by recent efforts to build up bone-like materials from collagen and hydroxyapatite.11 However, when coming to soft tissue, there is no available natural model in mammalians so that a ‘bio-inspired’ strategy based on the observation of other organisms may be fruitful.12,13 For instance, calcium carbonate phases are abundant in biominerals. However, despite their limited toxicity, their presence in soft tissue should be avoided to limit risks of pathological calcification.14 As an alternative, silica as found in diatoms, sponges, and other organisms was quite recently proposed as a promising partner to biopolymers for the formation of biomaterials.15
Of particular interest is the association of silica with type I collagen, as the major protein of mammalian tissues. Although the close association of these two components in nature is a matter of debate,16,17 they are independently the main constituents of several commercially-available biomaterials,18,19 raising great hopes for fruitful combinations. After more than 10 years of research in this area, a large variety of strategies have been successfully applied to associate collagen and silica, despite the intrinsic complexity of the biopolymer self-assembly process, of the inorganic polymerization dynamics, and of the biomineral interface. At this time, in vitro data and, more importantly, in vivo results are being gathered in order to obtain a deeper insight into the biological response of silica–collagen materials. Through a rational overview of currently-available data, this review aims at enlightening the converging evidences of synergetic effects within these protein-oxide constructs that raise great hopes for their future applications for soft and hard tissue repair.
2. Physiological and medical aspects of silicon and type I collagen
2.1 Silicon in mammalians
Silicon is the most prevalent element on the Earth, and crystalline silica in the form of quartz is the most abundant mineral in the Earth's crust. The study of the silica content in animal tissues and the effects of siliceous substances upon biological systems gained the attention of the scientific community over half a century ago.20 Both beneficial and toxicological effects of silicon were described. Considering detrimental effects, initial interest was focused on silicosis caused by dust inhalation.21 In parallel, several in vitro and in vivo studies have highlighted the importance of silicon for biological systems and the proper growth of animals.22 These pioneering works contributed to recognizing silicon as an essential nutritional element.23,24 Indeed, an increase of nearly 50% in growth rates in chicks was observed upon feeding with a diet containing a silicon supplement.25,26 In contrast, silicon deficiency in rats resulted in depressed growth and skull deformities. In this case, the addition of silicon in the diet also produced a 25% to 34% increase in growth rates.27 These earliest findings demonstrated the necessity of having silica in the body for proper growth and development.28,29Indeed, silicon is of remarkable importance for the structure and function of various tissues. Skeletal and other abnormalities involving the formation of the cartilage matrix and connective tissue were found to be associated with silicon deficiency in chicks.30 Moreover, the frontal bones from Si-deficient chicks had a significantly reduced collagen content.31 Tibia from Si supplemented chicks also had a significantly higher percentage and total amount of hexosamine and a higher percentage of collagen than deficient chicks.31 In both cases, it was reported that the formation of cartilage or bone organic matrix appeared to be more severely affected than the mineralization process. In addition, reduced amounts of collagen were found in silicon deficient chicks, suggesting a close relationship between silicon and collagen.29 In this way, it was suggested that silicon is involved in the development of the architecture of the fibrous elements of connective tissues and contributes to its structural integrity and mechanical strength.32 This specific importance of silicon in bone formation was supported by imaging ion microscopy, showing the presence of Si in osteoids and bones.33 Most recent studies also support the fact that the formation of the organic matrix of bone is more affected by silicon deficiency than the mineralization process, with silicon as a key element at the organic–inorganic interface.34 Furthermore, another study indicates that Si-supplemented yearlings may have decreased bone resorption, which may account for greater net bone formation. In this case, carboxy-terminal pyridinoline cross-linked telopeptide region of type I collagen concentrations was lower in supplemented yearlings on day 45 when compared to control yearlings.35 In parallel, silicon deprivation was demonstrated to decrease collagen formation in wounds and bone, together with the activity of ornithine transaminase, a key enzyme in proline synthesis, in liver.36
The bioavailability of silicon after the administration of a diet supplemented with orthosilicic acid was also investigated in calves. The results demonstrated that a 4.9% increase in the Si content of the diet leads to an increase of serum Si concentration of 70% compared to control animals. Collagen concentration in dermis was also significantly higher and a positive correlation was found between the Si concentration in serum and the collagen concentration in cartilage.37 The effect of silicon supplement on preventing bone mass loss induced by ovariectomy in rats was shown through the measurements of axial and peripheral bones. The Si inhibitory effect on bone mass loss as well as its stimulatory effect on bone formation were demonstrated. Both actions, namely, inhibition of resorption and stimulation of formation, infer that Si may have a potential therapeutic application.38 Dietary silicon intake was also positively associated with bone mineral density in men and premenopausal women.39
The removal pathway of silicon from the body has been also studied. The majority of absorbed Si is excreted in urine although some is taken up into tissues.40 Similarly, orthosilicic acid is readily absorbed from the gastrointestinal tract of humans and readily excreted in urine.41 It was reported that the median serum silicon value decreased from young adults (9.5 μmol L−1) to men over 60 years (8.5 μmol L−1) and then further above 74 years (7.70 μmol L−1). Higher and more variable values were obtained for women (18–29 years: 10.00 μmol L−1; 30–44 years: 11.10 μmol L−1; 45–59 years: 9.23 μmol L−1; >74 years: 8.00 μmol L−1).42 Other studies provide slightly higher41,43 or lower values.44
A series of in vitro investigations have focused on the interaction between soluble silica and relevant mammalian cells. Orthosilicic acid stimulates collagen type 1 synthesis and osteoblastic differentiation in human osteoblast-like cells in vitro.45,46 Gene expression of alkaline phosphatase and osteocalcin was also increased significantly,47–49 as well as the release of transforming growth factor β1 (TGF-β1), a cytokine that stimulates collagen production.50 Altogether, these data support the well-known stimulating effect of silicic acid on osteoblast-like cells,51 as observed from Si-containing cements.52 Silicates also significantly increase the amount of bone formed and the amount of bone attached to implant surfaces.53 Indeed, several studies demonstrated that silicon enhanced osteoblast adhesion, proliferation, differentiation, and gene expression.54–56 In addition to a direct biological role of orthosilicic acid,57 it is important to point out that cell–biomaterial interactions are largely governed by the stiffness and chemical composition of the substrate, together with surface topography, that all can be influenced by silicification.58–60 Finally, one important study has underlined the importance of silicic acid polymerization degree for its interaction with living cells, demonstrating that polysilicic acid could damage primary lung fibroblasts.61 Thus, although silica is generally accepted as having low toxicity, the biocompatibility of silicon and silica in its different forms (i.e. nanoparticles, gels, films) as a “new” class of biomaterial should be revisited.62
2.2 In vivo fate of silica-based materials
Colloidal silica has long been considered a safe additive in foods and pharmaceutical formulations for oral delivery.63–65 More recently, silica-based materials for in vivo applications have been mainly developed in two forms: nanoparticles and bulk hydrogels or xerogels. Indeed, although silica is generally accepted as having low toxicity, its behavior at the nanoscale has raised significant concerns, triggering extensive investigation of the biocompatibility of silica nanoparticles (SiNPs). In vitro studies have enlightened the influence of particle size, porosity, and surface charge on their cyto- and genotoxicity, which was also clearly dependent on the cell type used.66 The ability of silica particles to penetrate mammalian cells and to be excreted, with possible intracellular dissolution, has been reported.67,68In vivo data are more scarce and show rather wide biodistribution.69 In a particular study, the tolerability of negatively and positively charged SiNPs in acute single dose (107–5 × 108 SiNPs per animal) and subchronic multiple dose (108 SiNPs per animal per week for 4 weeks) administration demonstrated that SiNPs did not change either plasma levels of renal and hepatic biomarkers or plasma cytokines. The SiNPs did not lead to infiltration of leucocytes into liver, spleen, kidney, lung, brain, heart, and thyroid.70 Collectively, these data provide reasonable evidence for a safe administration of SiNPs.71 However, it is worth mentioning that selected internalization routes may have a strong impact on SiNPs in vivo toxicity, indicating low effect of intranasal, subcutaneous and intravenous injection but significant mortality after intraperitoneal infusion.72In a different approach, sol–gel silica materials have attracted a great deal of interest as functional materials for biomedical applications.73–78In vitro and in vivo studies have been performed to investigate the biodegradability and toxicity of silica matrices. For instance, in vivo tests of solid bioglass implants in the soft tissues of rats and rabbits for time periods of up to eight weeks were performed and proved silica to be non-toxic and biocompatible.79 In addition, the absence of a marked inflammatory response by polymorphonuclear leukocytes after implantation of sol–gel glasses was reported.80 At the same time, histological analysis revealed the formation of a thin fibrous capsule containing a few inflammatory cells at the tissue–implant interface of sol–gel coated discs implanted subcutaneously in rabbits, for 4 and 12 weeks.81 In the case of bioactive glass granules (45S5, 300–355 μm) implanted in the paraspinal muscle of rabbits it was observed that silicon was urinary excreted at 2.4 mg day−1 rate and no elevated concentration of silicon was found at the implant site or in the other organs after 24 weeks.82 The pathway for silica biodegradation includes erosion of the surface with formation of plasma-soluble silicic acid and its subsequent harmless excretion in a soluble form through the urine.83
Recent developments in the biomedical applications of silica are mainly related to the modification of traditional sol–gel procedures to design materials with optimized properties, including porosity, degradability rate and bioactivity.78 Besides all inorganic materials, many efforts are currently devoted to the elaboration of biohybrid or bionanocomposite materials9,84 associating silica with biological systems, being functional biomolecules,85 structural biopolymers86–88 or living cells (Fig. 1).89–92
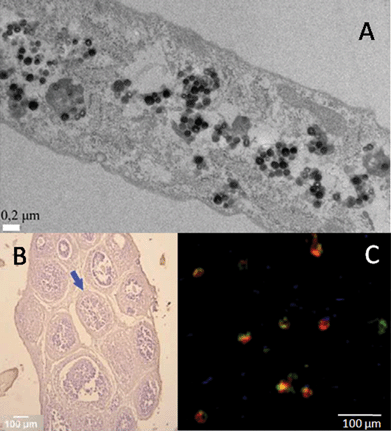 | ||
| Fig. 1 Mammalian cells’ interaction with silica. (A) TEM image of human dermal fibroblast cells after uptake of 200 nm silica nanoparticles (reprinted from ref. 67, Copyright (2012), with permission from Elsevier); (B) optical micrograph of sol–gel immobilized ovarian follicles, stained with hematoxylin (ref. 87–reproduced with permission from The Royal Society of Chemistry (RSC)); (C) fluorescence microscopy image of immobilized hybridoma cells. Living cells are red stained, while damaged cells appear green stained with the LIVE/DEAD kit (ref. 88 – reproduced with permission from The Royal Society of Chemistry (RSC)). | ||
2.3 A brief look at type I collagen
Collagens constitute nearly 30% of all proteins in mammals, representing the most abundant protein in animals. It is the major protein of connective tissue, tendons, ligaments, and cornea, and it forms the matrix of bones and teeth.93 Collagens are characterized by domains with repetitions of the tripeptide Gly-X-Y involved in the formation of trimeric collagen triple helices.94 Frequently, proline (Pro) and hydroxyproline (Hyp) follow each other, and about 10% of the molecule has the sequence Gly-Pro-Hyp. The most abundant group within the collagen types is the fibril-forming collagens (i.e. types I, II, III, V, and XI).95In vivo, type I collagen is synthesized intra-cellularly in the form of pro-collagen, i.e. triple helix structures with terminal propeptides that ensure solubility at neutral pH.96 After excretion, this propeptide is enzymatically cleaved so that the triple helices become insoluble and form fibrils. These fibrils organize to form structures of higher complexity as a function of the considered tissue, which have been identified as liquid crystal phases.97 Further enzymatic cross-linking, especially by lysyl oxidase, contributes to the stability of the material. This self-assembly process has been extensively studied in vitro. The influence of collagen concentration, pH, and ionic strength on fibrillogenesis and organization has been reported.98 As described below, these parameters are of primary importance in collagen-based biomaterials.
