O2-Protected diazeniumdiolate-modified silica nanoparticles for extended nitric oxide release from dental composites†
Alexis W. Carpentera, Katelyn P. Reigharda, Joseph E. Saavedrab and Mark H. Schoenfisch*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: schoenfisch@unc.edu
bBasic Science Program, SAIC Frederick, National Cancer Institute at Frederick, Frederick, Maryland 21702, USA
First published on 24th January 2013
Abstract
O2-Protected N-diazeniumdiolate-based silanes were grafted onto mesoporous silica nanoparticles to yield a scaffold with an NO payload of 2.4 μmol NO per mg and NO release half-life of 23 d. Adhesion of Streptococcus mutans to dental restorative materials doped with these NO-releasing particles was reduced by 3-log compared to controls.
Given the proven antimicrobial nature of nitric oxide (NO) and the use of silica particles in commercial oral-care products, NO-releasing silica nanoparticles may prove useful for combating plaque bacterial infections that plague dental restorative treatments. Indeed, 50–60% of restorative treatments result in secondary caries that necessitate replacement of the restoration.1 While dental composites modified with antimicrobial agents have been proposed to prolong the life-time of restorative materials, sustained release over the course of days–weeks has yet to be achieved.2–4
The gaseous and reactive nature of NO requires the use of NO donors (e.g., N-diazeniumdiolates, S-nitrosothiols, and metal nitrosyls) to achieve controlled therapeutic NO delivery.5N-diazeniumdiolates (NONOates) are among the most widely used NO donors as they are comprised of two molecules of NO covalently bound to an amine site, thus allowing for greater NO payloads compared to donors that store only one molecule of NO per donor site (e.g., S-nitrosothiols).6 Moreover, the NONOate functionality results in the spontaneous generation of NO in physiological solutions via a highly efficient proton-initiated decomposition. While advantageous, the labile nature of N-diazeniumdiolates often hinders the preparation of materials with prolonged release durations necessary for promoting implant integration. Modifying macromolecular scaffolds, such as silica nanoparticles, with NO donors allows for more localized NO-release payloads and greater tunability of NO-release kinetics by controlling water diffusion into the scaffold.6 Unfortunately, NO release from NONOate-based silica particles remains limited to only days at best.7,8 The stability of the NONOate can be increased by adding a protecting group at the O2-position. Release of NO then requires hydrolytic or metabolic cleavage of the protecting group prior to N-diazeniumdiolate decomposition.9,10 Indeed, O2-protected NONOates bound to low molecular weight structures exhibit half-lives on the order of days compared to seconds or minutes for unprotected analogs.9O2-substituted diazeniumdiolate NO donors have previously been used to covalently modify proteins and polymers to yield materials with extended NO release.9,11,12 To date, these structures have not been incorporated into silica nanoparticle scaffolds, which hold great promise in a wide range of biomedical applications.5
The lifetime of dental composites is often shortened due to the formation of plaque biofilms that degrade the material, increasing surface roughness and promoting further bacterial adhesion.13 Given that silica particles are a major component of commercial dental resin composites for strength and abrasion resistance,14 functionalized silica particles are excellent candidates to impart antimicrobial properties to the composite. Replacing the non-functionalized silica filler particles with silica particles capable of sustained NO release may result in improved antimicrobial dental composites with longer lifetimes.
Herein, mesoporous silica nanoparticles were functionalized with an O2-substituted N-diazeniumdiolate-modified organosilane synthesized by coupling an alkyl halide organosilane with O2-methoxymethyl 1-(piperazin-1-yl)diazen-1-ium-1,2-diolate. After evaluating the NO release from the resulting scaffolds, the particles were doped into a dental composite resin to yield a new class of antimicrobial restorative materials. The ability of NO release to decrease the number of viable adhered Streptococcus mutans, a leading cariogenic bacterium, to these materials over a 24 h period was evaluated in vitro.
