On–off switchable drug release from multi-responsive degradable poly(ether urethane) nanoparticles†
Yangyun
Wang
a,
Guolin
Wu
*a,
Xiaomeng
Li
a,
Yinong
Wang
a,
Hui
Gao
b and
Jianbiao
Ma
*ab
aKey Laboratory of Functional Polymer Materials of MOE, Institute of Polymer Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: guolinwu@nankai.edu.cn; Fax: +86 22 23502749; Tel: +86 22 23507746
bSchool of Chemistry and Chemical Engineering, Tianjin University of Technology, Tianjin 300191, P. R. China. E-mail: jbma@tjut.edu.cn; Fax: +86 22 23502749; Tel: +86 22 23507746
First published on 8th March 2013
Abstract
A novel on–off switchable drug-release system was developed based on a series of multi-responsive degradable poly(ether urethane)s. The multi-segmented poly(ether urethane)s were synthesized through a simple one-pot condensation polymerization of poly(ethylene glycol), 2,2′-dithiodiethanol, N-methyldiethanolamine and hexamethylene diisocyanate. The obtained amphiphilic copolymers could self-assemble into nanoparticles in aqueous solution, which were responsive to temperature, pH and redox potential with tailored phase-transition temperature. The whole process for the responsive behaviours of the poly(ether urethane) nanoparticles was confirmed by light transmission, dynamic light scattering, nuclear magnetic resonance and transmission electron microscopy. The nanoparticles could encapsulate hydrophobic drugs and showed a temperature-triggered accelerated and complete drug-release profile. The mechanism of the temperature-triggered multi-responsive accelerated drug release was also elucidated. These results presented the polymeric nanoparticles as an effective multi-responsive degradable nanocarrier to achieve on–off drug release.
Introduction
Due to profound impacts in medical fields, controlling drug-release technology has been extensively studied.1–3 Most of the previous work always focused on achieving a constant release of drug over a long period, irrespective of the patient's needs and changing physiological circumstances.4,5 However, for certain diseases, constant release is not the optimal method for drug delivery. For example, the periodicity of vascular endothelial growth factor (VEGF) molecules naturally released from breast cancer regulates cancer vascular permeability.6 Under this situation, on–off release of anti-tumor drugs corresponding with the VEGF expression will greatly improve the drug bioavailability.6,7 Similarly, for treatment of hormone-related diseases such as growth hormone and gonadotropin-releasing hormone, which are also secreted by the human body in an on–off fashion,8,9 it is desirable to explore on–off release systems to cure these diseases with better performance.An ideal on–off drug delivery carrier should release little or no drugs in the “off” state, and be reproducibly switchable to the “on” state without mechanical disruption, and then release a tunable dosage of drugs.10 To meet the above requirements, employing temperature-responsive polymers as drug carriers could be a promising way for the successful development of on–off switchable drug-delivery carriers.10,11 Recently, temperature-responsive polymeric systems encapsulating gold or iron oxide nanoparticles have been developed for on–off controlled drug release that exhibit sensitivity to non-invasive external triggers such as near-infrared radiation12–14 or oscillating magnetic fields.15–17 In these cases, the external energy can be converted into heating, and thus on–off switchable drug release can be triggered remotely to provide the proper dose magnitude and timing according to the patient's needs.18
However, single temperature-responsive carriers always result in transient drug release due to the slow polymer degradation and fast completion of phase transition, which cannot provide a high drug bioavailability.19,20 An alternative solution is to develop multi-responsive degradable polymeric carriers to achieve the complete drug release. Nevertheless, limited multi-responsive polymeric carriers are reported, not to mention the multi-responsive degradable ones.21–26 Thayumanavan et al. reported a triple-stimuli-responsive polymeric micelle which comprised an acid-sensitive hydrophobic core, a temperature-sensitive hydrophilic shell and a redox-sensitive interface.27 The system demonstrated the feasibility of combining different orthogonal triggers and provided a unique opportunity to release the encapsulated drug completely.
Recently, a series of poly(ether urethane) nanoparticles based on alternative hydrophilic and hydrophobic segments has been developed in our group, which exhibited reversible temperature responsiveness with the phase-transition temperature (Tp) adjusted in the range of 30–40 °C.28,29 Giving further credence to this approach, we have reported a series of dually responsive poly(ether urethane) nanoparticles which exhibited temperature-triggered redox-degradable behaviour.30 Inspired by these promising results, we envisaged the possibility of designing an effective on–off switchable drug-release system based on a series of segmented poly(ether urethane)s (denoted as SPU) by repeatedly incorporating both pH- and redox-responsive segments into the temperature-responsive poly(ether urethane) backbones. In this system, (1) temperature is designed to be used as the remote means of triggering on–off drug release, and (2) redox and pH responsiveness are used to enhance the release rate and bioavailability of the drug which only play a role in the ‘open state’ of drug release. The ability to use temperature to regulate multi-responsive behaviour in a single system is considered to be highly advantageous for future exploitation in biomedical fields. To the best of our knowledge, this is the first time an on–off switchable drug-release system based on multi-responsive polymers has been reported.
Experimental details
Chemicals and reagents
Poly(ethylene glycol) (PEG, Mw = 600, 1000, 1500 and 2000 Da), hexamethylene diisocyanate (HDI, 98%), 2,2′-dithiodiethanol (DiT), N-methyldiethanolamine (MDEA) and glutathione (GSH) were purchased from Sigma-Aldrich (Shanghai, China). Doxorubicin hydrochloride (Dox.HCl) was purchased from HuaFeng United technology Co. Ltd (Beijing, China). Tetrahydrofuran (THF) and 1,2-dichloroethane were distilled over sodium and calcium hydride, respectively. All other reagents were obtained from Tianjin Chemical Reagent Co. (Tianjin, China) and used without further purification.Synthesis
A certain amount of DiT (0.5 mmol) and MDEA (1.5 mmol) was dissolved in a mixture of 1,2-dichloroethane and THF (20 mL, CH2ClCH2Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF = 5:1). Then the mixture was added dropwise into the ready prepared PEG-diisocyanate (2 mmol) solution with stannous octanoate as the catalyst (0.5 wt%, with respect to the reactant). The reaction was performed at 80 °C under dry nitrogen atmosphere. After 24 h, an excess amount of methanol was added and stirred for 1 h to eliminate the stannous octanoate residue and oligomers. The production was then precipitated in diethyl ether. The resulting product was collected through filtration, followed by drying under vacuum to constant weight, affording a yield of over 80%.