2.4 Type I collagen-based biomaterials
Type I collagen is the most abundant component of the extracellular matrix, even when compared with other collagen types. This makes type I collagen an ideal component in many situations for the development of scaffold materials for enhanced cell adhesion and proliferation, since these properties are highly desirable for wound healing and tissue regeneration.99,100Basically, there are two main strategies to obtain collagen-based scaffolds. On the one hand, the decellularization process was utilized to remove intrinsic cells while maintaining the extracellular matrix components as scaffolds. Examples of these scaffolds include small intestinal submucosa, acellular dermis, amniotic membrane tissue, cadaveric fascia, and the bladder acellular matrix graft.101 However, incomplete elimination of the previous cell population that can result in an inflammation or infection may be a problem.102
On the other hand, it is possible to prepare collagen solutions and trigger their self-assembly to obtain fibrils or fibers that interact to form three-dimensional biomimetic matrices. In particular, hydrogels were early identified as promising materials for tissue repair.103 The initial method for producing collagen gels involves the neutralization of acidic low concentrated collagen solutions to form a stable hydrogel. The main advantage of these hydrogels is that it is possible to obtain cellularized materials, avoiding the need for additional colonization. Moreover, the cells act as a source of macromolecules and cytokines to promote wound healing. However, the initial method results in poor gel stiffness and therefore strong and fast cell-mediated contraction.104 The cells within the contracted collagen gels exhibit phenotypic modifications such as the loss of proliferation potential and apoptosis, leading to the biological failure of the implant.105,106 The induction of apoptosis in fibroblasts seems to be specific to contractile collagen gels, as it is not observed in anchored or high-density collagen gels that resist this effect.107 Though, it is important to develop new materials that would resist contraction, and therefore prolong the viability of entrapped fibroblasts.
One way to overcome this drawback was recently reported by Hélary et al.108 Instead of using diluted collagen solutions, typically 0.66 mg mL−1, they managed to control the self-assembly conditions and in this way increase the collagen concentration up to 5 mg mL−1 (Fig. 2A and B). Higher collagen concentration resulted in lower cell-mediated contraction, increased cell proliferation, and superior in vivo integration.109,110 More concentrated hydrogels can also be obtained by evaporation of the initial collagen solution followed by neutralization under ammonia vapors.111 This approach allows the preparation of hydrogels with concentrations as high as 40 mg mL−1 but is no longer compatible with direct 3D cell immobilization (Fig. 2C). Dense hydrogels with concentrations higher than 100 mg mL−1 were also recently obtained by injection, dialysis and combination of the two methods (Fig. 2D).111,112 These approaches have the advantage of mimicking the structure of living tissues, from the nanoscale to the macroscale. Moreover, they allow the enhancement of the mechanical properties without any cross-linking procedures.113 An alternative possibility is to prepare dense hydrogels by compression.114 Finally, collagen sponges made by freeze-drying techniques are also widely studied.115
 | ||
| Fig. 2 Collagen-based materials. Photographs of collagen gels at (A) 0.66 mg mL−1, (B) 3 mg mL−1, (C) 40 mg mL−1 and (D) 250 mg mL−1. (A–C) From (ref. 107 – reproduced with permission from The Royal Society of Chemistry (RSC)); (D) from (ref. 108 – reproduced with permission from The Royal Society of Chemistry (RSC)). | ||
A large number of studies have focused on the association of collagen-based materials with hydroxyapatite for bone repair applications.116–118 In this context, it is worth underlining recent reports about the mineralization of highly-ordered bone-like dense collagen hydrogels, exhibiting high mechanical strength.119 This suggests that biomimetic approaches that take into account both the organic and mineral components have high potential in biomaterials science.11
3. Processing routes and structural and mechanical properties of silica–collagen materials
A wide range of mechanically-demanding applications cannot be satisfied by well-established monophasic materials. However, mixtures of these can show remarkable property combinations that have established the development of composites. Started in the field of technical materials, composite science took some time to find its way into biomedical materials research. The typical characteristics of composites include the combination of two or more heterogeneous phases that differ in composition and form, and retain their identities and properties while interacting at their interface. From a structural point of view, composites can be fibrous (fibres in a matrix), laminar (layers of phases), particulate (particles or flakes in a matrix) or hybrids (combinations of former structures). As a result the composite provides improved specific or synergistic characteristics not obtainable by any of the original phases alone.120 Most composites studied in biomaterials research are based on a continuously distributed organic matrix phase with an embedded structural phase (inorganic particles, whiskers, fibres, lamellae, meshes). Usually the matrix phase provides elasticity and acts as a binder for the inorganic material that enhances the strength of the composite. The homogeneity is optimal when the size of the basic units of the structural phase and that of the matrix phase are in the same range. When one dimension of at least one of the basic units is ≤100 nm the term nanocomposite is used.121 As a result the interface area and cohesion between the phases increase, resulting in enhanced mechanical properties.122 Alternatively the term “hybrid” can be used when two phases are blended on the molecular scale using molecules, macromolecules, particles, or fibers. Inorganic phases are commonly formed in situ by molecular precursors which often tend to form clusters or particles potentially templated by the organic phase. In particular the sol–gel technique was identified to be an appropriate method for the conjugation of inorganic materials with biological systems, owing to the compatibility of experimental conditions.123 It is well known that the sol–gel route has more in common with biological processing than with conventional materials processing.124From a chemical, structural, and technological point of view, the silica and collagen systems provide ideal conditions for the formation of advantageous composites. As described below, the various silica–collagen composites presented in the literature were prepared by a number of methods leading to materials with a remarkable range of characteristic structures. Again the structure is one of the most crucial parameters determining the mechanical properties of a composite. In general, high porosity entails low mechanical strength while high mechanical strength can only be obtained with a compact structure. However porosity is also of tremendous importance in biomaterial properties. Hence, the following classification of published results on silica–collagen biomaterials, ranging from soft materials to load-bearing xerogels, enlightens the relationship between materials structure and function that determine their most adequate fields of application (Fig. 3).