Previously, Saavedra et al. described the preparation of piperazine-based methoxymethyl (MOM)-protected diazeniumdiolates that could be coupled to macromolecular structures through reaction with the free secondary amine on the piperazine ring.9 Since this seminal report, piperazine-based MOM-protected scaffolds with half-lives up to 3 weeks have been prepared.9,11,12 The incorporation of O2-protected NONOates into silica nanoparticles first required the synthesis of an O2-protected NONOate-modified organosilane. Ethoxycarbonyl piperazine was first N-diazeniumdiolated and subsequently alkylated with chloromethyl methyl ether. Removal of the ethoxycarbonyl group revealed MOM-Pip/NO with a free secondary amine. Full details of this synthesis have been previously described.9 A methoxymethyl protecting group was selected as it hydrolyzes slowly under physiological conditions (pH 7.4). Other protecting groups have been designed that require enzymatic cleavage; however, such methodologies are not suitable for dopant materials where access to enzymes would be low.
As shown in Scheme 1, the MOM-protected NONOate silane was synthesized by coupling MOM-Pip/NO (0.53 mmol) with an iodopropyltrimethoxysilane (0.58 mmol) with Hünig's base (0.79 mmol) in DMF (24 h, 60 °C). The successful formation of MOM-Pip/NO-TMS was indicated by a shift in the piperazine protons alpha to the secondary amine from 3.04 to 2.83 ppm (ESI†). The protons alpha to the halide also shifted from 3.57 to 2.57 ppm upon formation of MOM-Pip/NO-TMS. The reaction with iodopropyltrimethoxysilane resulted in a near complete reaction (94%). The successful formation of MOM-Pip/NO-TMS was also confirmed with ESI mass spectrometry. Excess Hünig's base was used to stabilize the MOM protecting group, which is unstable in acidic solutions. The stability of the MOM protecting group was confirmed by monitoring the singlet at 5.16 ppm that corresponds to the protons of the terminal methyl group.
 | ||
| Scheme 1 Synthesis of O2-methoxymethyl 1-(4-(3-(trimethoxysilyl)propyl)piperazin-1-yl)diazen-1-ium-1,2-diolate (MOM-Pip/NO-TMS). | ||
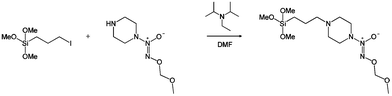
The incorporation of organic functionalities onto silica nanoparticles can be achieved via a surface-grafting method that involves condensing organosilanes with surface silanols on preformed particles.15 Typically, the preformed particles are synthesized from tetraalkoxysilane precursors, allowing for more uniform particles to be easily achieved. Particles with large organosilane concentrations can be prepared using mesoporous silica nanoparticles (MSN) due to their large specific surface area. Mesoporous silica nanoparticles were synthesized using micellular structures as templates for silica polymerization.16 Cetyltrimethylammonium bromide (290 mg) was dissolved in 50 mL of a 5 mM solution of ammonium hydroxide in water. After heating at 60 °C for 1 h, a 110 μL aliquot of tetraethoxysilane (TEOS) was added to the solution, stirred for 5 h, then a second 600 μL aliquot of TEOS was added. The solution was stirred for an additional 2 h, then allowed to age without stirring at 40 °C for 24 h. The resulting mesoporous silica nanoparticles were collected by centrifugation (3645g, 10 min) and washed with ethanol. The surfactant molecules were then removed using an acid wash to yield mesoporous silica particles. The reaction conditions used here (i.e., pH 11 and 40 °C) resulted in monodisperse rod-shaped silica nanoparticles with an aspect ratio of 3 (Fig. 1). Using Brauner–Emmett–Teller analysis of nitrogen adsorption–desorption isotherms, the MSNs were found to have a specific surface area of 1145 m2 g−1. O2-protected diazeniumdiolate-modified silica particles were subsequently prepared by condensing MOM-Pip/NO-TMS onto the MSNs by refluxing in tetrahydrofuran overnight. The successful addition of the MOM-Pip/NO-TMS into the silica network was confirmed using solid state 29Si nuclear magnetic resonance spectroscopy (SS 29Si NMR) with cross polarization and magic angle spinning (CP/MAS). As shown in Fig. 2, the resulting SS 29Si NMR spectra consisted of peaks at −110 and −60 ppm corresponding to the Q-band and the T-band, respectively. The Q-band results from 29Si bound to four functionalizable groups, representing silicon atoms originating from the TEOS precursor.17 The T-band results from 29Si bound to three hydrolyzable groups, corresponding to the MOM-Pip/NO-TMS precursor.17 Elemental analysis further confirmed MOM-Pip/NO-TMS modification as indicated by an increase in the carbon wt% from 8.97 to 17.26% and nitrogen wt% from 0.31 to 6.77%. Of note, the small carbon content present within the particle prior to modification is likely due to unreacted ethoxy ligands. The elemental analysis results indicate that 1.2 μmol MOM-Pip/NO-TMS were incorporated per mg of MSNs, which is comparable to the organosilane concentrations of previously reported amine-modified silica nanoparticles prepared via the Stöber method.7 The addition of MOM-Pip/NO-TMS did not affect particle size or morphology as determined by SEM.