:THF = 5:1). Then the mixture was added dropwise into the ready prepared PEG-diisocyanate (2 mmol) solution with stannous octanoate as the catalyst (0.5 wt%, with respect to the reactant). The reaction was performed at 80 °C under dry nitrogen atmosphere. After 24 h, an excess amount of methanol was added and stirred for 1 h to eliminate the stannous octanoate residue and oligomers. The production was then precipitated in diethyl ether. The resulting product was collected through filtration, followed by drying under vacuum to constant weight, affording a yield of over 80%.
:1, PEG/(MDEA + DiT) molar ratio = 1:1, 2:1 or 3:1) were dissolved in a mixture of 1,2-dichloroethane and THF (CH2ClCH2Cl:THF = 5:1). Then the mixture was added dropwise to another 1,2-dichloroethane solution with a certain amount of HDI (molar ratio of HDI to (PEG + DiT + MDEA) is 1:1). Random segmented poly(ether-urethane)s with different segment ratios were obtained through the same processes as above. The yield was over 85%.
Measurements
1H NMR spectra were recorded on a Varian UNITY-plus 400 NMR spectrometer using CDCl3 or DMSO-d6 as the solvent. Tetramethylsilane was used as the internal standard. Fourier transform infrared spectra (FT-IR) were measured using Bio-Rad FTS6000 spectrophotometer at room temperature. Polymer samples were prepared by dispersing the complexes well in KBr powder and compressing the mixtures to form a plate. The molecular weight and distribution of polymers were determined by gel permeation chromatography (GPC, Waters 2414 system Milford, MA) at 35 °C. THF was used as the eluent at a flow rate of 1.0 mL min−1. The average weights were calibrated with standard polystyrene samples.Preparation of the SPU nanoparticles
The poly(ether-urethane) nanoparticles were prepared by adding the amphiphilic polymers into distilled water, followed by sonication for 5 min to accelerate dissolution of the polymers in an ultrasonic bath and then stirred slowly for 24 h to form the polymer nanoparticles.The critical micelle concentration (CMC) of SPU2 is around 0.02 mg mL−1 at 25 °C, measured by fluorescence spectrometry using pyrene as a probe (Fig. S1 in the ESI†). The CMC of the other polymers have similar values. The concentration of polymer solutions used in the following measurements is 1 mg mL−1, which is much higher than their CMC.
Temperature responsiveness of the SPU nanoparticles
Light transmission of the polymer aqueous solution at different temperatures was determined using a UV-2450 (Shimadzu Co. Japan), equipped with a temperature-controllable cell. The measurements were performed with a wavelength of 480 nm, and the sample solutions were equilibrated for 15 min before each measurement during the cycle of heating and cooling (20–70 °C). The phase transition temperature (Tp) could be determined from the onset of the decrease in the transmittance of the polymer aqueous solution.The particle size and distribution (PDI) in response to temperature were determined by dynamic light scattering (DLS) using Zetasizer Nano ZS90 (Malvern Instruments, Southborough, MA). Measurements were carried out at special temperatures in the range from 20 to 70 °C during the heating and cooling process and equilibrated for 15 min before each measurement. In the present DLS study, the normalized field–field autocorrelation function |g(1)(τ)| (≡ [〈E(0) − E*(τ)〉/〈E(0)E*(0)〉]) of each copolymer solution at different temperature was measured at the scattering angle θ = 90°. For a polydisperse sample, g(1)(τ) is related to the distribution of the characteristic line width G(Γ) by:
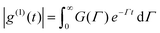 | (1) |
For a diffusive relaxation, Γ is related to the translational diffusion coefficient D by Γ = Dq2, when the solution is in dilute regime. In principle, G(Γ) can be calculated from the Laplace inversion according to eq (1). D is further related to the hydrodynamic radius Rh by Rh = kBT/(6πη0D). The well-accepted Laplace inversion CONTIN algorithm was used in the analysis of the DLS autocorrelation curves.32,33
pH responsiveness of the SPU nanoparticles
Copolymer aqueous solution at pH = 2 was prepared for the turbidity measurement. The titration was carried out by the stepwise addition of 0.1 N NaOH. The pH values were checked using a Sartorius PB-10 pH meter (Sartorius Instrument, Germany). The turbidity of the solution during the titration was measured by monitoring the absorbance at 480 nm using a UV-2450 spectrophotometer.The zeta potential, particle size and distribution in response to pH of the nanoparticles were also recorded using a Zetasizer Nano ZS90 Malvern instrument.
Redox responsiveness of the SPU nanoparticles
Changes of the particle sizes and scattering light intensity of the polymer aqueous solution in response to GSH (10 mM) versus time were monitored by DLS measurement using a Zetasizer Nano ZS90 Malvern instrument.Morphologies of the SPU nanoparticles
Morphologies of the blank and drug-loaded nanoparticles were examined by transmission electron microscopy (TEM). The digital images were recorded on a JEOL Microscope (Japan). A specimen suspension under different condition was prepared as follows: a drop of the suspension was placed onto a 200 mesh copper grid. Then the copper grids were dried at the corresponding temperature in the vacuum drying chamber. The obtained copper grid was further introduced for TEM measurement.Variable temperature and pH 1H-NMR of the SPU nanoparticles
Variable temperature 1H NMR (400 MHz) spectra were recorded on a thermo-regulated Bruker Avance 400 using solution of SPU2 (0.5 wt%) in D2O (99.9 D atom%). At each temperature, the samples or suspensions were equilibrated for 20 min before measurement. Variable pH 1H NMR spectra were performed in D2O at different pH value on the Varian UNITY-plus 400 NMR instrument.Preparation of the DOX-loaded SPU nanoparticles
DOX-loaded nanoparticles were prepared as follows. Briefly, DOX (5 mg) dissolved in DMF (5 mL) was stirred with 2 equiv. of triethylamine for 2 h. And then the SPU2 (25 mg) was added and stirred for 24 h. The mixture was dialyzed against deionized water under sink conditions for 72 h. Finally, DOX-loaded nanoparticles were lyophilized to give a red powder. The DOX contents in the polymers were measured by recording the fluorescence emission spectra at 610 nm (the excitation wavelength at 465 nm) in DMSO using a Shimadzu RF-5301PC spectrofluorophotometer. The drug-loading content (%) and entrapment efficiency (%) were calculated by the following equations: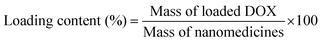
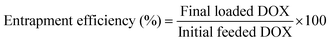
In vitro drug release from the SPU nanoparticles
The DOX-loaded SPU2 nanoparticles were dissolved in the respective PBS buffer (PBS 7.4, 6.0 or 5.0) to prepare the solutions with a concentration of 1 mg mL−1. A certain amount of the above sample was transferred into a screw-cap jar with or without GSH (10 mM). The solutions were placed in a shaking water bath (150 rpm) at different temperatures under sink conditions. At predetermined time intervals, fluorescence of different samples was recorded using a Shimadzu RF-5301PC spectrofluorophotometer with the excitation wavelength at 465 nm and the emission spectra at 610 nm.34The temperature-triggered on–off switchable drug-release studies were performed as follows: the “ON” state was triggered by placing the screw-cap jars with DOX-loaded SPU2 nanoparticles in phosphate-buffered saline (PBS, pH 6.0 or 7.4) in a 37 °C water bath. After incubation for a period of time, the above screw-cap jars was taken out and placed in a 25 °C water bath which was defined as the “OFF” state. This process was carried out for several cycles and fluorescence at different time intervals was recorded to get the “ON” and “OFF” profiles.