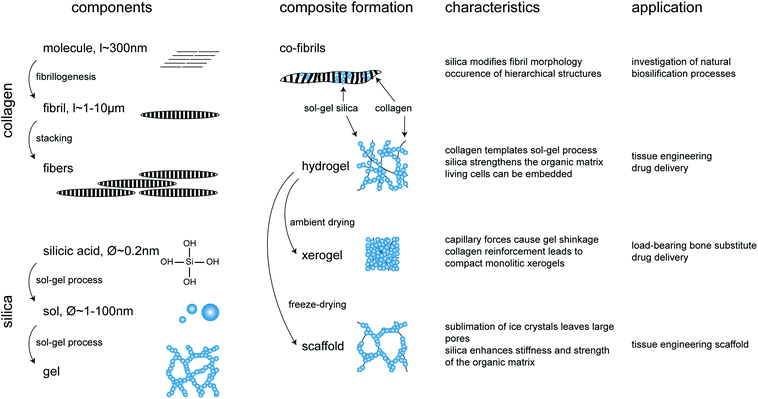 | ||
| Fig. 3 Overview of silica–collagen materials. Starting components, conditions of composite formation, main characteristics and potential applications. | ||
3.1 Solutions routes to soft materials
Associating silica species with collagen in diluted solutions constitutes optimal conditions to create a well-controlled bio-mineral interface at a molecular level, offering the possibility to observe silica-induced collagen fibrillogenesis modification and/or collagen-templated silica polymerization. In this context, Ono et al. pioneered the field in 1999 using collagen as a template for tetraethoxysilane (Si(OC2H5)4, TEOS) condensation.125 Two methods were described: either first growing type I collagen fibers by slow neutralization of a diluted acidic solution (3 mg mL−1, pH 3) with phosphate buffer (pH 6.86) over one month and soaking into TEOS or mixing the three components simultaneously. In both cases, silicification of collagen fibers occurred, as evidenced by the recovery of hollow silica fibers after calcination. This was attributed to the adsorption of the anionic silica oligomers onto the cationic collagen fibrils and further silica polymerization along the fibrils.As an alternative to alkoxides, Coradin et al.126 described the preparation of collagen–sodium silicate hybrids using a co-gelation process based on the exposure of an acidic mixture of both components to ammonia vapours. The influence of various sodium silicate and collagen concentrations on the morphology of the solids obtained suggested that the process is not only governed by the sodium silicate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :collagen ratio, but also by the initial concentrations of each component. The specific effect of sodium silicate on the kinetics of type I collagen fibril self-assembly was later investigated via turbidity profiles, silicic acid titration and transmission electron microscopy analysis.127 It was reported that sodium silicate at a concentration of 0.83 mM speeds up the collagen fibrillogenesis reaction, whereas at 10 mM, there was complete inhibition of this process. The effect of silica on collagen self-assembly was extended to other silica precursors including a silicon catecholate salt and silica nanoparticles.128 The turbidity profiles suggested that sodium silicate and silica nanoparticles (12 nm) containing solutions significantly modified the fibrillogenesis process at concentrations above 1 mM, whereas the silicon catecholate had a more limited effect. The reported data strongly suggest that the nature of the interaction between silica and collagen in solution was dependent on the form that the silicon is found, and thus on the silica precursor used. Another conclusion derived from this work was that silica precursors could be added only in limited amounts to collagen solutions whilst maintaining the self-assembly properties of the polymer.
:collagen ratio, but also by the initial concentrations of each component. The specific effect of sodium silicate on the kinetics of type I collagen fibril self-assembly was later investigated via turbidity profiles, silicic acid titration and transmission electron microscopy analysis.127 It was reported that sodium silicate at a concentration of 0.83 mM speeds up the collagen fibrillogenesis reaction, whereas at 10 mM, there was complete inhibition of this process. The effect of silica on collagen self-assembly was extended to other silica precursors including a silicon catecholate salt and silica nanoparticles.128 The turbidity profiles suggested that sodium silicate and silica nanoparticles (12 nm) containing solutions significantly modified the fibrillogenesis process at concentrations above 1 mM, whereas the silicon catecholate had a more limited effect. The reported data strongly suggest that the nature of the interaction between silica and collagen in solution was dependent on the form that the silicon is found, and thus on the silica precursor used. Another conclusion derived from this work was that silica precursors could be added only in limited amounts to collagen solutions whilst maintaining the self-assembly properties of the polymer.
This aspect was further evidenced in a series of studies devoted to the preparation of collagen–silica hydrogels. In a first step, a diluted (0.66 mg mL−1) type I collagen solution in acidic conditions was mixed with sodium silicate and then neutralized with NaOH. It was found that silicate concentration above 5 mM inhibited fibrillogenesis (Fig. 4),129 while, in the same conditions, nanoparticles 12 nm and 80 nm in diameter could be added up to 10 mM.130 Interestingly, when collagen concentration was increased to 3 mg mL−1, silicate addition up to 10 mM did not prevent fibrillogenesis.131 This can be explained considering that fibrillogenesis can occur only if the fraction of collagen molecules interacting with silicic acids is small compared to the whole collagen content, as already proposed for gelatin–silica systems.132 Further attempts to use 5 mg mL−1 solutions led to the development of an alternative preparation route where phosphate buffer was replaced by Tris–HCl to avoid the influence of phosphate ions on collagen fibrillogenesis.133 In these conditions, it was possible to increase silica concentration up to 25 mM for both silicate and colloidal silica precursors. From a structural point of view, the presence of silica particles did not modify either the collagen fibrous structure or its mechanical properties, i.e. storage modulus was ca. 100 Pa for a 5 mg mL−1 collagen concentration with or without added nanoparticles. Silicates were more effective in improving the stiffness of the collagen network (ca. 1 kPa). Eglin et al. also prepared Bioglass-containing collagen hydrogel composites at inorganic:collagen weight ratios of 10:1.134 The obtained white hydrogels were flexible and showed enough mechanical strength to be easily manipulated.
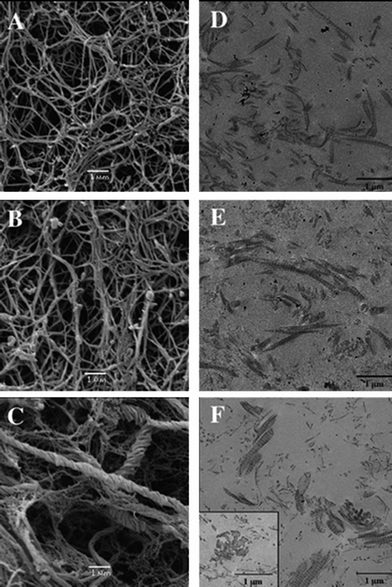 | ||
| Fig. 4 Silicified collagen hydrogels. SEM and TEM images of collagen hydrogels (0.66 mg mL−1) (A and D, respectively) and silicified collagen with 1 mM (B and E, respectively) and 5 mM (C and F, respectively) sodium silicate after incubation for 14 days (ref. 125 – reproduced with permission from The Royal Society of Chemistry (RSC)). | ||
Heinemann et al. described a strategy to mimic the natural process of biosilicification under ambient conditions.135 For this purpose, collagen from sponges and fibrillar calf skin collagen have been used as templates for silicification. Tetramethoxysilane (Si(OCH3)4, TMOS) was hydrolyzed and mixed with fibrillar collagen under neutral conditions. The presence of silica aggregates attached to single collagen fibrils was observed by scanning electronic microscopy (SEM) as well as by atomic force microscopy (AFM). Moreover, the measurement of the concentration of free primary amine groups of collagen and un-reacted silicic acid suggested the direct interaction between the negatively-charged silica species and the protonated positively-charged amine groups of the collagen, confirming what Ono et al. initially postulated.125 As a result, gelation time decreased noticeably with increasing concentration of collagen as well as TMOS. FTIR analysis of collagen–silica nanohybrids obtained by mixing solvent-denatured collagen with hydrolyzed TEOS was also reported.136 It was suggested that hydrogen bonds exist between the silanol groups of the silica and carboxyl, hydroxyl and amino groups of the collagen.
Two alternative strategies were very recently reported. In the first one, a functional silane, 3-glycidoxypropyl trimethoxysilane (GPTMS), was mixed with a diluted collagen solution in acidic conditions, then cast and dried.137 This strategy is similar to previous reports,5 where the epoxy group of GPTMS should form a covalent bond with the protein backbone while the silanol groups can be involved in the hydrogel cross-linking. In a completely different approach, it was attempted to grow collagen networks from the surface of silica nanoparticles.138 In this case, it was necessary to introduce sulfonate groups on the particle surface in order to favour collagen triple helices adsorption via electrostatic interactions in acidic conditions. Noticeably, the preservation of the typical patterned structure of the collagen fibrils was considered as a key indication of the compatibility of the silicification process with collagen biochemistry.
3.2 Collagen materials hardening through silicification
Altogether, the previous data indicate that silicic acid interacts strongly with collagen, limiting the possibility to associate these components at high concentration from solution mixtures. As a result, the mechanical properties of the silicified materials remain in the range of a few kPa as a maximum. Improving these values implies increasing the collagen concentration and/or mineralization rate.One possibility relies on the preparation of the collagen materials followed by the introduction of a silica source. Although collagen hydrogels or sponges exhibit high porosity that should allow easy diffusion of small molecules, it is important to ensure that the silica monomer does not react too strongly with the surface of collagen fibrils or fibers in order to obtain homogeneous silicification. A first approach, based on a vapor-phase technique, was described by Carturan et al. who applied the Biosil process to make a sol–gel silica coating on a collagen layer. According to this process a gaseous flux of silicon alkoxides was passed over the collagen membranes. Elemental analysis of the dried silicified collagen solid indicated a linear dependence of Si content with exposure time. The success of this method was attributed to the presence of hydroxyl groups in protein structures that favours hydrogen bonds with gaseous Si-alkoxides, leading to its surface deposition.139 In terms of mechanical properties a maximum increase of about 50% in the original elastic modulus was obtained by the deposition of a very thin sol–gel silica layer on the protein surface. A similar method based on the diffusion of a volatile alkoxide was successfully applied for the preparation of collagen–silica hybrids with long-range organization by the replication of the collagen chiral nematic organization present in concentrated solutions (Fig. 5).140 Unfortunately, no precise mechanical characterization of these materials is available. As an alternative to vapour phase impregnation, a solution route was recently reported that involves a stabilized form of silicic acid, choline–silicic acid complexes, to infiltrate collagen sponges.141 This stabilization allows the incorporation of 50 wt% of silica, resulting in an increase of the modulus of toughness from 0.1 kPa to more than 150 kPa.
 | ||
| Fig. 5 Silicified collagen liquid crystalline phases. Polarized light optical photographs of (a) the concentrated collagen solution and (b) the collagen–silica hybrid (crossed polarizers (P and A)) (bars = 5 μm); TEM micrographs of the silica-concentrated collagen solution hybrid (c) before (bar = 1 μm) and (d) after calcination (bar = 50 nm) (ref. 136 – reproduced with permission from The Royal Society of Chemistry (RSC)). | ||
As underlined above, the detrimental effect of the silica source mainly occurs at the stage of fibrillogenesis. Hence, an interesting strategy to associate collagen and silica at high concentration and high ratio is to use a pre-fibrillated collagen suspension rather than a triple helices solution as the “precursor” of the composite structure.142 It was shown that polymerizing silicic acid acted as a cross-linker and influenced the structure of the scaffolds since the morphology of separated collagen fibrils – similar to Desimone's hydrogels129 – was maintained also after freeze-drying. Addition of hydroxyapatite particles independently or in combination with silica was also studied. The mineralization did not disturb the open and interconnected porosity with pore sizes varying in the range of 100–200 μm despite increased apparent density. In the dry state, silica had no stiffening effect since the 0.4 MPa Young's modulus was similar to reference collagen scaffolds. Nevertheless, the presence of 12.5 wt% or 25 wt% silica resulted in structural stabilization, so that the scaffolds maintained their shape during chemical cross-linking and PBS immersion. In the wet state, silica-containing collagen scaffolds exhibited highly elastic behaviour during cyclic compression. The effects of silica and hydroxyapatite on the scaffolds’ mechanical properties were demonstrated to be partially comparable and could be combined. The influence of calcium and silica on the tensile strength and the breaking elongation of collagen films formed from mixtures of each precursor was also analyzed.143 The addition of sodium silicate resulted in an increase of both mechanical parameters. Furthermore it was confirmed that the strength of films that contain silica is higher than that of films that contain equivalent amounts of calcium.