 | ||
| Fig. 1 Scanning electron micrographs of mesoporous silica nanoparticles synthesized via a surfactant templated approach. Scale bar = 1 μm. | ||
 | ||
| Fig. 2 Solid state CP/MAS 29Si NMR spectrum of MOM-Pip/NO-modified MSNs. The T-band (ca. −60 ppm) indicates silicon atoms bound to three oxygens (i.e., MOM-Pip/NO-TMS), and the Q-band (ca. −100 ppm) represents silicon atoms bound to four oxygens (i.e., TEOS). | ||
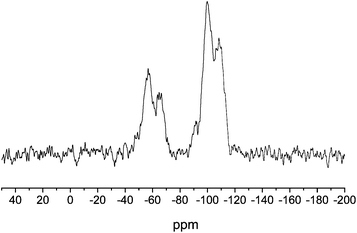
Nitric oxide storage and release from the MOM-Pip/NO-modified particles were evaluated by elemental analysis and the Griess assay, respectively.18 The MOM-Pip/NO-modified particles were characterized by a total NO payload of 2.4 μmol NO per mg. As shown in Fig. 3, the rate of NO generation from the MOM-Pip/NO-modified particles was slow and continued for several weeks at 0.5–1.0 pmol mg−1 s−1. The half-life of NO release was ultimately determined to be 23 d, which is significantly longer than any other NO-releasing silica particle to date.6 The longest previously reported NO release half-life from silica nanoparticles was 4.2 h from Stöber silica particles containing N-diazeniumdiolate-modified (3-trimethoxysilyl)diethylenetriamine.8 Thus, NO-releasing silica particles with sustained NO release was achieved by covalently incorporating an O2-protected N-diazeniumdiolate NO donor within the silica network.
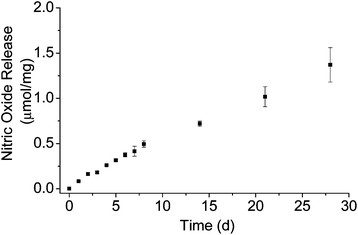 | ||
| Fig. 3 Nitric oxide release from MOM-Pip/NO-MSNs in PBS at 37 °C over 4 weeks. Total NO storage was 2.5 μmol mg−1 mesoporous silica. | ||
Nablo et al. previously showed that the release of NO from xerogel-coated silicon rubber implants significantly decreases subcutaneous Staphylococcus aureus infections in vivo.19 A number of subsequent studies have demonstrated the ability of NO-releasing surfaces to decrease the adhesion of both Gram positive and Gram negative bacteria.20–23 To date, most evaluations have employed short exposure periods (0.5–2 h) in non-nutrient conditions, which are less representative of the complex environment present in vivo. To assess the utility of MOM-Pip/NO-modified particles as dopants for antimicrobial dental restorative materials, we sought to evaluate the ability of these materials to resist bacterial adhesion over 24 h exposure times under nutrient conditions (10 vol% BHI in PBS). Streptococcus mutans was chosen as the test microbe as it has been implicated as a causative cariogenic bacterium.24
Nitric oxide release from the particle-doped composites was evaluated in 10 vol% BHI in PBS over 24 h to mimic the bacterial adhesion assays. The total NO released from MOM-Pip/NO-doped composites in this media over 24 h was 6.7 ± 0.1 nmol. The NO release flux from the composite was low (0.1–0.4 pmol cm−2 s−1) as a result of NO scavenging by proteins present in the nutrient broth and was not unexpected.25 The NO release from MOM-Pip/NO-doped composites continually increased during the 24 h exposure period due to the slow decomposition rate of the protected N-diazeniumdiolate (ESI†). The MOM-Pip/NO-doped composites exhibited NO fluxes below that previously reported to be the minimum required to inhibit adhesion of Gram negative Pseudomonas aeruginosa.20,21