Results and discussion
Synthesis and characterization
In this work, we have designed a multi-responsive system based on the following rules: (1) different diols and HDI have been used to synthesize the linear segmented poly(ether urethane)s. The balance between the hydrophilic and hydrophobic segments is used to switch the Tp of these polymers. (2) The disulfide-containing diols (DiT) have been incorporated into the polymers repeatedly to serve as redox-responsive segments. It can cleave the polymers into oligomers in the presence of a mild reducing agent, such as GSH. (3) The tertiary amine-containing diols (MDEA) in the backbones are pH-responsive. The hydrophobic tertiary amine converts to a protonized hydrophilic one in acidic environment, leading to swelling or even dissolution of the nanoparticles.The segmented poly(ether urethane)s could be easily synthesized in two different ways. Scheme 1 illustrates the synthesis route of PEG-alt-(MDEA/DiT) copolymers based on condensation reaction between different molecular weight PEG-diisocyanates and two diols with pH and redox responsive groups. The parent PEG-diisocyanates with different molecular weights were prepared by coupling reaction using 1 equiv. PEG and 2 equiv. HDI. A typical 1H NMR spectrum of PEG-diisocyanates is shown in Fig. 1(A). The 1HNMR results confirm the formation of the PEG-diisocyanate. Scheme 2 illustrates the synthesis route of PEG-co-(MDEA/DiT) copolymers based on one-pot condensation reaction between DiT, MDEA, PEG and HDI. The second approach is more convenient to adjust their hydrophilic/hydrophobic balance based on different monomer feedings, which is crucial for the control of its phase-transition temperature. All synthesized multi-block copolymers consisted of PEG and HDI as hydrophilic and hydrophobic segments, respectively. Disulfide bonds and tertiary amines served as stimuli-responsive moieties. The synthesized polymers were characterized by 1HNMR, FT-IR and GPC. A typical SPU2 1HNMR spectrum of the obtained polymers is shown in Fig. 1(B). In addition to the typical peaks of PEG-diisocyanate, the characteristic peaks corresponding to the MDEA and DiT segments were observed in Fig. 1(B). The representative FT-IR spectra of the multi-block poly(ether urethane) SPU2, and PEG-diisocyanate, DiT and MEDA are shown in Fig. 2. For the segmented copolymer, the bands at 1715 and 3331 cm−1 are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H stretching of urethane groups, respectively. The absence of absorbance at 2272 cm−1 indicates no isocyanate groups in the resulting polymer. The typical GPC trace of PEG and the corresponding SPU are shown in Fig. S2 in the ESI.† A unimodal GPC peak was observed for the obtained polymer. The characterization results of all synthesized segmented copolymers are collected in Table 1.
O and N–H stretching of urethane groups, respectively. The absence of absorbance at 2272 cm−1 indicates no isocyanate groups in the resulting polymer. The typical GPC trace of PEG and the corresponding SPU are shown in Fig. S2 in the ESI.† A unimodal GPC peak was observed for the obtained polymer. The characterization results of all synthesized segmented copolymers are collected in Table 1.
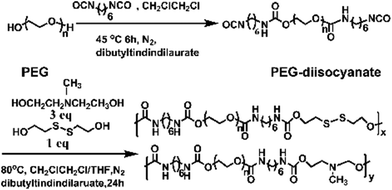 | ||
| Scheme 1 Synthesis of the PEG-alt-(MDEA/DiT) copolymers. | ||
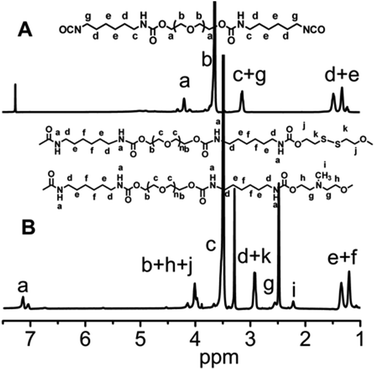 | ||
| Fig. 1 (A) 1H NMR spectrum of PEG-diisocyanate (Mw = 1000) in CDCl3. (B) 1H NMR spectrum of the segmented poly(ether urethane) SPU2 in DMSO-d6. | ||
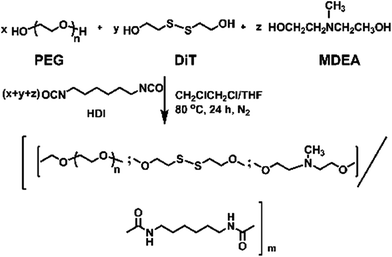 | ||
| Scheme 2 Synthesis of the PEG-co-(MDEA/DiT) copolymers. | ||
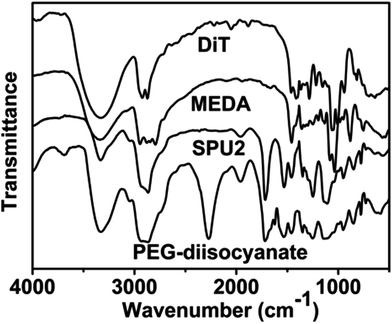 | ||
| Fig. 2 FTIR spectra of MDEA, DiT, PEG-diisocyanate (Mw = 1000) and the segmented poly(ether urethane) SPU2. | ||
| Sample code | Copolymer composition | PEG Mn |
R (MDEA:DiT)c |
R
1 (PEG:(MDEA + DiT))d |
Polymer | ||
|---|---|---|---|---|---|---|---|
|
M
ne |
PDIf |
T
pg (°C) |
|||||
|
a PEG-alt-(MDEA/DiT) copolymers based on PEG-diisocyanate and (MDEA/DiT).