3.3 Load-bearing compact xerogels
The need of innovative biomaterials for bone substitution applications stimulated the development of load-bearing silica–collagen biomaterials with high mechanical strength. In a series of studies, Heinemann et al. demonstrated that hydrogels obtained from highly concentrated mixtures of silicic acid and fibrillar collagen suspensions can be considered as intermediates that can be dried in ambient conditions or high relative humidity to obtain monolithic composite xerogels. Indeed, the shrinkage process is determined by the balance of capillary forces and gel strength,144 the first one depending on the nature of the hydrogel liquid phase that can be substituted during the process.145 The hierarchical organization of silica and marine or bovine collagen in the resulting xerogels turned out to be remarkably similar to those found in the siliceous basal spicules of marine glass sponges,135 underlining the biomimetic character of the preparation procedure.Pure silica xerogels exhibited a brittle amorphous structure that was characterized by a close arrangement of single particles.146 Using collagen as a template and incorporating it into disc-like samples allowed the investigation of the influence of the organic phase on the structure of silica xerogels.146 It was shown that denatured collagen (gelatin) did not yield sufficient composite characteristics because the drying stress exceeded the gel strength (Fig. 6A). In contrast, collagen fibers, composed of bundles of single fibrils, induced large pores and therefore hindered the formation of a dense gel network. Silica templating by using separate collagen fibrils turned out to yield optimal xerogel composite characteristics. In all cases the matrix phase was silica while up to 40 wt% fibrillar collagen was discontinuously and randomly dispersed. The fibrils act as fiber-like reinforcement and have to be pulled out from the silica matrix during gel fracturing, which in turn influences the mechanical properties (Fig. 6B and C). Increasing collagen content in the silica xerogels resulted in decreased Young's modulus whereas compressive strength and strain at fracture clearly increased up to about 200 MPa and 11% respectively.88 The maximum strength and strain values were obtained for 20–30% collagen, close to the organic–inorganic ratio of native bone. Additional phases, i.e. hydroxyapatite particles, reduce the mechanical properties since their chemical interaction with silica is weak. The mechanical properties of the silica–collagen–calcium phosphate xerogel system could be adjusted by the ratio of components and could range between that of human cancellous and cortical bone.
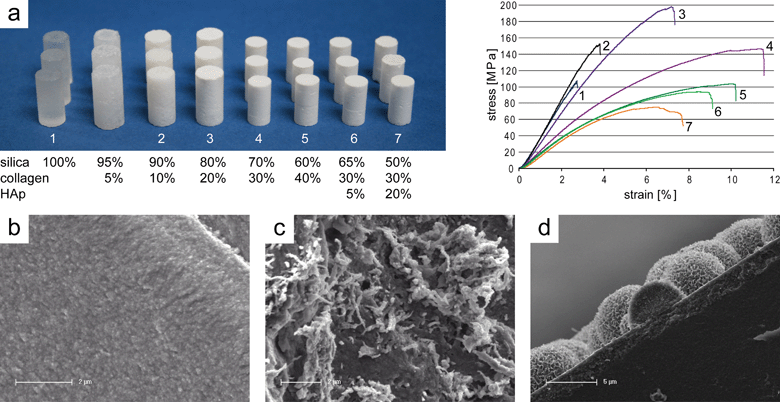 | ||
| Fig. 6 Silica–collagen xerogels. (a) Photograph of machined xerogel samples (diameter about 5–6 mm) with varying composition and corresponding stress–strain curves recorded during compression tests (reprinted from ref. 85, Copyright (2011), with permission from Elsevier); SEM images of (b) pure silica xerogel and (c) composite xerogel with 70% silica and 30% bovine collagen (reprinted from ref. 142, Copyright (2009), with permission from Elsevier); (d) SEM imaging cross-section of apatite deposited on the surface of a bioactive silica–collagen xerogel during immersion in SBF. | ||
4. In vitro cellular studies
The immobilization of mammalian cells in suitable matrices that can preserve their viability and capability to produce therapeutic molecules has gained attention in recent years for the development of bioartificial organs.147–151 In addition, the development of hybrids and nanocomposites has been recognized as a promising strategy to fulfil the complex requirements of scaffolds for cell-based tissue engineering applications.In the first case, it was demonstrated that the SiO2 layer deposited on collagen membranes from gaseous alkoxide precursors behaved as an efficient barrier to the diffusion of high molecular weight protein macromolecules, contributing to overcome the lack of immunoisolation inherent to porous collagen hydrogels required to design bioartificial organs. In parallel the production of bilirubin monoconjugate, ammonia removal, and urea and diazepam metabolites production were similar in controls and entrapped hepatocytes.152,153
More recently, the possibility to prepare silica–collagen hybrid hydrogels in conditions compatible with the encapsulation of human dermal fibroblasts was reported.129 After encapsulation, the number of metabolically active fibroblasts was larger in hybrid gels than in pure collagen gels. The highest survival rate was obtained in the presence of 1 mM sodium silicate and the lower one was for hybrid materials with 5 mM sodium silicate concentration. This was attributed to the fact that the structure of the hybrids was not modified by low amounts of sodium silicate, while with 5 mM sodium silicate, rope-like twisted bundles of collagen fibrils whose average diameter was ca. 400 nm were observed (Fig. 4). At this concentration, the remodeling activity of the cells, as monitored by the gelatin hydrolysis activity of the MMP-2 enzyme, was higher. This situation was attributed to the large silica–collagen bundles which may not favor cellular adhesion. In addition, an important release of silicic acid from the hybrid gels was measured over the first week post-encapsulation followed by a slower dissolution until day 21, in agreement with the high solubility of silica in biological media.154–156 Fibroblasts were also immobilized in silica–collagen bionanocomposite hydrogels obtained by addition of silica nanoparticles to a protein suspension followed by neutralization.130 When compared to collagen hydrogels or silica–collagen hybrids, these bionanocomposite materials showed lower surface contraction and higher viability of entrapped cells.131 A low level of gelatinase MMP-2 enzyme expression was also found in the nanocomposites. The Si release was significantly lower than in hybrids. The effect of various silica nanoparticles on nanocomposite formation and cell behavior was also reported.133 The observed variations between silicified collagen materials were considered in terms of mechanical properties and chemistry/topology of the internal pores that are of considerable importance in 3D environments.157–160
Several studies demonstrated sol–gel glasses to be highly biocompatible towards cells in the bone remodelling process, mainly osteoblasts and its precursors.161 The positive results were attributed to silica and its degradation products.162 Moreover, Si incorporation was observed to occur preferentially in the collagenous phase.163,164 In this context, it was reported that silica–collagen composites exhibit in vitro osteoinductive properties when exposed to simulated body fluid solution (SBF) (Fig. 6D). They allow the formation of apatite whereas this is not possible with their components alone.134 In parallel, silica xerogels immersed in SBF were observed to be highly bioactive and that apatite formation ability was reduced with increasing collagen percentage up to 30 wt% in the substrate, which can be a result of the decreased number of available hydroxyl groups due to interaction with collagen.146 The bioactivity increased again when calcium phosphates were embedded as a third component. Andrade et al. also reported that bioactive glass-coated collagen fibers exhibit in vitro bioactivity, improving the calcium and phosphate precipitation on the collagen surface when immersed in a simulated body fluid. Moreover, rat primary osteoblasts cultured on this sample were able to produce collagen and expressed higher levels of alkaline phosphatase.165
In a similar approach Wang et al. reported that the rate of hydroxyapatite formation could be significantly improved by the addition of hyaluronic acid and phosphatidylserine to bioglass–collagen composites. Moreover, MC3T3-E1 cells attached and spread on the surface of these scaffolds.166 Seeded human mesenchymal stem cells presented significantly higher levels of alkaline phosphatase activity and osteocalcin, osteopontin and alkaline phosphatase gene expression.167 It is worth mentioning that collagen, hyaluronic acid and phosphatidylserine are components of the extracellular matrix and are involved in the specific adhesion and proliferation of cells. Another composite material, prepared from calcium phosphate/SiO2 granules (BONITmatrix®) linked to a dense collagen mesh, was evaluated for compatibility with endothelial cells. This study demonstrated the importance of serum protein adsorption on the material surface to favour their adhesion and proliferation.168
In terms of a tissue engineering approach, Heinemann et al. studied the capability of human bone marrow stromal cells (hBMSC) to adhere, differentiate and invade into macroporous scaffolds based on fibrillar collagen mineralized by 25 wt% silica and/or hydroxyapatite.142 They demonstrated that silica slightly retards the osteogenic differentiation of hBMSC without being detrimental to cytocompatibility. The open porosity allowed cells to migrate throughout the scaffolds while maintaining their viability, both confirmed by MTT staining and confocal laser scanning (cLSM) microscopy (Fig. 7A–C).