Despite the low NO release, composites doped with 1 wt% MOM-Pip/NO particles were characterized by a 3-log decrease in the number of viable adhered bacteria compared to undoped controls (Fig. 4). Hetrick et al. reported a minimum flux of 20 pmol cm−2 s−1 required to inhibit adhesion of P. aeruginosa to xerogels films.21 However, Dobmeier and Schoenfisch previously noted a 50% reduction in adhesion of P. aeruginosa to xerogel microarrays with lower NO fluxes (1.0 pmol cm−2 s−1).20 Both prior studies were conducted in nutrient-free media at <2 h exposure times. To evaluate the advantage of the sustained NO release scaffolds, composites doped with unprotected (and thus faster NO-releasing) N-diazeniumdiolate-modified AHAP/NO particles were also prepared. While the NO release from MOM-Pip/NO-doped composites continually increased during the 24 h exposure period due to the slow decomposition rate of the protected N-diazeniumdiolate, the AHAP/NO-doped composites reached a maximum NO flux within the first few minutes, and the NO levels gradually decreased over the 24 h exposure time. As a result, despite the larger total NO release from the AHAP/NO-doped composites (19.2 ± 0.04 nmol), the faster rate of NO release proved less effective at preventing adhesion of viable S. mutans to composites (Fig. 4). Of note, there was no significant difference in the surface roughness as a result of doping NO-releasing particles into the composites (ESI†). Moreover, composites doped with non-NO-releasing piperazine-modified silica particles did not exhibit a statistically significant difference compared to controls (undoped), indicating the decreased adhesion was the result of the sustained NO release (ESI†). The results presented here indicate that the NO flux required to decrease the number of viable bacteria adhered to a surface is dependent on the substrate, bacterial strain, and exposure conditions (e.g., time and media).
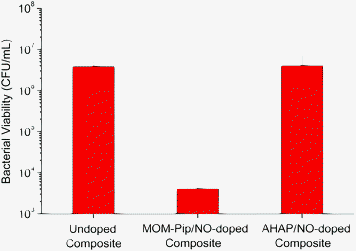 | ||
| Fig. 4 Number of viable S. mutans adhered to control or NO-releasing particle-doped composites following 24 h incubation in 10 vol% BHI in PBS. | ||
While silica nanoparticles have distinct advantages for the therapeutic delivery of NO, limited NO-release durations remain a major hindrance for their use in many biomedical applications. Herein, we demonstrated that NO-releasing silica particles with sustained NO release could be achieved by employing O2-protected N-diazeniumdiolate chemistry. The O2-protected N-diazeniumdiolate silane-modified silica particles exhibited an NO release half-life of 3 weeks, the longest duration achieved from a silica particle scaffold to date. As a result of the work presented here, the potential applications of NO-releasing silica nanoparticles may now include those requiring longer release durations at low NO fluxes.
Preliminary therapeutic utility of the MOM-Pip/NO-modified silica nanoparticles was indicated using particle-doped dental composites. The release of NO from the MOM-Pip/NO-doped composites effectively decreased the number of adhered viable S. mutans compared to control (undoped) composites. This report is the first employing NO release to prevent adhesion of plaque bacteria to a substrate. To evaluate the full potential of NO-releasing dental composites, future studies should evaluate longer bacterial exposure times and whether NO inhibits plaque biofilm formation. The effectiveness of NO-releasing composites in environments mimicking the oral cavity should also be evaluated as the lower pH will result in faster NO release, thus potentially influencing bacterial adhesion/viability. Greater NO payloads may be achieved by forming composites with larger NO-releasing silica nanoparticle concentrations. Indeed, unmodified silica particles comprise 70–90 wt% of commercial dental composites to provide increased modulus, strength, and abrasion resistance.26 Designing new composites where NO-releasing silica particles are used to provide both strength and antiplaque properties may yield improved composites with respect to resistance to biofilm formation. The effect of the NO-releasing particle dopants on the properties of the polymer composites should also be evaluated in future work.