b PEG-co-(MDEA/DiT) copolymers based on PEG, HDI and (MDEA/DiT).
c MDEA:DiT molar ratio in feed.
d PEG:(MDEA + DiT) molar ratio in feed.
e Determined by GPC in THF.
f Polydispersity index.
g
T
p was determined from the onset of the decrease in the transmittance of the polymer aqueous solution or from the onset of the increase in the hydrodynamic diameter of the polymer aqueous solution.
|
|||||||
| SPU1 | PEG600-alt-(MDEA–DiT)a | 600 | 3:1 |
1:1 |
12352 |
1.76 | 25 |
| SPU2 | PEG1000-alt-(MDEA–DiT)a | 1000 | 3:1 |
1:1 |
32685 |
1.43 | 35 |
| SPU3 | PEG1500-alt-(MDEA–DiT)a | 1500 | 3:1 |
1:1 |
19556 |
1.52 | 55 |
| SPU4 | PEG2000-alt-(MDEA–DiT)a | 2000 | 3:1 |
1:1 |
32069 |
1.61 | 60 |
| SPU5 | PEG1000-co-(MDEA–DiT) (2:1)b |
1000 | 3:1 |
2:1 |
23542 |
1.49 | 40 |
| SPU6 | PEG1000-co-(MDEA–DiT) (3:1)b |
1000 | 3:1 |
3:1 |
69531 |
1.49 | 45 |
Multi-responsive behaviour of the SPU nanoparticles
The SPU nanoparticles were designed to respond to temperature, redox potential and pH value. First, SPU1 was taken as an example to preliminarily test their environmental stimuli responsiveness (Fig. 3). An aqueous solution of SPU1 (1 mg mL−1, 10 mL) was prepared, which was transparent at 20 °C. The solution was divided into two equal aliquots. One of the SPU1 solutions was heated to 40 °C: a turbid and homogeneous dispersion was observed and this process was thermally reversible. When an amount of GSH was added into the turbid solution, it changed immediately to be transparent. The other 5 mL SPU1 solution was titrated with a 0.1 N NaOH solution. During the titration, the clear solution turned cloudy with the increase of the pH value. When the pH value of the solution was decreased to the initial state, the cloudy solution returned to transparent again. All these phenomena preliminarily indicate that the polymer has good reversible temperature and pH responsiveness. However, the transparent solution treated by GSH cannot return to the turbid dispersion again due to the damage of the poly(ether urethane) backbones.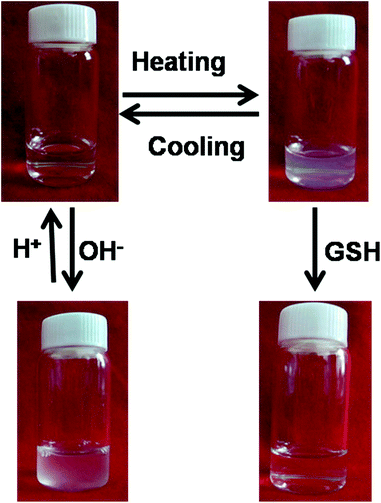 | ||
| Fig. 3 Photographs showing an aqueous solution of SPU1 under different conditions. | ||
Temperature-responsive aggregation of the SPU nanoparticles
To study the temperature-responsive aggregation behaviour of the polymeric nanoparticles under different temperatures, temperature-dependent light transmittance measurements and hydrodynamic diameter measurements for SPU1 were carried out using a UV-vis spectrometer and dynamic light scattering (Fig. 4A). A typical transmittance vs. temperature curve for SPU1 in aqueous solution is shown in Fig. 4Aa. The phase-transition temperature could be determined from the onset of decrease in the transmittance of the polymer aqueous solution, which is 25 °C for SPU1. When the temperature was raised above Tp (25 °C), the transmittance of SPU1 solution decreased sharply and the transition temperature range was narrow. DLS measurements (Fig. 4Ab) showed the hydrodynamic diameter started to increase from about 200 to 900 nm upon temperature increasing from 20 to 70 °C and the transition temperature also appeared at 25 °C. These results are caused by the aggregation of SPU particles with increasing temperature.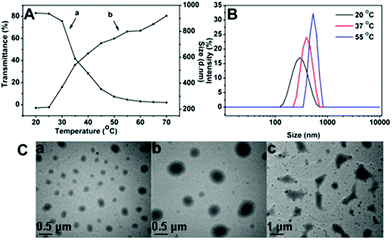 | ||
| Fig. 4 (A) Temperature-dependence of the transmittance (a) and the hydrodynamic diameter (b) of the SPU1 aqueous solution. (B) The size change of SPU1 under different temperatures in aqueous solution. (C) TEM graphs for SPU1 nanoparticles under different temperatures in aqueous solution: (a) 20 °C; (b) 37 °C; (c) 55 °C. | ||
To further understand the temperature-responsive aggregation of the SPU nanoparticles, the size distribution of the SPU1 in aqueous solution under different temperatures is also given in Fig. 4B. At 37 or 55 °C, which was above its Tp of the copolymer, a sharp increase in the average effective diameter of nanoparticles was presented compared with at 20 °C, which is below its Tp. This is because above the Tp the hydrogen bonds between PEG and water molecules are weaker and a part of water molecules are expelled from the polymer chains and the interaction between the hydrophobic units of the polymers becomes stronger and results in the aggregation of the polymer nanoparticles and turbidity of the solution. The behaviour of the temperature-responsive aggregation of SPU1 nanoparticles at the corresponding temperature was further investigated using TEM images (Fig. 4C). The morphologies of the polymer particles at different stages of the aggregation process during heating were observed. Almost uniform-sized spherical nanoparticles of 200 nm were found at 20 °C, below its Tp (Fig. 4Ca). When above Tp, large aggregations were formed with a diameter of about 400 and 600 nm at 37 and 50 °C (Fig. 4Cb and c), respectively. These results are in good accordance with the DLS measurements.