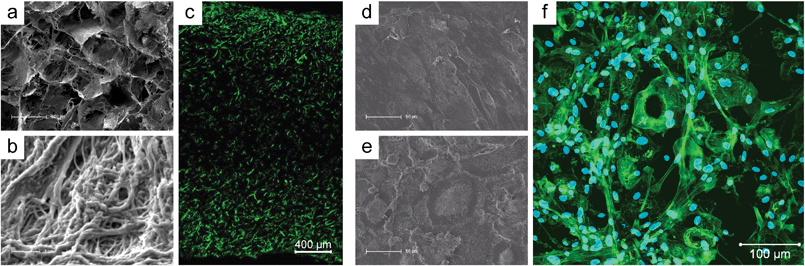 | ||
| Fig. 7 In vitro evaluation of silica–collagen composite scaffolds and composite xerogels. (a, b) SEM cross-sectional view of collagen-based scaffolds with 25 wt% silica and (c) cLSM images of the same scaffold after 28 days of cultivating hMSC-derived osteoblasts on this biomaterial. Cytoskeletal actin was stained with AlexaFluor 488-Phalloidin and is visualized green (reprinted with permission from ref. 138, Copyright (2011), American Chemical Society; SEM images after 28 days of osteoblast/osteoclast co-cultivation on (d) low bioactive silica–collagen xerogel and (e) highly bioactive silica–collagen xerogel with integrated calcium phosphate phases (reprinted from ref. 166, Copyright (2013), with permission from Elsevier); (f) 3D reconstruction from cLSM image stacks after 28 days of osteoblast/osteoclast co-cultivation on silica–collagen–calcium phosphate composite xerogel. The actin skeletons and cell nuclei are visualized in green and blue, respectively. | ||
The combination of silica with fibrillar bovine collagen also enabled the preparation of monolithic xerogel discs suitable for long-term (up to 42 days) cell culture experiments, allowing the evaluation of the response of hBMSC-derived osteoblasts145 and human monocyte-derived osteoclasts not only in mono-culture146 but also using these cells as a novel human co-culture model.169,170 It was demonstrated that the silica–collagen xerogels support adhesion, proliferation, and osteogenic differentiation of human mesenchymal stem cells, as confirmed by cLSM immunofluorescence, DNA measurement, and biochemical analysis of alkaline phosphatase activity (Fig. 7D–F).145 Furthermore, cultivation on the xerogels allowed migration of monocytes to form multinuclear osteoclasts exhibiting all characteristic morphological features.146 Interestingly, these cells were tested positive for tartrate-resistant acid phosphatise (TRAP) type 5b, a lysosomal enzyme only produced by active osteoclasts.
Osteoblast/osteoclast co-culture experiments indicated that biphasic silica–collagen xerogels provide the best conditions for bone formation.170 These samples supported adhesion, differentiation, and proliferation of osteoblasts and allowed osteoclastogenesis as well as cell-mediated degradation directly on the xerogels. In addition, calcium phosphate-containing triphasic xerogels entailed enhanced bioactivity and calcium-deficient cell culture conditions which favoured osteoclasts over osteoblasts. The complex cell–xerogel interaction was monitored via a large number of biochemical markers and gene expression profiles. As a conclusion calcium phosphate was considered as a dose-dependent agent embedded in the silica–collagen xerogel influencing the osteoblast/osteoclast ratio. This is an important point concerning the application of these materials in environments afflicted with systemic diseases such as osteoporosis.
5. In vivo studies
In vivo studies also reported the advantages of using hybrid and nanocomposite silica–collagen materials as biological dressings. The in vivo integration of concentrated (3 mg mL−1 collagen content) silicified hydrogels during the vascular inflammatory phase was recently investigated and confirmed significant fibroblast colonization and endothelial cells organization in open tubular structures (indicative of vascularization) while the infiltration of macrophages was very little (Fig. 8).131 However, for diluted silicified-collagen materials (0.66 mg mL−1 collagen content), more macrophages were found at the implantation site. Such a difference was attributed to the fact that these materials are more easily hydrolyzed by metalloproteases, such as MMP2, leading to proteolytic fragments that affect multiple functions and properties of inflammatory and immune cell.171,172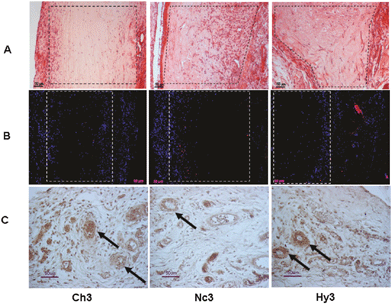 | ||
| Fig. 8 In vivo fate of silicified concentrated collagen hydrogels. Histological and immunohistological studies 8 days post-surgery. Pure collagen (3 mg mL−1, Ch3), nanocomposite (Nc3), and hybrid (Hy3) scaffolds were implanted subcutaneously. Sections were stained with hematoxylin–eosin (line A), detection of macrophages (CD68 marker) (line B), and endothelial cells (RECA-1 marker) (line C). Reprinted with permission from ref. 127. Copyright (2011), American Chemical Society. | ||
In the field of bone repair, xerogels composed of 70% silica and 30% bovine type I collagen were tested in 12-week-old rats for full circumferential femur defects.173 The ductile character of the material enabled the surgeon to individually tailor the final shape of the xerogels to the defect size directly during surgical procedure. Histological, immunohistochemical, and nano-computer tomography (CT) analyses confirmed excellent biocompatibility and indicated newly formed blood vessels surrounding the bone substitution material. Xerogel degradation occurred by the direct transport of silica particles as well as collagen bundles from the implant surface to the surrounding tissue.
In vivo bone-regenerative potential of a bioactive glass–collagen–hyaluronic acid–phosphatidylserine (BG–COL–HYA–PS) composite scaffold with mesenchymal stem cells (MSCs) was investigated in a rat bone defect model (femur). HrGFP-labeled MSCs were cultured for 2 weeks on the BG–COL–HYA-PS scaffold before implantation into the defect and a cell-free scaffold and an untreated defect were used as controls. The regeneration process was evaluated by histology, X-ray analysis and mechanical rigidity experiments. The results revealed that BG–COL–HYA–PS scaffold exhibited a low inflammatory response and foreign body response within 3 weeks. After 6 weeks, the inflammatory response disappeared following scaffolds resorption and formation of new bone. Compared to the controls, the introduction of MSCs into the porous scaffold dramatically enhanced the efficiency of new bone formation, the biomechanical properties of the femur and thus the healing of the bone defect. In addition, the transplanted MSCs could survive for at least 3 weeks.174,175
The in vivo controlled drug delivery potential of silica–collagen materials was also recently evaluated. Three-dimensional collagen scaffolds infiltrated with silicon complexes were found osteoconductive and up-regulated the expressions of osteogenesis- and angiogenesis-related genes more significantly than non-silicified collagen scaffolds. In addition, these scaffolds reversibly bind SDF-1, which plays a pivotal role in mobilization and homing of stem cells to injured tissues, and can therefore be used for the sustained release of this chemokine. When implanted subcutaneously in a mouse model, SDF-1-loaded silicified collagen scaffolds stimulate the formation of ectopic bone and blood capillaries within the scaffold.176
6. Conclusions and perspectives
A wide variety of silica species could be used for the silicification of soluble collagen, fibrils, fibers, films, gels or scaffolds in order to obtain materials with different compositions and structures that would meet the requirements of various biomedical applications. Since silica and collagen turned out to be ideal partners for composite formation, advantageous synergistic effects in material properties can be obtained. In all cases, the combination conditions of the organic and inorganic materials need to be carefully analyzed, since the properties of the resulting materials depend on the nature and concentration of each one as well as the ratio in which they are combined. The ability to control the structure of these materials from the nanoscale to the macroscale and the possibility to generate appropriate environments for living cells is one of the most important factors to be considered in future. The driving forces for these developments consider that both phases, individually or within a composite, are highly suitable to implement biologically-relevant functions in the biomaterial, especially degradability, bioactivity and local delivery of drugs. From this point of view, silica–collagen based biomaterials can be considered as a modular platform which can be tested, modified, and adjusted for a remarkably wide range of desirable applications. Especially we believe that the combination of collagen networks with silica and hydroxyapatite is particularly promising for bone repair materials whereas drug-loaded silica–collagen hydrogels may prove particularly fruitful as biological dressings.Acknowledgements
S.H. gratefully acknowledges the Deutsche Forschungsgemeinschaft Grant TRR79-TP-M03 for financial support. M.F.D acknowledges the support of grants from the University of Buenos Aires UBACYT20020110100081 and from Agencia Nacional de Investigaciones Científicas y Técnicas BID 1728/OC-AR PICT 1783. M.F.D. and T.C. thank the Argentina–France MINCYT-ECOS-Sud (project A12S01) and CONICET-CNRS programs for financial support. They also wish to express their profound gratitude to M.M. Giraud-Guille and C. Hélary, from LCMCP, for sharing their expertise in the field of collagen bio-chemistry over the last few years.References
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- J. Kopecek, Biomaterials, 2007, 28, 5185–5192 CrossRef CAS.
- J. Kopecek and J. Yang, Polym. Int., 2007, 56, 1078–1098 CrossRef CAS.
- M. F. Butler, Y. F. Ng and P. D. A. Pudney, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3941–3953 CrossRef CAS.
- A. Fatimi, J. Francois Tassin, S. Quillard, M. A. V. Axelos and P. Weiss, Biomaterials, 2008, 29, 533–543 CrossRef CAS.
- J. J. Sperinde and L. G. Griffith, Macromolecules, 2000, 33, 5476–5480 CrossRef CAS.