Acknowledgements
This work was supported by a National Science Foundation grant (DMR 1104892). A.W.C. gratefully acknowledges a graduate research fellowship from Eastman Chemical Company (Kingsport, TN). This project was also funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The authors thank Dr. Luiz Pimenta of the UNC School of Dentistry for providing dental composite materials and equipment.Notes and references
- V. Deligeorgi, I. A. Mjor and N. H. F. Wilson, Prim. Dent. Care, 2001, 8, 5–11 CrossRef CAS.
- N. Beyth, I. Yudovin-Farber, M. Perez-Davidi, A. J. Domb and E. I. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22038–22043 CrossRef CAS.
- L. Cheng, M. D. Weir, S. M. Xu, J. M. Antonucci, A. M. Kraigsley, N. J. Lin, S. Lin-Gibson and X. Zhou, J. Dent. Res., 2012, 28, 561–572 CAS.
- C. Fan, L. Chu, H. R. Rawls, B. K. Norling, H. L. Cardenas and K. Whang, Dent. Mater., 2011, 27, 322–328 CrossRef CAS.
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- J. H. Shin, S. K. Metzger and M. H. Schoenfisch, J. Am. Chem. Soc., 2007, 129, 4612–4619 CrossRef CAS.
- J. H. Shin and M. H. Schoenfisch, Chem. Mater., 2008, 20, 239–249 CrossRef CAS.
- J. E. Saavedra, M. N. Booth, J. A. Hrabie, K. M. Davies and L. K. Keefer, J. Org. Chem., 1999, 64, 5124–5131 CrossRef CAS.
- J. E. Saavedra, T. M. Dunams, J. L. Flippen-Anderson and L. K. Keefer, J. Org. Chem., 1992, 57, 6134–6138 CrossRef CAS.
- M. M. Reynolds, J. E. Saavedra, B. M. Showalter, C. A. Valdez, A. P. Shanklin, B. K. Oh, L. K. Keefer and M. E. Meyerhoff, J. Mater. Chem., 2010, 20, 3107–3114 RSC.
- J. A. Hrabie, J. E. Saavedra, P. P. Roller, G. J. Southan and L. K. Keefer, Bioconjugate Chem., 1999, 10, 838–842 CrossRef CAS.
- H. J. Busscher, M. Rinastiti, W. Siswomihardjo and H. C. van der Mei, J. Dent. Res., 2010, 89, 657–665 CrossRef CAS.
- M. H. Chen, J. Dent. Res., 2010, 89, 549–560 CrossRef CAS.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673–2685 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Deliv. Rev., 2008, 60, 1278–1288 CrossRef CAS.
- K. Albert and E. Bayer, J. Chromatogr., 1991, 544, 345–370 CrossRef CAS.
- P. N. Coneski and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3753–3758 RSC.
- B. J. Nablo, H. L. Prichard, R. D. Butler, B. Klitzman and M. H. Schoenfisch, Biomaterials, 2005, 26, 6984–6990 CrossRef CAS.
- K. P. Dobmeier and M. H. Schoenfisch, Biomacromolecules, 2004, 5, 2493–2495 CrossRef CAS.
- E. M. Hetrick and M. H. Schoenfisch, Biomaterials, 2007, 28, 1948–1956 CrossRef CAS.
- P. N. Coneski, K. S. Rao and M. H. Schoenfisch, Biomacromolecules, 2010, 11, 3208–3215 CrossRef CAS.
- D. A. Riccio, P. N. Coneski, S. P. Nichols, A. D. Broadnax and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2012, 4, 796–804 CAS.
- C. Poggio, C. R. Arciola, F. Rosti, A. Scribante, E. Saino and L. Visai, Int. J. Artif. Organs, 2009, 32, 671–677 Search PubMed.
- E. M. Hetrick, J. H. Shin, N. A. Stasko, C. B. Johnson, D. A. Wespe, E. Holmuhamedov and M. H. Schoenfisch, ACS Nano, 2008, 2, 235–246 CrossRef CAS.
- J. Stansbury and C. Bowman, Mater. Matters, 2010, 5, 73–81 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NO release, Atomic Force Microscopy images, surface roughness data, control bacterial adhesion assays. See DOI: 10.1039/c3bm00153a |
| This journal is © The Royal Society of Chemistry 2013 |