The size distribution of SPU2 (Tp, 35 °C) in aqueous solution at different temperatures is also illustrated in Fig. S3 in the ESI.† At 25 °C, a single peak around 200 nm was observed. With an increase of temperature to 45 °C, a new distribution above 1000 nm appeared which indicates of the aggregation of nanoparticles into microparticles at this temperature. It is known that increase in the length of the hydrophilic segments or ratio of hydrophilic/hydrophobic segments would increase the Tp of the polymer.35,36 From the Fig. S4 and S5 in the ESI,†Tp was indeed strongly affected by the polymer composition. The SPU2–SPU4 containing longer hydrophilic PEG segments (Mw = 1000, 1500 or 2000 Da) and the SPU5 and SPU6 with a higher ratio of hydrophilic/hydrophobic segments exhibited higher Tp compared with SPU1. The heating–cooling cycle of SPU2 aqueous solution was also conducted (Fig. S6 in the ESI†). The results showed that the cooling curve was very close to the heating curve, indicating a good reversibility of the temperature-responsive phase transition process. The reversibility of the temperature responsiveness for the nanoparticles during the heating–cooling cycling was also investigated via light scattering. The DLS results show excellent reversibility similar to the turbidity/transmission results. A typical hydrodynamic radius distribution and corresponding TEM images of blank SPU2 nanoparticles at 25 °C during the heating and cooling process are shown in Fig. S7B and S7C in the ESI.† The corresponding field–field autocorrelation functions of blank SPU2 nanoparticles were also given to support the DLS results (Fig. S7A in the ESI†). The hydrodynamic radius distribution was calculated by using the well-accepted Laplace inversion CONTIN algorithm. These results reveal that the phase transition of PEG analogues is much more uniform than that of poly(N-isopropylacrylamide) (PNIPAM), which has a significant hysteresis observed in the cooling process.37
To assess whether such a transition in response to temperature would result in the concomitant release of the encapsulated agents, DOX-loaded SPU2 nanoparticles were prepared to perform release experiments. The mixture of SPU2 and DOX solution was dialyzed against the deionized water in the dark until the water outside the dialysis tube exhibited negligible fluorescence emission of DOX. This dialysis process guaranteed the removal of the non-entrapped DOX molecules. The size of drug-loaded nanoparticles is an important factor affecting their stability, in vivo distribution, cellular internalization, and the intracellular fate. By dynamic light scattering (DLS) measurements (Fig. 5A and B), the average hydrodynamic diameter of the blank SPU2 nanoparticles in an aqueous solution was about 230 nm, while the size of the DOX-loaded SPU2 nanoparticles was about 300 nm. The increase in average size of the particles is caused by the drug molecules that have been entrapped. TEM micrographs revealed that the nanoparticles had an almost spherical morphology with an average size of about 250 nm for the blank nanoparticles (Fig. 5C) and about 300 nm for the DOX-loaded nanoparticles (Fig. 5D). The drug loading content and entrapment efficiency were calculated to be 15.6% and 91.2%, respectively.
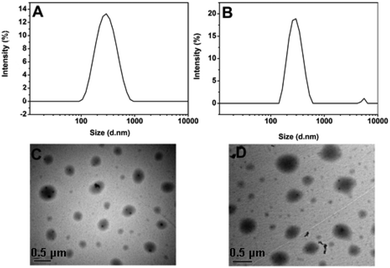 | ||
| Fig. 5 (A) and (C) DLS and TEM images of blank SPU2 nanoparticles. (B) and (D) DLS and TEM images of DOX-loaded SPU2 nanoparticles. | ||
The drug-loaded polymeric solution was treated under different temperatures (25, 37 and 50 °C) at pH = 7.4. The release of DOX from the nanoparticles was monitored as a function of time and is depicted in Fig. 6. At 25 °C, a slow release of DOX was observed and only about 24% of the drug was released in 2 days. Therefore, below the Tp, the release rate is very sluggish. At 37 °C, around Tp, an accelerated drug release was observed compared with that at 25 °C and 33% of the drug was released in 2 days. When temperature was increased above Tp at 50 °C, the drug-release rate increased dramatically and more than 40% of DOX was released out in 10 h. This suggests the faster release kinetics of DOX at higher temperature above Tp with concomitant aggregation of nanoparticles. While below the Tp, the release rate is very sluggish. It should be noted that, as shown in Fig. 6, the drug release was shut down in a short period of time with the completion of phase transition. After 10 hours only a very small amount of drug was released continuously. Therefore, temperature can be used as a switch to trigger the drug release in this system. However, single temperature-induced drug delivery cannot provide a high drug bioavailability.