- J. J. Sperinde and L. G. Griffith, Macromolecules, 1997, 30, 5255–5264 CrossRef CAS.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS.
- C. Aimé and T. Coradin, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 669–680 CrossRef.
- P. X. Ma, Adv. Drug Delivery Rev., 2008, 60, 184–198 CrossRef CAS.
- Y. Wang, T. Azais, M. Robin, A. Vallée, C. Catania, P. Legriel, G. Pehau-Arnaudet, F. Babonneau, M. M. Giraud-Guille and N. Nassif, Nat. Mater., 2012, 11, 724–733 CrossRef CAS.
- M. A. Meyers, P. Y. Chen, M. I. Lopez, Y. Seki and A. Y. M. Lin, J. Mech. Behav. Biomed. Mater., 2011, 4, 626–657 CrossRef.
- M. A. Meyers, P. Y. Chen, A. Y. M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53, 1–206 CrossRef CAS.
- C. M. Giachelli, Am. J. Pathol., 1999, 154, 671–675 CrossRef CAS.
- T. Coradin, J. Allouche, M. Boissière and J. Livage, Curr. Nanosci., 2006, 2, 219–230 CrossRef CAS.
- H. C. Schroder, X. Wang, W. Tremel, H. Ushijima and W. E. G. Müller, Nat. Prod. Rep., 2008, 25, 455–474 RSC.
- H. Ehrlich, R. Deutzmann, E. Brunner, E. Cappellini, H. Koon, C. Solazzo, Y. Yang, D. Ashford, J. Thomas-Oates, M. Lubeck, C. Baessmann, T. Langrock, R. Hoffmann, G. Worheide, J. Reitner, P. Simon, M. Tsurkan, A. V. Ereskovsky, D. Kurek, V. V. Bazhenov, S. Hunoldt, M. Mertig, D. V. Vyalikh, S. L. Molodtsov, K. Kummer, H. Worch, V. Smetacek and M. J. Collins, Nat. Chem., 2010, 2, 1084–1088 CrossRef CAS.
- R. Parenteau-Bareil, R. Gauvin, S. Cliche, C. Gariépy, L. Germain and F. Berthod, Acta Biomater., 2011, 7, 3757–3765 CrossRef CAS.
- A. Y. Au, R. Y. Au, J. L. Demko, R. M. McLaughlin, B. E. Eves and C. G. Frondoza, J. Biomed. Mater. Res. A, 2010, 94, 380–388 Search PubMed.
- E. M. Carlisle, Nutr. Rev., 1975, 33, 257–261 CrossRef CAS.
- G. Erdogdu and V. Hasirci, Environ. Res., 1998, 78, 38–42 CrossRef CAS.
- E. M. Carlisle, Ciba Found. Symp., 1986, 121, 123–139 CAS.
- E. M. Carlisle, Nutr. Rev., 1982, 40, 193–198 CrossRef CAS.
- E. M. Carlisle, Sci. Total Environ., 1988, 73, 95–106 CrossRef CAS.
- E. M. Carlisle, Science, 1970, 167, 279–280 CAS.
- E. M. Carlisle, Science, 1972, 178, 619–621 CAS.
- K. Schwarz and D. B. Milne, Nature, 1972, 239, 333–334 CrossRef CAS.
- K. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 1608–1612 CrossRef CAS.
- E. M. Carlisle, Fed. Proc., 1974, 33, 1758–1766 CAS.
- E. M. Carlisle, J. Nutr., 1976, 106, 478–484 CAS.
- E. M. Carlisle, J. Nutr., 1980, 110, 352–359 CAS.
- K. Schwarz and S. C. Chen, Fed. Proc., 1974, 33 Search PubMed.
- W. J. Landis, D. D. Lee and J. T. Brenna, Calcif. Tissue Int., 1986, 38, 52–59 CrossRef CAS.
- N. B. Matsko, N. Žnidaršič, I. Letofsky-Papst, M. Dittrich, W. Grogger, J. Štrus and F. Hofer, J. Struct. Biol., 2011, 174, 180–186 CrossRef CAS.
- K. J. Lang, B. D. Nielsen, K. L. Waite, G. M. Hill and M. W. Orth, J. Anim. Sci., 2001, 79, 2627–2633 CAS.
- C. D. Seaborn and F. H. Nielsen, Biol. Trace Elem. Res., 2002, 89, 251–261 CrossRef CAS.
- M. R. Calomme and D. A. Vanden Berghe, Biol. Trace Elem. Res., 1997, 56, 153–165 CrossRef CAS.
- H. Rico, J. L. Gallego-Lago and E. R. Hernandez, Calcif. Tissue Int., 2000, 66, 53–55 CrossRef CAS.
- R. Jugdaohsingh, K. L. Tucker, N. Qiao, L. A. Cupples, D. P. Kiel and J. J. Powell, J. Bone Miner. Res., 2004, 19, 297–307 CrossRef CAS.
- S. Sripanyakorn, R. Jugdaohsingh, R. P. H. Thompson and J. J. Powell, Nutr. Bull., 2005, 30, 222–230 CrossRef.
- D. M. Reffitt, R. Jugdaohsingh, R. P. H. Thompson and J. J. Powell, J. Inorg. Biochem., 1999, 76, 141–147 CrossRef CAS.
- E. Bissé, T. Epting, A. Beil, G. Lindinger, H. Lang and H. Wieland, Anal. Biochem., 2005, 337, 130–135 CrossRef.
- R. Jugdaohsingh, S. H. C. Anderson, K. L. Tucker, H. Elliott, D. P. Kiel, R. P. H. Thompson and J. J. Powell, Am. J. Clin. Nutr., 2002, 75, 887–893 CAS.
- H. Robberecht, R. Van Cauwenbergh, V. Van Vlaslaer and N. Hermans, Sci. Total Environ., 2009, 407, 4777–4782 CrossRef CAS.
- L. J. Cardenas, A. Takeuchi, K. Tsuru, S. Matsuya and K. Ishikawa, IFMBE Proc., 2010, 27, 207–210 CrossRef.
- Q. Zhao, J. Qian, H. Zhou, Y. Yuan, Y. Mao and C. Liu, Biomed. Mater., 2010, 5 CAS.
- D. M. Reffitt, N. Ogston, R. Jugdaohsingh, H. F. J. Cheung, B. A. J. Evans, R. P. H. Thompson, J. J. Powell and G. N. Hampson, Bone, 2003, 32, 127–135 CrossRef CAS.
- S. Ni, J. Chang, L. Chou and W. Zhai, J. Biomed. Mater. Res. B Appl. Biomater., 2007, 80, 174–183 CrossRef.
- S. Zou, D. Ireland, R. A. Brooks, N. Rushton and S. Best, J. Biomed. Mater. Res. B Appl. Biomater., 2009, 90, 123–130 CAS.
- P. E. Keeting, M. J. Oursler, K. E. Wiegand, S. K. Bonde, T. C. Spelsberg and B. L. Riggs, J. Bone Miner. Res., 1992, 7, 1281–1289 CrossRef CAS.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757–2774 CrossRef CAS.
- G. Mestres, C. Le Van and M. P. Ginebra, Acta Biomater., 2012, 8, 1169–1179 CrossRef CAS.
- M. J. Coathup, S. Samizadeh, Y. S. Fang, T. Buckland, K. A. Hing and G. W. Blunn, J. Bone Joint Surg.–Ser. A, 2011, 93, 2219–2226 CrossRef.
- I. S. Byun, S. K. Sarkar, M. Anirban Jyoti, Y. K. Min, H. S. Seo, B. T. Lee and H. Y. Song, J. Mater. Sci.: Mater. Med., 2010, 21, 1937–1947 CrossRef CAS.
- E. Landi, J. Uggeri, S. Sprio, A. Tampieri and S. Guizzardi, J. Biomed. Mater. Res. A, 2010, 94, 59–70 CrossRef.
- M. Wiens, X. Wang, U. Schlomacher, I. Lieberwirth, G. Glasser, H. Ushijima, H. C. Schröder and W. E. G. Müller, Calcif. Tissue Int., 2010, 87, 513–524 CrossRef CAS.
- P. Han, C. Wu and Y. Xiao, Biomater. Sci., 2013, 1, 379–392 RSC.
- J. D. Bass, E. Belamie, D. Grosso, C. Boissiere, T. Coradin and C. Sanchez, J. Biomed. Mater. Res. A, 2010, 93, 96–106 Search PubMed.
- F. Gentile, R. La Rocca, G. Marinaro, A. Nicastri, A. Toma, F. Paonessa, G. Cojoc, C. Liberale, F. Benfenati, E. di Fabrizio and P. Decuzzi, ACS Appl. Mater. Interfaces, 2012, 4, 2903–2911 CAS.
- M. G. Bellino, S. Golbert, M. C. De Marzi, G. J. A. A. Soler-Illia and M. F. Desimone, Biomater. Sci., 2013, 1, 186–189 RSC.
- D. S. Linthicum, Tissue Cell, 2001, 33, 514–523 CrossRef CAS.
- A. M. Mebert, D. E. Camporotondi, M. L. Foglia, G. S. Alvarez, P. L. S. Orihuela, L. E. Diaz and M. F. Desimone, J. Biomater. Tissue Eng., 2013, 3, 108–121 Search PubMed.
- M. Hrast and A. Obreza, Vloga silicijevih spojin v živih organizmih, 2010, 61, 37–41 Search PubMed.
- M. Manzano, M. Colilla and M. Vallet-Reg, Expert Opin. Drug Delivery, 2009, 6, 1383–1400 CrossRef CAS.
- M. Vallet-Regí, F. Balas, M. Colilla and M. Manzano, Drug Metab. Lett., 2007, 1, 37–40 CrossRef.
- S. Quignard, S. Masse and T. Coradin, in Intracellular Delivery: Fundamentals and Applications, Fundamental Biomedical Technologies, ed. A. Prokop, Springer Science, 2011, pp. 333–361 Search PubMed.