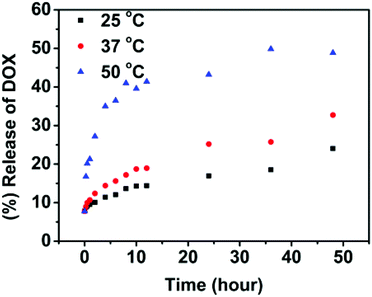 | ||
| Fig. 6 In vitro release of doxorubicin from the SPU2 nanoparticles in PBS (pH 7.4) at different temperatures. | ||
The pH-responsive disassembly of the SPU nanoparticles
The backbone of poly(ether urethane)s is comprised of ternary amino moieties, which can accept protons in response to environmental pH. Under certain pH conditions, the ternary amino functional groups presented along the backbones of the SPU undergo ionisation that leads to a conformational change in the polymers resulting in swelling or dissolution. To further investigate the pH responsiveness of the nanoparticles, the nanoparticle transmittance and zeta potential of blank nanoparticles were measured at different pH conditions (Fig. 7A). As shown in Fig. 7Aa, when the pH of the SPU1 solution was more than 7.1 the transmittance of the solution increased with the pH increase due to gradual precipitation of the aggregated particles. When the pH values were in the range of 7.1–6.3, the transmittance of the solution increased slowly with the pH decrease. The reason may be that a small quantity of ternary amino groups were protonated, resulting in the slight increase of polymer molecular polarity, so that the nanoparticles transformed from dense to swollen structures. When the pH values dropped to 5.7, the transmittance of the solution increased sharply with the decrease of pH, indicating that the pH-responsive segments were mostly hydrophilic which lead to the disassembly or even dissolution of the SPU particles.38 The zeta-potentials of SPU1 solution gave a similar result which is shown in Fig. 7Ab. With pH decrease, zeta potential first increased rapidly, and then reached a plateau at pH 5.7, attributed to the tertiary amine moieties being ionized fully.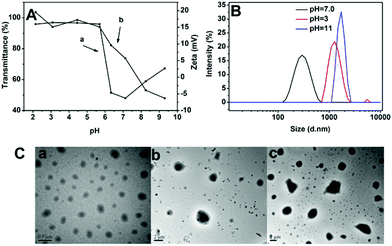 | ||
| Fig. 7 (A) pH-dependence of the transmittance (a) and zeta potential (b) of SPU1 aqueous solution under different pH values at 20 °C. (B) The size change of SPU1 under different pH at 20 °C. (C) TEM graphs of SPU1 under different pH at 20 °C: (a) pH = 7.0; (b) pH = 3; (c) pH = 11. | ||
The SPU nanoparticles were also monitored using DLS measurements. The SPU1 solution (1 mg mL−1) was dissolved in phosphate buffer with different pH values (pH = 3, 7.0 or 11). As shown in Fig. 7B, the size of the nanoparticle increased at both higher or lower pH conditions compared with the physiological condition (pH 7.4). Fig. 7C shows the corresponding TEM images of the nanoparticles, which also showed larger particles under the treatment of H+ or OH−. However, the mechanisms of particle size increase at these two conditions are different. When nanoparticles were in acidic conditions, particle size increased from 282 nm (Fig. 7Ca) to 1455 nm (Fig. 7Cb) and scattering intensity (kcps) decreased sharply from 166 to 67 (not shown). This is because at acidic condition ternary amino moieties in MDEA segments are protonated and changed to become more hydrophilic, leading to the nanoparticles transforming from dense to swollen structures or even dissolving. When the solution was in basic conditions, particle size increased from 282 nm to 1736 nm (Fig. 7Cc) and scattering intensity (kcps) increased from 166 to 250 (not shown). This is because at basic condition ternary amino moieties are completely deprotonated, resulting in increasing of intermolecular hydrophobic interactions and aggregation of the nanoparticles.
In vitro drug-release studies of the DOX-loaded nanoparticles were performed under physiological conditions (pH 7.4) and slightly acidic environments (pH = 6.0) at 37 °C to simulate the tumor microenvironment. The release profiles of DOX are shown in Fig. 8. The drug release from nanoparticles at pH = 7.4 was considerably slow, with a release of about 33% in 48 h. On the contrary, the drug release was faster at pH 6.0, with approximately 51% of the drug released within 48 h for pH 6.0. The change in pH from 7.4 to 6.0 leads to swelling or disassembly of nanoparticles, thereby causing the release of the enclosed drug.
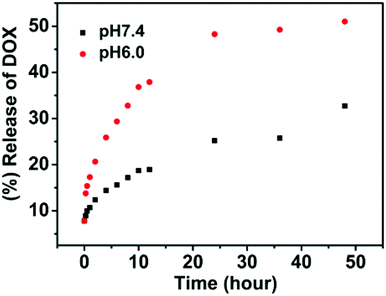 | ||
| Fig. 8 In vitro release of doxorubicin from SPU2 nanoparticles at different pH at 37 °C. | ||
Redox-responsive dissociation of the SPU nanoparticles
For polyurethanes, the main pathways of bond cleavage are dependent on hydrolysis with the degradation time ranging from weeks to months, which leads to limited applications as temporary biomaterials. A redox-responsive cleavage mechanism was used to develop rapid degradable materials in this system. Disulfide linkages were incorporated into the SPUs, which are sufficiently stable in the extracellular environment and in vivo circulation, but could be rapidly degraded under a reductive environment through disulfide cleavage on a time scale from minutes to hours. Thus the redox-responsive carriers can provide an accelerated dissociation of the carriers and more complete release of encapsulated agents in cancer cells.The size change of SPU1 nanoparticles in response to GSH was monitored by DLS measurements. Briefly, GSH was added to 1.5 mL of nanoparticle solution in PBS (10 mM, pH 7.4) to yield a final GSH concentration of 10 mM, which is a typical intracellular concentration. At different time intervals, the light-scattering intensity and the nanoparticle sizes were measured using DLS at 37 °C. The light-scattering intensity decreased sharply after treatment with 10 mM GSH for 5 min, while the SPU1 nanoparticles maintain similar scattering intensity for 1 h without the GSH treatment (Fig. 9A). In the presence of GSH, the quick dissociation of the nanoparticles and bimodal and broad size distribution were observed instead of uniform and narrow size distribution at the same temperature (37 °C) (Fig. 9B). TEM images for SPU1 nanoparticles treated with GSH are shown in Fig. 9C and irregularly shaped nanoparticles of varying sizes were found due to the dissolution of polymeric particles. As the disulfide bonds are broken, the particles dissociate and dissolve. As a result, the turbid solution changed to be transparent (Fig. 3). The bimodal and broad size distribution is probably due to the remaining debris of the dissociated particles. The result is in good agreement with the drastic decrease in scattering intensity.
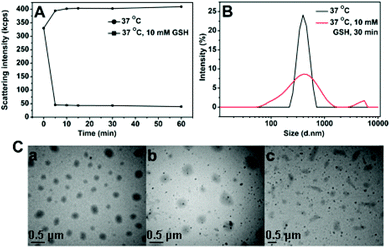 | ||
| Fig. 9 (A) Redox-triggered scattering intensity changes of SPU1 aqueous solution under different conditions. (B) Reduction-triggered changes of SPU1 nanoparticle size distributions. (C) TEM graphs of SPU1 nanoparticles treated with GSH in aqueous solution: (a) 37 °C without GSH, (b) 37 °C with 10 mM GSH for 1 h, (c) 37 °C with 10 mM GSH for 2 h. | ||
Along the same lines, the release of DOX from nanoparticles was investigated under the effect of glutathione. In Fig. 10, at a concentration of 10 mM glutathione, it showed a persistent increase over a long time. About 80% of DOX was released in 2 days. And only 32% release was observed without GSH over the same period of time. This is because as the disulfide cleaves under GSH, polymers are degraded and the encapsulated drugs are released. Note that the intracellular glutathione concentrations have been estimated to be as high as 10 mM, while the concentration is even higher in tumor cells. It suggests that the polymeric nanoparticles respond to redox stimulus well and are suitable to be used as redox-responsive release carriers.
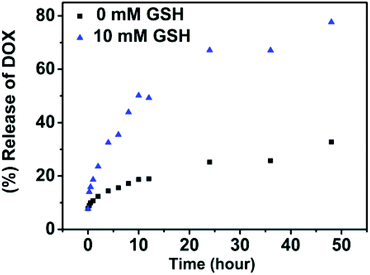 | ||
| Fig. 10 In vitro release of doxorubicin from SPU2 nanoparticles at different concentrations of GSH at 37 °C and pH = 7.4. | ||
The purpose of this study is to integrate multiple stimuli-responsive properties into a single macromolecular carrier, which could provide an opportunity to fine-tune the release kinetics of encapsulated agents and obtain a desired release profile at specific circumstances through the synergistic effect of these stimuli. First, the responsiveness of the nanoparticles to pH, temperature and redox potential has been confirmed independently. The release kinetics of DOX from the polymer nanoparticles was found to be incomplete or slow when a temperature, pH or redox potential stimulus was applied by itself. In this system, all these three stimuli-responsive functionalities were incorporated into a single particle. To test the synergistic effect of these stimuli, the DOX-encapsulating SPU2 nanoparticles were subjected to glutathione (10 mM) in acetate buffer (pH 6.0) at 37 °C. The release profile of DOX was monitored and plotted against time (Fig. 11). A dramatic increase in the release rate of DOX was observed and almost 100% of the drug was released within 48 h. In aqueous solution, poly(ether urethane)s are dispersed as nanoparticles, where HDI, MDEA and DiT segments serve as cores and PEG segments serve as hydrophilic shells. Hydrophobic drugs could be loaded into the core of the nanoparticles via hydrophobic interactions. When above Tp, the PEG segments dehydrate and shrink to the hydrophobic cores and the nanoparticles aggregate, and more disulfide bonds and ternary amino moieties are exposed to react with GSH and H+, respectively. The pH-induced particle disassembly would allow more GSH to go into the particles to react with disulfide linkages. All these accelerate the particle dissociation and cause a fast and complete drug release at higher temperature.
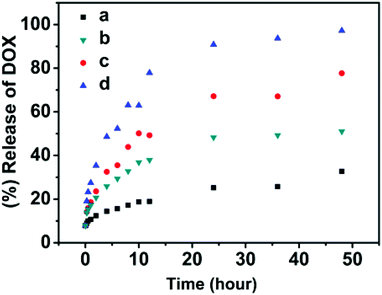 | ||
| Fig. 11 In vitro release of doxorubicin from the SPU2 nanoparticles under different conditions: (a) 37 °C, pH = 7.4; (b) 37 °C, pH = 6.0; (c) 37 °C, pH = 7.4, 10 mM GSH; (d) 37 °C, pH = 6.0, 10 mM GSH. | ||
Variable-temperature and -pH 1H NMR
To further verify the above interpretation, SPU2 was further investigated by variable temperature and pH 1H NMR. As shown in Fig. 12A, all of the signals were visible at temperatures from 25 to 55 °C, and moved to lower field with increasing temperature. This is caused by the gradual breakdown of polymer–water hydrogen bonding. The peaks (h) and (g) (end methylene groups in MDEA and DiT) became higher with increasing temperature. This predicates the freeze of PEG segments and the release of disulfides and ternary amino moieties as temperature increased. The broadening peaks (g), (i) and (k) in MDEA and DiT were observed at 37 °C, which was around its Tp. This suggests that these protons were in a complex environment during the transition process. And these peaks became sharp at 55 °C after the phase transition. The variable-pH 1H NMR spectra are shown in Fig. 12B. In the 1H NMR spectra at higher pH values such as pH = 11, all special peaks of MDEA and DiT in 1H NMR were negligible because MDEA and DiT segments were connected with hydrophobic segments leading to encapsulation in the particles. With the decrease in the pH value, the MDEA segments become hydrophilic due to the protonation of ternary amino moieties. Therefore, more MDEA signals are observed at lower pH values.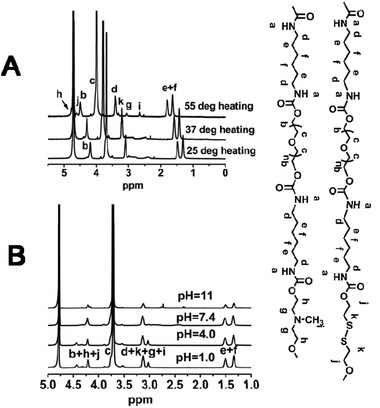 | ||
| Fig. 12 (A) Variable-temperature 1H NMR spectra of SPU2 at pH = 7.4. (B) Variable-pH 1H NMR spectra of SPU2 at 25 °C. | ||
On–off switchable drug release from the SPU nanoparticles
From the 1H NMR data, it can be determined that the amphiphilic SPU assemble into nanoparticles with responsive groups (tertiary amine and disulfide bond) buried inside the particles and PEG segments stretching outside when temperature is below the Tp. The two functionalities are slowly released with increasing temperature, especially when temperature is above the Tp. To evaluate drug-release applications, a stepwise temperature approach was employed to investigate the temperature-switchable drug-release behaviour of the multi-responsive degradable SPU2. In Fig. 13, experimental drug-release data from SPU2 (Tp, 35 °C) is displayed for a system in which the temperature was repeatedly switched between 37 °C (above the Tp) and 25 °C (below the Tp) under pH = 6.0 and 10 mM GSH. The resulting release profile has a clear on–off pulsatile character. It can be seen that the release (the slope of the total drug-release curve) was fast above Tp and negligible below Tp. The reason is that when the temperature is below the Tp the tertiary amine and disulfide bond are buried in the hydrophobic core, and it would take a long time for the H+ and GSH to enter the core of the particles to react with the responsive groups and the drug can only be released through the diffusion, which is very slow. However, when temperature is above the Tp the responsive groups will be released, leading to fast dissociation of the nanoparticles. Consequently, the encapsulated drug can be released quickly. The temperature switch of this polymer would allow fine spatiotemporal control of drug release: the drug is released only at the heating site, during the heating period. In synergy with the specific redox and pH conditions, it is possible to obtain a finely controlled and completely on-demand drug release.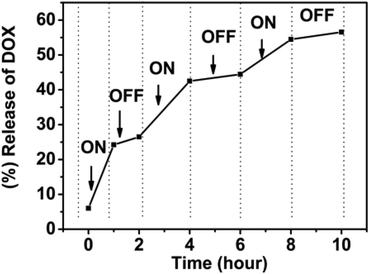 | ||
| Fig. 13 In vitro pulsatile release of doxorubicin from multi-responsive SPU2 nanoparticles under pH = 6.0, 10 mM GSH where the temperature is varied between 25 and 37 °C. | ||
Conclusions
A series of novel multi-segmented poly(ether urethane)s were synthesized via a facile one-pot method. They could self-assemble into nanoparticles in aqueous solution and could be used to encapsulate hydrophobic guest molecules. The poly(ether urethane) nanoparticles showed good response to temperature, pH and redox potential and the Tp of the obtained poly(ether urethane) nanoparticles can be precisely tuned by the length of PEG segments or the hydrophilic/hydrophobic segment ratios. The results showed this series of polymers could be used to fabricate nanoparticle platforms for repeated and effective on–off drug release, in which temperature was utilized as a switch to regulate the release of drug, while redox and pH-responsive behaviours were used to accelerate the drug release which only worked in the ‘on’ state. We envision that this temperature-controlled multi-responsive degradable polymeric system will lead to a new generation of on–off drug delivery vehicles and stimulate the development of unique and clinically applicable therapies.Acknowledgements
This work was financially supported by the NSFC (No. 51203079) and the PhD Programs Foundation for New Teachers of Education Ministry of China (No. 20090031120012).Notes and references
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- M. Biondi, F. Ungaro, F. Quaglia and P. A. Netti, Adv. Drug Delivery Rev., 2008, 60, 229–242 CrossRef CAS.