- S. Quignard, G. Mosser, M. Boissière and T. Coradin, Biomaterials, 2012, 33, 4431–4442 CrossRef CAS.
- I. Stayton, J. Winiarz, K. Shannon and Y. Ma, Anal. Bioanal. Chem., 2009, 394, 1595–1608 CrossRef CAS.
- R. Kumar, I. Roy, T. Y. Ohulchanskky, L. A. Vathy, E. J. Bergey, M. Sajjad and P. N. Prasad, ACS Nano, 2010, 4, 699–708 CrossRef CAS.
- T. Tanaka, B. Godin, R. Bhavane, R. Nieves-Alicea, J. Gu, X. Liu, C. Chiappini, J. R. Fakhoury, S. Amra, A. Ewing, Q. Li, I. J. Fidler and M. Ferrari, Int. J. Pharm., 2010, 402, 190–197 CrossRef CAS.
- M. L. Foglia, G. S. Alvarez, P. N. Catalano, A. M. Mebert, L. E. Diaz, T. Coradin and M. F. Desimone, Recent Pat. Biotechnol., 2011, 5, 54–61 CrossRef CAS.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045–4055 CrossRef CAS.
- M. F. Desimone, G. S. Alvarez, M. L. Foglia and L. E. Diaz, Recent Pat. Biotechnol., 2009, 3, 55–60 CrossRef CAS.
- A. Leonard, P. Dandoy, E. Danloy, G. Leroux, C. F. Meunier, J. C. Rooke and B. L. Su, Chem. Soc. Rev., 2011, 40, 860–885 RSC.
- A. P. Chiriac, L. E. Nita, I. Neamtu and M. T. Nistor, Recent Pat. Mater. Sci., 2011, 4, 224–237 CrossRef CAS.
- D. Avnir, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- T. Coradin and J. Livage, Acc. Chem. Res., 2007, 40, 819–826 CrossRef CAS.
- M. Vallet-Regí, M. Colilla and B. González, Chem. Soc. Rev., 2011, 40, 596–607 RSC.
- J. Wilson, G. H. Pigott, F. J. Schoen and L. L. Hench, J. Biomed. Mater. Res., 1981, 15, 805–817 CrossRef CAS.
- G. Palumbo, L. Avigliano, G. Strukul, F. Pinna, D. Del Principe, I. D'Angelo, M. Annicchiarico-Petruzzelli, B. Locardi and N. Rosato, J. Mater. Sci.: Mater. Med., 1997, 8, 417–421 CrossRef CAS.
- M. Gerritsen, A. Kros, V. Sprakel, J. A. Lutterman, R. J. M. Nolte and J. A. Jansen, Biomaterials, 2000, 21, 71–78 CrossRef CAS.
- W. Lai, P. Ducheyne, J. Garino and C. M. Flaitz, Mater. Res. Soc. Symp. Proc., 2000, 599, 261–266 CrossRef CAS.
- W. Lai, J. Garino and P. Ducheyne, Biomaterials, 2002, 23, 213–217 CrossRef CAS.
- E. Ruiz-Hitzky, P. Aranda, M. Darder and M. Ogawa, Chem. Soc. Rev., 2011, 40, 801–828 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS.
- T. Coradin, M. Boissière and J. Livage, Curr. Med. Chem., 2006, 13, 99–108 CrossRef CAS.
- R. Esquembre, S. N. Pinto, J. A. Poveda, M. Prieto and C. R. Mateo, Soft Matter, 2012, 8, 408–417 RSC.
- S. Heinemann, T. Coradin, H. Worch, H. P. Wiesmann and T. Hanke, Compos. Sci. Technol., 2011, 71, 1873–1880 CrossRef CAS.
- M. Blondeau and T. Coradin, J. Mater. Chem., 2012, 22, 22335–22343 RSC.
- E. J. A. Pope, K. Braun and C. M. Peterson, J. Sol-Gel Sci. Technol., 1997, 8, 635–639 CAS.
- P. N. Catalano, N. S. Bourguignon, G. S. Alvarez, C. Libertun, L. E. Diaz, M. F. Desimone and V. Lux-Lantos, J. Mater. Chem., 2012, 22, 11681–11687 RSC.
- M. F. Desimone, M. C. De Marzi, G. S. Alvarez, I. Mathov, L. E. Diaz and E. L. Malchiodi, J. Mater. Chem., 2011, 21, 13865–13872 RSC.
- M. G. Patino, M. E. Neiders, S. Andreana, B. Noble and R. E. Cohen, Implant Dent., 2002, 11, 280–285 CrossRef.
- K. Gelse, E. Pöschl and T. Aigner, Adv. Drug Delivery Rev., 2003, 55, 1531–1546 CrossRef CAS.
- A. M. Ferreira, P. Gentile, V. Chiono and G. Ciardelli, Acta Biomater., 2012, 8, 3191–3200 CrossRef CAS.
- D. J. S. Hulmes, J. Struct. Biol., 2002, 137, 2–10 CrossRef CAS.
- M. M. Giraud Guille, G. Mosser, C. Helary and D. Eglin, Micron, 2005, 36, 602–608 CrossRef CAS.
- P. De Sa Peixoto, A. Deniset-Besseau, M. C. Schanne-Klein and G. Mosser, Soft Matter, 2011, 7, 11203–11210 RSC.
- R. A. Brown and J. B. Phillips, Int. Rev. Cytol., 2007, 262, 75–150 CrossRef CAS.
- I. Jones, L. Currie and R. Martin, Br. J. Plast. Surg., 2002, 55, 185–193 CrossRef CAS.
- J. Hodde, Tissue Eng., 2002, 8, 295–308 CrossRef CAS.
- J. Hodde and M. Hiles, Acta Chir. Belg., 2007, 107, 641–647 CAS.
- E. Bell, H. P. Ehrlich, D. J. Buttle and T. Nakatshji, Science, 1981, 211, 1052–1054 CAS.
- E. Bell, B. Ivarsson and C. Merrill, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1274–1278 CrossRef CAS.
- J. Fluck, C. Querfeld, A. Cremer, S. Niland, T. Krieg and S. Sollberg, J. Invest. Dermatol., 1998, 110, 153–157 CrossRef CAS.
- E. Hadjipanayi, V. Mudera and R. A. Brown, J. Tissue Eng. Regener. Med., 2009, 3, 77–84 CrossRef CAS.
- C. C. Berry, J. C. Shelton and D. A. Lee, J. Tissue Eng. Regener. Med., 2009, 3, 43–53 CrossRef CAS.
- C. Helary, I. Bataille, A. Abed, C. Illoul, A. Anglo, L. Louedec, D. Letourneur, A. Meddahi-Pellé and M. M. Giraud-Guille, Biomaterials, 2010, 31, 481–490 CrossRef CAS.
- C. Helary, A. Abed, G. Mosser, L. Louedec, A. Meddahi-Pelle and M. M. Giraud-Guille, J. Tissue Eng. Regener. Med., 2011, 5, 248–252 CrossRef CAS.
- C. Helary, M. Zarka and M. M. Giraud-Guille, J. Tissue Eng. Regener. Med., 2012, 6, 225–237 CrossRef CAS.
- M. M. Giraud Guille, C. Helary, S. Vigier and N. Nassif, Soft Matter, 2010, 6, 4963–4967 RSC.
- Y. Wang, J. Silvent, M. Robin, F. Babonneau, A. Meddahi-Pelle, N. Nassif and M. M. Giraud Guille, Soft Matter, 2011, 7, 9659–9664 RSC.
- S. Vigier, C. Catania, B. Baroukh, J. L. Saffar, M. M. Giraud-Guille and M. L. Colombier, Tissue Eng., Part A, 2011, 17, 889–898 CrossRef CAS.
- E. A. Abou Neel, U. Cheema, J. C. Knowles, R. A. Brown and S. N. Nazhat, Soft Matter, 2006, 2, 986–992 RSC.
- H. Schoof, J. Apel, I. Heschel and G. Rau, J. Biomed. Mater. Res., 2001, 58, 352–357 CrossRef CAS.
- P. N. Sisco, C. G. Wilson, E. Mironova, S. C. Baxter, C. J. Murphy and E. C. Goldsmith, Nano Lett., 2008, 8, 3409–3412 CrossRef CAS.
- Y. Cao, Y. Zhou, Y. Shan, H. Ju and X. Xue, Adv. Mater., 2006, 18, 1838–1841 CrossRef CAS.
- A. R. Boccaccini and J. J. Blaker, Expert Rev. Med. Devices, 2005, 2, 303–317 CrossRef CAS.
- N. Nassif, F. Gobeaux, J. Seto, E. Belamie, P. Davidson, P. Panine, G. Mosser, P. Fratzl and M. M. Giraud Guille, Chem. Mater., 2010, 22, 3307–3309 CrossRef CAS.
- G. Kickelbick, in Hybrid Materials. Synthesis, Characterization, and Applications, ed. G. Kickelbick, Wiley-VCH, Weinheim, 2007, pp. 1–48 Search PubMed.
- C. K. Chan, T. S. Kumar, S. Liao, R. Murugan, M. Ngiam and S. Ramakrishnan, Nanomedicine, 2006, 1, 177–188 CrossRef CAS.
- F. Watari, A. Yokoyama, M. Gelinsky and W. Pompe, in Interface Oral Health Science 2007, ed. M. Watanabe and O. Okuno, Springer Japan, Tokyo, 2008, pp. 139–147 Search PubMed.
- G. Carturan, R. Dal Toso, S. Boninsegna and R. Dal Monte, J. Mater. Chem., 2004, 14, 2087–2098 RSC.
- T. Coradin and J. Livage, Acc. Chem. Res., 2007, 40, 819–826 CrossRef CAS.