- S. Ghosh, J. Chem. Res., 2004, 2004, 241–246 CrossRef.
- S. Freiberg and X. X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS.
- S. You and W. Li, Med. Hypotheses, 2008, 71, 141–147 CrossRef CAS.
- R. K. Jain, Nat. Med., 2001, 7, 987–989 CrossRef CAS.
- J. J. T. Santini, A. C. Richards, R. Scheidt, M. J. Cima and R. Langer, Angew. Chem., Int. Ed., 2000, 39, 2396–2407 CrossRef CAS.
- A. C. R. Grayson, I. S. Choi, B. M. Tyler, P. P. Wang, H. Brem, M. J. Cima and R. Langer, Nat. Mater., 2003, 2, 767–772 CrossRef CAS.
- Y. Chen, R. Wang, J. Zhou, H. Fan and B. Shi, React. Funct. Polym., 2011, 71, 525–535 CrossRef CAS.
- T. Hoare, J. Santamaria, G. F. Goya, S. Irusta, D. Lin, S. Lau, R. Padera, R. Langer and D. S. Kohane, Nano Lett., 2009, 9, 3651–3657 CrossRef CAS.
- A. M. Alkilany, L. B. Thompson, S. P. Boulos, P. N. Sisco and C. J. Murphy, Adv. Drug Delivery Rev., 2012, 64, 190–199 CrossRef CAS.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS.
- M. Pernia Leal, A. Torti, A. Riedinger, R. La Fleur, D. Petti, R. Cingolani, R. Bertacco and T. Pellegrino, ACS Nano, 2012, 6, 10535–10545 CAS.
- N. S. Satarkar and J. Z. Hilt, J. Controlled Release, 2008, 130, 246–251 CrossRef CAS.
- S. A. Rovers, R. Hoogenboom, M. F. Kemmere and J. T. F. Keurentjes, Soft Matter, 2012, 8, 1623–1627 RSC.
- A. Chan, R. P. Orme, R. A. Fricker and P. Roach, Adv. Drug Delivery Rev., 2012 DOI:10.1016/j.addr.2012.07.007.
- Y. Cheng, J. Hao, L. A. Lee, M. C. Biewer, Q. Wang and M. C. Stefan, Biomacromolecules, 2012, 13, 2163–2173 CrossRef CAS.
- C. S. Brazel, Pharm. Res., 2008, 26, 644–656 Search PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- Y.-g. Jia, X. X. Zhu, L.-y. Liu and J. Li, Langmuir, 2012, 28, 4500–4506 CrossRef CAS.
- K. Wang, D.-S. Guo, X. Wang and Y. Liu, ACS Nano, 2011, 5, 2880–2894 CrossRef CAS.
- X. J. Loh, M.-H. Tsai, J. d. Barrio, E. A. Appel, T.-C. Lee and O. A. Scherman, Polym. Chem., 2012, 3, 3180–3188 RSC.
- J. Dong, Y. Wang, J. Zhang, X. Zhan, S. Zhu, H. Yang and G. Wang, Soft Matter, 2013, 9, 370–373 RSC.
- Z. Y. Qiao, R. Zhang, F. S. Du, D. H. Liang and Z. C. Li, J. Controlled Release, 2011, 152, 57–66 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS.
- H. Fu, H. Gao, G. Wu, Y. Wang, Y. Fan and J. Ma, Soft Matter, 2011, 7, 3546–3452 RSC.
- X. Sun, H. Gao, G. Wu, Y. Wang, Y. Fan and J. Ma, Int. J. Pharm., 2011, 412, 52–58 CrossRef CAS.
- Y. Wang, G. Wu, X. Li, J. Chen, Y. Wang and J. Ma, J. Mater. Chem., 2012, 22, 25217–25226 RSC.
- Y.-E. Fang and X. Zhao, J. Appl. Polym. Sci., 1998, 68, 75–82 CrossRef CAS.
- T. Ngai and C. Wu, Macromolecules, 2003, 36, 848–854 CrossRef CAS.
- A. Niu, C. Li, Y. Zhao, J. He, Y. Yang and C. Wu, Macromolecules, 2001, 34, 460–464 CrossRef CAS.
- W. Cao, Y. Li, Y. Yi, S. Ji, L. Zeng, Z. Sun and H. Xu, Chem. Sci., 2012, 3, 3403–3408 RSC.
- Y. Y. Choi, J. H. Jang, M. H. Park, B. G. Choi, B. Chi and B. Jeong, J. Mater. Chem., 2010, 20, 3416–3421 RSC.
- F. Wang, A. Klaikherd and S. Thayumanavan, J. Am. Chem. Soc., 2011, 133, 13496–13503 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- C. Y. Zhang, Y. Q. Yang, T. X. Huang, B. Zhao, X. D. Guo, J. F. Wang and L. J. Zhang, Biomaterials, 2012, 33, 6273–6283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm00188a |
| This journal is © The Royal Society of Chemistry 2013 |