- Y. Ono, Y. Kanekiyo, K. Inoue, J. Hojo, M. Nango and S. Shinkai, Chem. Lett., 1999, 475–476 CrossRef CAS.
- T. Coradin, M. M. Giraud-Guille, C. Helary, J. Livage and C. Sanchez, Mater. Res. Soc. Symp. Proc., 2002, 726, 79–83 CAS.
- D. Eglin, T. Coradin, M. M. Giraud Guille, C. Helary and J. Livage, Bio-Med. Mater. Eng., 2005, 15, 43–50 CAS.
- D. Eglin, K. L. Shafran, J. Livage, T. Coradin and C. C. Perry, J. Mater. Chem., 2006, 16, 4220–4230 RSC.
- M. F. Desimone, C. Helary, G. Mosser, M.-M. Giraud-Guille, J. Livage and T. Coradin, J. Mater. Chem., 2010, 20, 666–668 RSC.
- M. F. Desimone, C. Helary, I. B. Rietveld, I. Bataille, G. Mosser, M. M. Giraud-Guille, J. Livage and T. Coradin, Acta Biomater., 2010, 6, 3998–4004 CrossRef CAS.
- M. F. Desimone, C. Helary, S. Quignard, I. B. Rietveld, I. Bataille, G. J. Copello, G. Mosser, M. M. Giraud-Guille, J. Livage, A. Meddahi-Pelle and T. Coradin, ACS Appl. Mater. Interfaces, 2011, 3, 3831–3838 CAS.
- T. Coradin, S. Bah and J. Livage, Colloids Surf., B, 2004, 35, 53–58 CrossRef CAS.
- S. Quignard, G. J. Copello, C. Aimé, I. Bataille, C. Hélary, M. F. Desimone and T. Coradin, Adv. Eng. Mater., 2012, 14, B51–B55 CrossRef.
- D. Eglin, S. Maalheem, J. Livage and T. Coradin, J. Mater. Sci.: Mater. Med., 2006, 17, 161–167 CrossRef CAS.
- S. Heinemann, H. Ehrlich, C. Knieb and T. Hanke, Int. J. Mater. Res., 2007, 98, 603–608 CrossRef CAS.
- C. J. Garcia-Valdes, G. Hernandez-Padron, M. V. Garcia-Garduno and V. M. Castano, E-Polymers, 2009, 72, 1–7 Search PubMed.
- S. Chen, S. Chinnathambi, X. Shi, A. Osaka, Y. Zhu and N. Hanagata, J. Mater. Chem., 2012, 22, 21885–21892 RSC.
- C. Aime, G. Mosser, G. Pembouong, L. Bouteiller and T. Coradin, Nanoscale, 2012, 4, 7127–7134 RSC.
- V. M. Sglavo, G. Carturan, R. Dal Monte and M. Muraca, J. Mater. Sci., 1999, 34, 3587–3590 CrossRef CAS.
- D. Eglin, G. Mosser, M. M. Giraud-Guille, J. Livage and T. Coradin, Soft Matter, 2005, 1, 129–131 RSC.
- L. N. Niu, K. Jiao, Y. P. Qi, C. K. Y. Yiu, H. Ryou, D. D. Arola, J. H. Chen, L. Breschi, D. H. Pashley and F. R. Tay, Angew. Chem., Int. Ed., 2011, 50, 11688–11691 CrossRef CAS.
- S. Heinemann, C. Heinemann, M. Jager, J. Neunzehn, H. P. Wiesmann and T. Hanke, ACS Appl. Mater. Interfaces, 2011, 3, 4323–4331 CAS.
- M. Chirita, J. Bionic Eng., 2008, 5, 149–158 CrossRef.
- J. Zhong and D. C. Greenspan, J. Biomed. Mater. Res., 2000, 53, 694–701 CrossRef CAS.
- S. Heinemann, C. Heinemann, H. Ehrlich, M. Meyer, H. Baltzer, H. Worch and T. Hanke, Adv. Eng. Mater., 2007, 9, 1061–1068 CrossRef.
- S. Heinemann, C. Heinemann, R. Bernhardt, A. Reinstorf, B. Nies, M. Meyer, H. Worch and T. Hanke, Acta Biomater., 2009, 5, 1979–1990 CrossRef CAS.
- G. Orive, R. M. Hernandez, A. R. Gascon, R. Calafiore, T. M. Chang, P. De Vos, G. Hortelano, D. Hunkeler, I. Lacik, A. M. Shapiro and J. L. Pedraz, Nat. Med., 2003, 9, 104–107 CrossRef CAS.
- G. Orive, R. M. Hernandez, A. R. Gascon, M. Igartua and J. L. Pedraz, Trends Biotechnol., 2002, 20, 382–387 CrossRef CAS.
- G. Orive, R. M. Hernandez, A. R. Gascon, R. Calafiore, T. M. Chang, P. de Vos, G. Hortelano, D. Hunkeler, I. Lacik and J. L. Pedraz, Trends Biotechnol., 2004, 22, 87–92 CrossRef CAS.
- H. Uludag, P. de Vos and P. A. Tresco, Adv. Drug Delivery Rev., 2000, 42, 29–64 CrossRef CAS.
- M. Vallet-Regí, C. R. Chim., 2010, 13, 174–185 CrossRef.
- L. Armanini, G. Carturan, S. Boninsegna, R. Dal Monte and M. Muraca, J. Mater. Chem., 1999, 9, 3057–3060 RSC.
- M. Muraca, M. T. Vilei, G. E. Zanusso, C. Ferraresso, S. Boninsegna, R. Dal Monte, P. Carraro and G. Carturan, Artif. Organs, 2002, 26, 664–669 CrossRef.
- R. Viitala, M. Jokinen and J. B. Rosenholm, Int. J. Pharm., 2007, 336, 382–390 CrossRef CAS.
- J. D. Bass, D. Grosso, C. Boissiere, E. Belamie, T. Coradin and C. Sanchez, Chem. Mater., 2007, 19, 4349–4356 CrossRef CAS.
- K. Finnie, D. Waller, F. Perret, A. Krause-Heuer, H. Lin, J. Hanna and C. Barbé, J. Sol-Gel Sci. Technol., 2009, 49, 12–18 CrossRef CAS.
- M. Gardel and S. Ulrich, J. Phys.: Condens. Matter, 2010, 22 Search PubMed.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- Z. Feng, M. Yamato, T. Akutsu, T. Nakamura, T. Okano and M. Umezu, Artif. Organs, 2003, 27, 84–91 CrossRef CAS.
- M. Karpov, M. Laczka, P. S. Leboy and A. M. Osyczka, J. Biomed. Mater. Res. A, 2008, 84, 718–726 CrossRef.
- A. M. Pietak, J. W. Reid, M. J. Stott and M. Sayer, Biomaterials, 2007, 28, 4023–4032 CrossRef CAS.
- A. K. Lynn, R. E. Cameron, S. M. Best and W. Bonfield, Transactions – 7th World Biomaterials Congress, 2004, 30 Search PubMed.
- A. K. Lynn, T. Nakamura, N. Patel, A. E. Porter, A. C. Renouf, P. R. Laity, S. M. Best, R. E. Cameron, Y. Shimizu and W. Bonfield, J. Biomed. Mater. Res. A, 2005, 74, 447–453 CrossRef CAS.
- Â. L. Andrade, P. Valério, A. M. De Goes, M. D. F. Leite and R. Z. Domingues, J. Biomed. Mater. Res. B Appl. Biomater., 2007, 83, 481–489 CrossRef.
- Y. Wang, C. Yang, X. Chen and N. Zhao, Macromol. Mater. Eng., 2006, 291, 254–262 CrossRef CAS.
- C. Xu, Y. Wang, X. Yu, X. Chen, X. Li, X. Yang, S. Li, X. Zhang and A. P. Xiang, J. Biomed. Mater. Res. A, 2009, 88, 264–273 CrossRef.
- B. W. Thimm, R. E. Unger, H. G. Neumann and C. J. Kirkpatrick, Biomed. Mater., 2008, 3, 15007 CrossRef.
- C. Heinemann, S. Heinemann, H. Worch and T. Hanke, Eur. Cell Mater., 2011, 21, 80–93 CAS.
- S. Heinemann, C. Heinemann, S. Wenisch, V. Alt, H. Worch and T. Hanke, Acta Biomater., 2013, 9, 4878–4888 CrossRef CAS.
- A. K. Lynn, I. V. Yannas and W. Bonfield, J. Biomed. Mater. Res., Part B, 2004, 71B, 343–354 CrossRef CAS.
- T. L. Adair-Kirk and R. M. Senior, Int. J. Biochem. Cell Biol., 2008, 40, 1101–1110 CrossRef CAS.
- V. Alt, D. V. Kogelmaier, K. S. Lips, V. Witt, S. Pacholke, C. Heiss, M. Kampschulte, S. Heinemann, T. Hanke, U. Thormann, R. Schnettler and A. C. Langheinrich, Acta Biomater., 2011, 7, 3773–3779 CrossRef CAS.
- C. Xu, P. Su, Y. Wang, X. Chen, Y. Meng, C. Liu, X. Yu, X. Yang, W. Yu, X. Zhang and A. P. Xiang, J. Biomed. Mater. Res., Part A, 2010, 95A, 495–503 CrossRef CAS.
- C. Xu, P. Su, X. Chen, Y. Meng, W. Yu, A. P. Xiang and Y. Wang, Biomaterials, 2011, 32, 1051–1058 CrossRef CAS.
- L. N. Niu, K. Jiao, Y. P. Qi, S. Nikonov, C. K. Y. Yiu, D. D. Arola, S. Q. Gong, A. El-Marakby, M. R. O. Carrilho, M. W. Hamrick, K. M. Hargreaves, A. Diogenes, J. H. Chen, D. H. Pashley and F. R. Tay, FASEB J., 2012, 26, 4517–4529 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |