Hydrogels formed by oxo-ester mediated native chemical ligation†
Iossif
Strehin
a,
Dmitri
Gourevitch
b,
Yong
Zhang
b,
Ellen
Heber-Katz
b and
Phillip B.
Messersmith
*a
aBiomedical Engineering Department, Materials Science and Engineering Department, Chemical and Biological Engineering Department, Chemistry of Life Processes Institute, Institute for Bionanotechnology in Medicine, Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Evanston, IL 60208, USA. E-mail: philm@northwestern.edu; Fax: +1 (847)491-4928; Tel: +1 (847)467-5273
bMolecular and Cellular Oncogenesis Program, The Wistar Institute, Philadelphia, PA 19104, USA
First published on 1st March 2013
Abstract
Oxo-ester mediated native chemical ligation (OMNCL) is a variation of the more general native chemical ligation reaction that is widely employed for chemoselective ligation of peptide fragments. While OMNCL has been used for a variety of peptide ligations and for biomolecular modification of surfaces, it is typically practiced under harsh conditions that are unsuitable for use in a biological context. In this report we describe the use of OMNCL for polymer hydrogel formation, in vitro cell encapsulation, and in vivo implantation. Multivalent polymer precursors containing N-hydroxysuccinimide (NHS) activated oxo-esters and N-cysteine (N-Cys) endgroups were chemically synthesized from branched poly(ethylene glycol) (PEG). Hydrogels formed rapidly at physiologic pH upon mixing of aqueous solutions of NHS and N-Cys functionalized PEGs. Quantitative 1H NMR experiments showed that the reaction proceeds through an OMNCL pathway involving thiol capture to form a thioester intermediate, followed by an S-to-N acyl rearrangement to yield an amide cross-link. pH and temperature were found to influence gelation rate, allowing tailoring of gelation times from a few seconds to a few minutes. OMNCL hydrogels initially swelled before contracting to reach an equilibrium increase in relative wet weight of 0%. This unique behavior impacted the gel stiffness and was attributed to latent formation of disulfide cross-links between network-bound Cys residues. OMNCL hydrogels were adhesive to hydrated tissue, generating a lap shear adhesion strength of 46 kPa. Cells encapsulated in OMNCL hydrogels maintained high viability, and in situ formation of OMNCL hydrogel by subcutaneous injection in mice generated a minimal acute inflammatory response. OMNCL represents a promising strategy for chemical cross-linking of hydrogels in a biological context and is an attractive candidate for in vivo applications such as wound healing, tissue repair, drug delivery, and tissue engineering.
Introduction
When it was first described in 1992,1 native chemical ligation (NCL) revolutionized peptide synthesis by providing a facile, chemoselective synthetic method for preparation of large peptides and functional proteins from short fragments. In NCL, an unprotected N-terminal cysteine (Cys) of one peptide reacts with the thioester-activated C-terminus of another peptide to form a thioester intermediate that rearranges via an S-to-N acyl migration to yield an amide bond linking the two fragments together (Scheme 1).2,3 NCL leads to minimal epimerization and byproduct formation and is highly selective to the N-terminal Cys, allowing the use of unprotected, post-translationally modified, and non-naturally occurring amino acids. NCL has been used to synthesize ion channel proteins such as KcsA,4 the plant protein crambin,3 glycosylated proteins such as monocyte chemotactic protein-3,5 and other difficult or otherwise impossible protein sequences.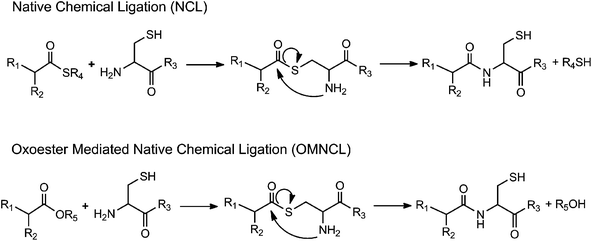 | ||
| Scheme 1 Generalized reaction schemes for native chemical ligation (NCL) and oxo-ester mediated native chemical ligation (OMNCL). | ||
Several research groups in the biomaterials community have explored NCL for preparation of functional materials.6–12 These studies focused on the use of NCL for synthesis of collagen mimetic biomaterials,9 chemical modification of polymers10,11 and self-assembled peptide scaffolds,8 and modification of substrate surfaces.6 In principle, the chemoselectivity of NCL is attractive for in vitro and in vivo use, allowing chemical reactions to proceed with specificity in a complex biological milieu, preserving the bioactivity of endogenous compounds and facilitating the targeting of therapeutic or diagnostic molecules to specific biomolecular targets such as cell surface proteins and components of the extracellular matrix. Our group has been developing polymer hydrogels cross-linked via NCL,7,12 for potential in vitro and in vivo applications. The general strategy involves the reaction of a thioester-derivatized polymer with a second polymer containing N-terminal Cys residues. Mixing of the two polymer precursors under mild aqueous conditions led to gel network formation via NCL without the need for added catalysts.7 Later, we extended this strategy to the formation of gels for in vitro cell encapsulation, incorporating polymer-bound IL-1 receptor inhibitory peptides that provided an immunoprotective effect to entrapped insulin secreting cells.12
Despite these recent advances, several aspects of the NCL reaction remain challenging for use in a biological setting. For example, standard NCL conditions employ the use of strong reducing agents that may be harmful in living systems. Furthermore, the slow rate of NCL cross-linking,7 the hydrolytic instability of the thioester, and the adverse biological effects of the thiol leaving group13 remain obstacles to future in vivo applications of NCL.
Several modifications of the NCL reaction have been introduced in an effort to expand the utility of the method.14–16 Danishefsky and coworkers described the use of oxo-esters in NCL (Scheme 1), first through an indirect approach involving o-thiophenolic ester17 and followed later by a direct approach utilizing p-nitrophenyl (pNP) activated C-terminal ester.18 Termed oxo-ester mediated NCL (OMNCL), this approach enables high efficiency reactions even with bulky C-terminal amino acids, although disadvantages include hydrolytic susceptibility of the pNP ester and challenges associated with direct solid phase synthesis of pNP ester peptides.19 Weissenborn et al. described OMNCL on oxo-ester activated surfaces and found 2,3,4,5,6-pentafluorophenyl (PFP) to be more efficient than pNP and N-hydroxysuccinimide (NHS) activating agents.20
Here we describe polymer hydrogel formation via OMNCL between branched polymer precursors containing NHS activated ester and N-Cys endgroups.21 The focus of this initial report was to elucidate the cross-linking mechanism and to provide preliminary assessments of mechanical properties, cytocompatibility and in vivo acute inflammatory response. Mixing of NHS and N-Cys polymer precursors led to gel formation within seconds, and quantitative NMR studies revealed the cross-linking mechanism to be OMNCL. In addition to characterizing the bulk mechanical and adhesive properties of the hydrogels, we performed the first in vitro and in vivo studies of OMNCL hydrogels, showing favorable biological response in cytotoxicity assays and in a subcutaneous implant model. The OMNCL hydrogel strategy overcomes many of the earlier limitations of NCL, including cytotoxicity of thiol leaving groups and slow reaction kinetics, and represents a promising strategy for chemical cross-linking of hydrogels in a biological context.
Materials and methods
Materials
PEG-OH (8 Arm, MW 20082) and PEG-NH2 (8 Arm, MW 19715) were purchased from JenKem Technology USA Inc. (Allen, TX, USA). Glutaric anhydride, N,N-diisopropylethylamine (DIEA), 1,2-ethanedithiol (EDT), triisopropylsilane (TIS), L-cysteine (L-Cys), S-methyl-L-Cysteine, N-acetyl-L-Cysteine, glutaric acid, dibasic sodium phosphate (Na2HPO4), potassium chloride (KCl), sodium chloride (NaCl), N-hydroxysuccinimide (NHS) and ethidium homodimer-1 were purchased from Sigma (St. Louis, MO, USA). The coupling reagent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) was purchased from TCI America (Portland, OR, USA). Boc-Cys(Trt)-OH and Calcein AM were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (BOP) was purchased from Advanced ChemTech (Louisville, KY, USA). Porcine pericardium was purchased from Animal Technologies, Inc. (Tyler, TX, USA). The 3T3-swiss albino fibroblasts were purchased from the American Type Culture Collection center (Manassas, VA, USA). Trifluoroacetic acid (TFA) and β-mercaptoethanol were purchased from Merck KGaE (Darmstadt, Germany). Hematoxylin and eosin were purchased from Leica Microsystems (Buffalo Grove, IL, USA). Picro-sirius red was purchased from Poly Scientific (Bay Shore, NY, USA). Monobasic potassium phosphate (KH2PO4) and permount mounting medium were purchased from Fisher Scientific (Waltham, MA, USA). Dulbecco's phosphate buffered saline (pH 7.0) without calcium or magnesium (Life Technologies, Grand Island, NY, USA) was used to prepare hydrogels and included 2.67 mM KCl, 137.93 mM NaCl, 1.47 mM KH2PO4 and 8.06 mM Na2HPO4 (PBS). Hydrogels prepared with this buffer and used in vitro were incubated in excess PBS (pH 7.0) or culture medium following gelation (not more than 15 minutes of gelation). NMR, pH measurements, and part of the gelation kinetics experiments were done with phosphate buffer (100 mM PBS) which was prepared from PBS substituted with additional KH2PO4 and Na2HPO4 and included 2.67 mM KCl, 137.93 mM NaCl, 15 mM KH2PO4 and 85 mM Na2HPO4. Other concentrations of PBS were prepared by diluting 100 mM PBS with saline (2.67 mM KCl, 137.93 mM NaCl).Synthesis of glutaric acid terminated 8 arm PEG (P8G)
Glutaric acid terminated PEG was synthesized by dissolving 8-arm PEG-OH (20 g, 7.97 mmol OH) and glutaric anhydride (4.54 g, 39.84 mmol) in chloroform (20 mL). Pyridine (3.21 mL, 39.84 mmol) was added dropwise, and the reaction mixture was refluxed at 80 °C for 24 hours under inert air. The product was diluted with MeOH (240 mL), precipitated at −20 °C for 1 hour, and centrifuged at −5 °C. The supernatant was discarded, and the MeOH wash procedure was repeated twice more. Following diethyl ether precipitations (180 mL), the product was dried under high vacuum overnight to afford a white powder (97.6% yield, 100% conversion). 1H NMR (500 MHz, CDCl3), δ, ppm: δ 4.24 (2H, t, terminal PEG CH2), δ 3.64 (1823H, m, backbone PEG CH2), δ 2.43 (2H, t, C2 of glutaric acid), δ 2.39 (2H, t, C4 of glutaric acid), δ 1.95 (2H, p, C3 of glutaric acid).Synthesis of N-hydroxysuccinimide terminated 8 arm PEG (P8NHS)
NHS terminated 8 arm PEG was synthesized by dissolving P8G (20 g, 7.62 mmol), NHS (8.77 g, 76.20 mmol) and EDC (14.61 g, 76.20 mmol) in DMSO (50 mL). After 30 minutes of stirring at room temperature, the product was diluted with MeOH (500 mL), precipitated at −20 °C for 1 hour, and spun down at −5 °C. The supernatant was discarded, and the MeOH wash procedure was repeated twice more. Following diethyl ether precipitations (480 mL), the product was dried under high vacuum overnight to afford a white powder (80.6% yield, 97.4% conversion). 1H NMR (500 MHz, CDCl3), δ, ppm: δ 4.24 (2H, t, terminal PEG CH2), δ 3.64 (1961H, m, backbone PEG CH2), δ 2.84 (4H, m, NHS protons), δ 2.71 (2H, t, C4 of glutaric acid), δ 2.49 (2H, t, C2 of glutaric acid), δ 2.06 (2H, p, C3 of glutaric acid).Synthesis of cysteine terminated 8 arm PEG (P8Cys)
Cysteine terminated 8 arm PEG was synthesized using BOP as a coupling reagent. In one reaction vessel, PEG-NH2 (13.4 g, 5.44 mmol NH2) was dissolved in DMF (25 mL) and DIEA was added dropwise (947 μL, 5.44 mmol). In a separate reaction vessel, Boc-Cys(Trt)-OH (10.0 g, 21.75 mmol) and BOP (9.62 g, 21.75 mmol) were dissolved in DMF (25 mL) and DIEA (3.79 mL, 21.75 mmol) was added dropwise. Five minutes after adding DIEA, the BOP solution was combined with the PEG solution and the coupling reaction was allowed to proceed at room temperature for 18 hours. Following precipitation in cold diethyl ether (280 mL), the product was re-dissolved in MeOH (70 mL) and precipitated in cold diethyl ether once more (280 mL). The cysteine was deprotected with TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIS:EDT (210 mL, 95:2.5:2.5) cleavage solution at room temperature for 4 hours. TFA was evaporated under low pressure, and the product was precipitated in cold diethyl ether (160 mL). P8Cys was dissolved in MeOH (120 mL), precipitated at −20 °C overnight, and centrifuged at −5 °C. The supernatant was decanted and the MeOH precipitation was repeated twice more. Following diethyl ether precipitations (120 mL), the product was dried under high vacuum overnight to afford a white powder (50% yield, 86% endgroup conversion). 1H NMR (500 MHz, acetic acid-d4), δ, ppm: δ 4.42 (1H, t, α-C cysteine), δ 3.69 (1872H, m, backbone PEG CH2), δ 3.13 (2H, d, CH2 cysteine).
:TIS:EDT (210 mL, 95:2.5:2.5) cleavage solution at room temperature for 4 hours. TFA was evaporated under low pressure, and the product was precipitated in cold diethyl ether (160 mL). P8Cys was dissolved in MeOH (120 mL), precipitated at −20 °C overnight, and centrifuged at −5 °C. The supernatant was decanted and the MeOH precipitation was repeated twice more. Following diethyl ether precipitations (120 mL), the product was dried under high vacuum overnight to afford a white powder (50% yield, 86% endgroup conversion). 1H NMR (500 MHz, acetic acid-d4), δ, ppm: δ 4.42 (1H, t, α-C cysteine), δ 3.69 (1872H, m, backbone PEG CH2), δ 3.13 (2H, d, CH2 cysteine).
NMR analysis of reaction between L-Cys and P8NHS
All reactions were done at room temperature. First, 100 mM PBS (pH adjusted to either 6.0 or 7.0) was lyophilized, redissolved in the same volume of D2O, and then used to prepare solutions of L-cysteine (57 mM) and P8NHS (10 w/v%, 38 mM NHS ester). The two solutions were mixed in a 1:1 v/v ratio and reaction kinetics followed using 1H NMR in a Varian Inova 500 MHz NMR. Similar experiments were carried out in pure D2O as described above by mixing solutions of L-Cys (29 mM) and P8NHS (1.25% w/v, 5 mM NHS ester).
The relative abundance (RA) of polymer species during the OMNCL reaction between P8NHS and L-Cys was determined by integrating the triplets associated with the protons bound to the C2 and C4 carbons of the terminal glutarate linker. In this reaction four polymer species are possible (P8NHS, P8G, P8G-TE-Cys, P8G-AM-Cys), each possessing slightly different chemical shifts of the C2 and C4 protons depending on the composition of the terminal group (Fig. S1 and Table S1†). The chemical shifts for the protons associated with the C2 and C4 carbons of glutarate of P8G and P8NHS were determined from spectra of the synthesized intermediate and final product (see above), whereas the chemical shifts of thioester (P8G-TE-Cys) and amide (P8G-AM-Cys) linked products were estimated from spectra obtained by reaction of P8NHS with N-acetyl-L-cysteine and S-methyl-L-cysteine, respectively. Integrated peak values were used in the equations below to calculate the relative abundance of each species present during the reaction of P8NHS and L-Cys.
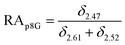 | (1) |
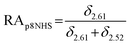 | (2) |
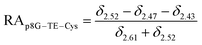 | (3) |
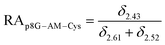 | (4) |
pH measurement
PBS (100 mM PBS and dilutions of the buffer) was used to prepare 38 mM L-cysteine and 10% (w/v) P8NHS (38 mM NHS ester). The two solutions were mixed in a 1:1 v/v ratio and the pH of the reaction mixture was followed over time using a standard pH meter (Accumet Titration Controller, Model 150, Fisher Scientific, Waltham, MA, USA) and electrode (Beckman 511275-AB Electrode, Beckman, Pasadena, CA, USA).
Gelation time
The time to form a hydrogel was measured using a previously described protocol.22 Briefly, 10% (w/v) P8NHS and 10% (w/v) P8Cys were prepared in phosphate buffered saline (PBS, 100 mM PBS, or dilutions of 100 mM PBS). The two solutions were then mixed in a 1:1 (v/v) ratio and pipetted up and down using a standard 2–200 μL pipette tip. The time at which the material blocked the pipette tip was designated as the gelation time. Temperature was controlled within the range 4–60 °C through the use of a water bath.
Hydrogel swelling
PBS was used to prepare 10% (w/v) P8NHS and 10% (w/v) P8Cys and the two solutions were mixed in a 1:1 (v/v) ratio to form cylindrical 70 μL hydrogels (5 mm diameter × 3.5 mm height). The gels were allowed to set for 15 minutes and then transferred to PBS. The wet weights of the gels were measured at various time points, and buffer was replaced at least 5 times a week. At 5 hours, half of the hydrogels (n = 5) were exposed to reducing agent (0.2 M β-mercaptoethanol in PBS) and swelling was monitored until equilibrium was reached for both groups. At 303 hours the hydrogels equilibrated in PBS were exposed to reducing agent and swelling was monitored until 447 hours, after which the hydrogels were transferred back to PBS and swelling was followed until equilibrium was reached. The hydrogel swelling experiments were all carried out under room temperature and with five replicates per group. Relative wet weight was calculated as defined in eqn (5), where w0 is the initial wet weight and wt is the wet weight of the hydrogel at time t.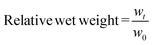 | (5) |
Mechanical testing
The compressive moduli and adhesive strength of the hydrogel (10% w/v, 1:1 w/w P8NHS:P8Cys, prepared in PBS) were measured at room temperature with a Syntech Model # 20G screw actuation testing machine equipped with 250 g and 1000 lb load cells. Compressive moduli were measured using cylindrical hydrogels (6.5 mm height × 8.5 mm diameter) that were swollen for 5, 75 or 150 hours in PBS. The hydrogels were compressed along their axes between two flat plates, and the equilibrium stress was recorded for strains between 1 and 10%. The Young's modulus was calculated by measuring the slope of the linear portion of the stress vs. strain curve.
The adhesive strength of the hydrogel was measured using a lap shear test adapted from ASTM standard F2255-05. Unprocessed porcine pericardium (2.5 cm × 3 cm) was adhered to aluminum fixtures using a cyanoacrylate based adhesive and then covered with a PBS moistened paper. After 1 hour, 100 μL of hydrogel precursor was placed on one tissue surface and a second tissue surface brought into contact (2.5 cm × 1.25 cm overlap) such that the tissue sections were glued together. After 10 minutes of curing, the tissue and glue were covered with PBS moistened paper towel for an additional 50 minutes. The glued tissue was pulled apart at 5 mm min−1 in tensile shear, and the peak stress and tissue overlap area were used to calculate the adhesive strength of the material.
Cytotoxicity
Cytotoxicity was evaluated using two methods. For the first assay, the guidelines found in ISO standards 10993-05 and 10993-12 were used. Briefly, 3T3 fibroblasts were exposed to polymer precursor or hydrogel extract. Extracts were prepared by suspending a 200 μL hydrogel in 1 mL of culture medium (n = 3); alternatively, P8NHS was dissolved directly in culture medium to yield a 5% (w/v) solution (n = 3). These solutions were then incubated at 37 °C, 5% CO2 and >90% RH for 24 hours, diluted to various concentrations using fresh medium, and 100 μL of each dilution added to a subconfluent monolayer of 3T3 fibroblasts in 96 well plates (n = 3). The cells were incubated in the presence of the conditioned medium at 37 °C, 5% CO2 and >90% RH for 24 hours. After washing with PBS the cells were exposed for 3 hours to 0.4% neutral red prepared in DMEM. The cells were again washed with PBS and destained using 1% glacial acetic acid, 50% ethanol and 49% ddH2O. Following 10 minutes of agitation, absorbance at 540 nm was used to quantify viability. Culture medium and 0.2% SDS were used as a negative and positive control and were incubated along with the extracts at 37 °C, 5% CO2 and >90% RH for 24 hours prior to addition to the subconfluent monolayer of cells. Per the requirements stated in the ISO standard, IC50 of the positive control SDS was found to be within the acceptable range, confirming the validity of the assay.In the second viability assay, cells were suspended in the P8Cys component dissolved in PBS and mixed with the P8NHS component dissolved in PBS to yield a 7% (w/v) hydrogel containing 1:1 (w/w) ratio of P8Cys to P8NHS. The hydrogels were allowed to set for 1 to 2 minutes and then incubated in culture medium at 37 °C and 5% CO2. After 24 hours, cell viability was quantified using calcein AM and ethidium homodimer-1. Cells were stained for 15 minutes in culture medium substituted with 4 μM calcein AM and 4 μM ethidium homodimer-1. Images were acquired using a fluorescent microscope equipped with a 485 ± 10 nm optical filter for calcein AM (live cells) and a 530 ± 12.5 nm optical filter for ethidium homodimer-1 (dead cells). The images were merged and processed using ImageJ (National Institute of Health, Bethesda, MD).
In vivo studies
B10.BR male mice were obtained from the Jackson Laboratories (Bar Harbor, ME, USA). The mice were housed in the Animal Facility of the Wistar Institute and all treatments were approved by the Wistar IACUC. At 10 weeks of age, mice were injected subcutaneously at the base of the neck with 100 μL of 10% (w/v) 1:1 (w/w) ratio of P8Cys to P8NHS hydrogel prepared in PBS. Six weeks later, mice were euthanized and tissue together with the hydrogel were removed and fixed in 4% buffered formalin for 24 hours. Tissue was then washed, dehydrated through serial ethanol washes, cleared with xylene and embedded in paraffin overnight. Tissue sections of 5–10 μm thickness were cut and mounted on superfrost slides (VWR, Radnor, PA, USA).
Tissue sections were cleared, rehydrated, and then stained with hematoxylin and eosin or with picro-sirius red, dehydrated, cleared with xylene and coverslipped with Permount mounting media. Staining was visualized using an Olympus (AX70) microscope (Olympus America, Center Valley, PA, USA) in bright field for H&E and under polarized light for Picro-Sirius Red. Images were recorded using a Spot camera with bounded software.
Statistical analysis
One-way ANOVA was used to detect significant effects among groups. Tukey's multiple comparison tests were used to detect significant differences between groups, and a p-value ≤ 0.05 was considered significant.Results
Polymer synthesis and hydrogel formation
P8NHS was synthesized with an overall yield of ∼95% through a two-step reaction involving addition of a glutaric acid linker to an 8-arm PEG followed by activation of terminal acid groups with NHS. P8Cys was synthesized in one step from amine terminated 8-arm PEG and N,S-protected Cys amino acid followed by deprotection and purification for a yield of 50%. Within ∼20 seconds of mixing P8Cys and P8HNS the solution became noticeably viscous and solidified to form a stiff hydrogel (Fig. 1). Under the same conditions, a hydrogel formed within ∼240 seconds when P8Cys was replaced with the 8-arm PEG-NH2 polymer used to synthesize P8Cys, suggesting the reaction mechanism between P8Cys and P8NHS involves thiol capture followed by a S-to-N acyl rearrangement (OMNCL) rather than a direct reaction between the activated ester of P8NHS and the terminal amine of P8Cys.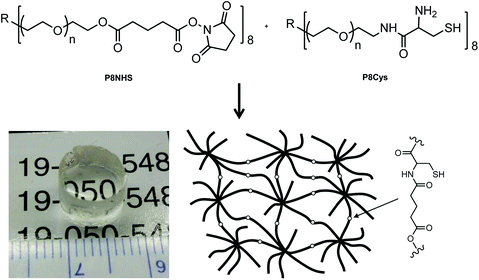 | ||
| Fig. 1 The two polymer precursors P8NHS and P8Cys react in aqueous solution via OMNCL to yield polymer hydrogels with network cross-links as shown at bottom right. | ||
Mechanism of hydrogel formation
In model experiments designed to elucidate the reaction mechanism, the buffered and unbuffered reaction between P8NHS and L-Cys were analyzed by 1H NMR. We determined the relative abundance of four major polymer species with time during the reaction: the NHS activated polymer precursor (P8NHS), hydrolysis product (P8G), the thioester formed by thiol capture of L-Cys by P8NHS (P8G-TE-Cys), and the amide linked product (P8G-AM-Cys) (Fig. 2A). Analysis of the 1H NMR spectra acquired for up to 120 h after mixing revealed clear chemical shift changes corresponding to the appearance and disappearance of the reactants, intermediates and products (Fig. S2†). Spectral analysis using peak assignments obtained from reference spectra (Fig. S1†) and calculation of relative abundance according to eqn (1)–(4) revealed the temporal progression of the reaction (Fig. 2B).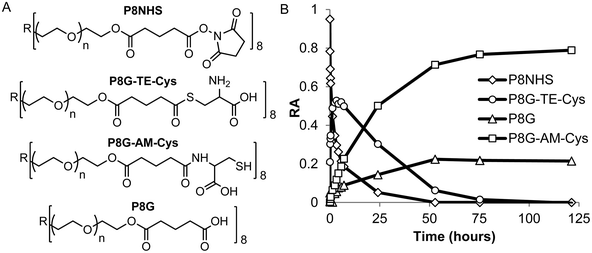 | ||
| Fig. 2 Quantitative 1H NMR analysis of the model reaction between P8NHS and L-Cys in D2O. Chemical structures (A) and relative abundance (B) of polymer species observed during the reaction. | ||
Upon mixing P8NHS and L-Cys, rapid disappearance of P8NHS was accompanied by rapid emergence of the thioester intermediate P8G-TE-Cys. P8G-TE-Cys reached a maximum value after several minutes and slowly started to disappear thereafter and was found in only trace amounts after 75 h (Fig. 2B). Concomitant with disappearance of P8G-TE-Cys, the amide linked product P8G-AM-Cys emerged slowly over the first hour and represented over ∼80% of species present in the reaction mixture after 75 h. P8G appeared in minor but detectable amounts, present at less than ∼20% at all time points. Similar trends were observed when the reaction of P8NHS and L-Cys was performed in the presence of phosphate buffer at pH 6, albeit with significantly faster kinetics (Fig. 3A and Fig. S3†). At pH 7, the reaction was essentially complete within 5 minutes (Fig. 3B). Under the same conditions, control experiments showed that hydrolysis of P8NHS was insignificant within this timeframe (Fig. S4†). At pH 7, when L-Cys was replaced with S-methyl-L-cysteine, the reaction was significantly slower (Fig. 3C) despite the lower pKa of the amino group of S-methyl-L-cysteine (8.7523vs. 10.78 for L-Cys23). In a separate experiment at pH 7, P8NHS was reacted with equal concentrations of both L-Cys and L-Gly (amine pKa 9.623), yielding 80% P8G-AM-Cys, 15% P8G-AM-Gly and 5% P8G (Fig. S5†). Interestingly, mixtures of 10% (w/v) P8NHS (38 mM NHS ester) and 19 mM L-Cys were observed to form stable gels after 2 hours of incubation (100 mM PBS, pH 7.0), although these gels liquefied in the presence of β-mercaptoethanol but not PBS (Fig. S6†), implying cross-linking via disulfide bond formation.
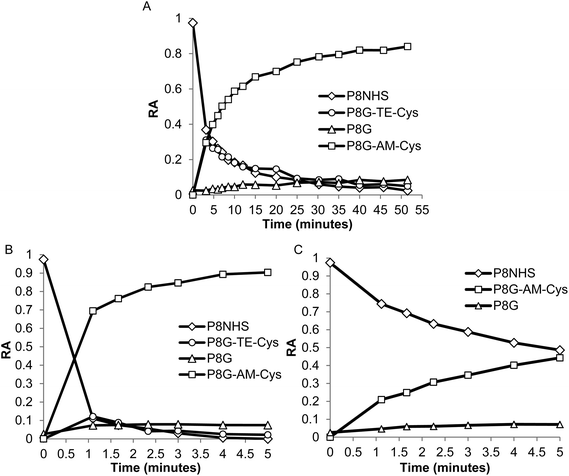 | ||
| Fig. 3 Quantitative 1H NMR analysis of the reaction between P8NHS and L-Cys in buffered D2O. (A, B) Relative abundance of polymer species formed during reaction of P8NHS with L-Cys at (A) pH 6.0 and (B) pH 7.0, indicating that the reaction proceeds more quickly at higher pH. (C) Relative abundance of polymer species formed during the reaction of P8NHS with S-methyl-L-cysteine at pH 7.0, illustrating significantly slower reaction kinetics when the thiol group is protected. | ||
Physical characterization of hydrogels
Buffer concentration, pH and temperature were found to strongly influence the rate of gel formation. As the phosphate buffer concentration (initial pH 7.3) was increased from 10 to 80 mM, gel time decreased from ∼28 s to less than 9 s, respectively (Fig. S7†). As we could not measure the pH within the hydrogel itself, instead we measured the pH of a P8NHS–L-Cys reaction mixture under equivalent conditions in order to ascertain pH changes during the reaction. As shown in Fig. S8† the pH was found to decrease with time during the reaction. Reaction solutions with higher buffer concentration yielded smaller pH changes, with buffer concentrations of 80 mM exhibiting a pH drop of 0.8 pH units during the reaction. Hence, higher buffer concentration led to faster gelation and a lower drop in pH, implying that the reaction is accelerated at high pH.These findings led us to select a buffer concentration of 100 mM for gel kinetic studies under different pH conditions. The pH dependence of gelation time for a 10% w/v mixture of P8NHS and P8Cys in 100 mM PBS is shown in Fig. 4. It can be seen in this figure that gel time changed significantly within the pH range 6–8, ranging from ∼50 s at pH 6 to <10 s at pH 8. Finally, we determined the temperature dependence of gelation time, which showed that gel formation was accelerated by ∼9 s when the temperature was changed from room to body temperature (Fig. S9†).
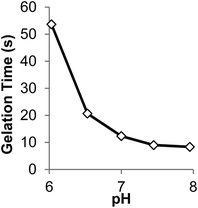 | ||
| Fig. 4 The effect of initial pH on gelation time of 10% w/v hydrogels prepared in 100 mM PBS at room temperature with P8Cys and P8NHS (1:1 w/w). | ||
Several types of mechanical characterizations were undertaken on hydrogels formed from P8NHS with P8Cys. Swelling experiments were performed by immersing hydrogel samples in excess PBS and measuring the weight changes as a function of time. OMNCL hydrogels increased in relative wet weight by approximately 21% in the first several hours and then slowly contracted over a period of many hours to a final increase in relative wet weight of approximately 0% (Fig. 5A). The young's modulus of the hydrogel was measured at 5, 75 and 150 h and was found to increase from 128 to 182 kPa during this time (Fig. 5B). The latent modulus increase is unlikely to be attributed to additional cross-linking by the OMNCL mechanism, as model 1H NMR studies showed that at pH 7 the reaction was mostly complete after 5 minutes (Fig. 3B). The observed swelling and modulus changes are unlikely to reflect mass changes induced by hydrolytic degradation of the gels, as NMR analysis of ester hydrolysis (Fig. S10†) and a preliminary evaluation of gel degradation at pH 7.0 showed little mass loss over a 12 week period (Fig. S11†).
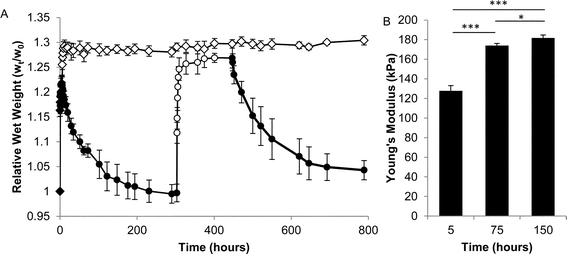 | ||
| Fig. 5 Physical characterization of OMNCL hydrogels formed by mixing equal volumes of 10% (w/v) P8NHS and 10% (w/v) P8Cys in PBS. (A) Swelling of OMNCL hydrogels in 10 mM PBS (closed symbols) or 10 mM PBS substituted with 0.2 M β-ME (open symbols). The two sets of hydrogels (diamonds and circles, n = 5 per set) varied by the sequence in which they were incubated in PBS or β-ME. In one case (circles), the hydrogels were incubated in PBS followed by β-ME and then PBS again. In the second case (diamonds), the hydrogels were incubated in PBS for the first few hours and thereafter in β-ME. (B) Young's moduli (n = 4) at various time points for OMNCL hydrogels incubated in PBS. *p < 0.05, ***p < 0.001. | ||
Suspecting therefore that gel shrinkage was the result of disulfide bond formation, fully equilibrated hydrogels (>300 hours swelling in PBS) were transferred into 0.2 M β-mercaptoethanol in PBS, whereupon the hydrogels increased in relative wet weight by approximately 27% (Fig. 5A). After approximately 140 hours in reducing agent, swollen samples were transferred back into PBS and swelling was observed to decrease once again, implying the re-formation of disulfide bonds.
Finally, the adhesive strength of the OMNCL hydrogel to unprocessed porcine pericardium was measured in lap shear similar to the protocol described in ASTM standard F2255-05. Tissue surfaces were glued together using the OMNCL hydrogel and then pulled apart after one hour post gelation, yielding a lap shear adhesive strength of 46 ± 8 kPa.
Cytotoxicity
The cytotoxicity of the hydrogel was tested using two methods. The first method was conducted in accordance with ISO standards 10993-5 and 12, from which it was concluded that the OMNCL hydrogel as well as P8HNS polymer (5% w/v solution) were not toxic to cells as viability remained well above 90% (Fig. 6A). It was not possible to analyze P8Cys precursor in this cytotoxicity assay, as a 5% (w/v) solution of P8Cys in cell culture medium solidified within 3 hours, presumably via disulfide bond formation. In the second test, cells were encapsulated in OMNCL hydrogels and viability was quantified after 24 hours using calcein AM and ethidium homodimer-1. In this assay we observed that 87 ± 7% of the encapsulated cells remained viable 1 day following encapsulation (Fig. 6B).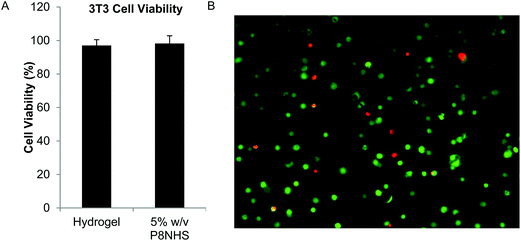 | ||
| Fig. 6 In vitro cytocompatibility of OMNCL hydrogels. (A) Quantitative analysis of 3T3 fibroblast viability after 24 hours in conditioned medium, conducted in accordance with ISO standards 10993-05 and 10993-12. Cell culture medium included either extract from P8NHS/P8Cys hydrogel or 5% w/v P8NHS. (B) 3T3 fibroblasts encapsulated in OMNCL hydrogels and stained with calcein AM (green, live cells) and ethidium homodimer-1 (red, dead cells). Image analysis indicated 87 ± 7% of cells remained viable after 24 hours of encapsulation. | ||
In vivo studies
Subcutaneous injections of OMNCL hydrogel were administered to B10.BR male mice, and after 6 weeks the implants were extracted and processed for histological analysis. The results showed the remnants of an acute inflammatory response typical of PEG based hydrogels. H&E stained histological sections showed that the surrounding tissue was intact. A low number of inflammatory cells were observed in the vicinity of the implant, and a thin fibrous capsule formed around the hydrogel (Fig. 7A,B), as indicated by positive staining for collagen fibrils with Picro-Sirius red under polarized light illumination (Fig. 7C).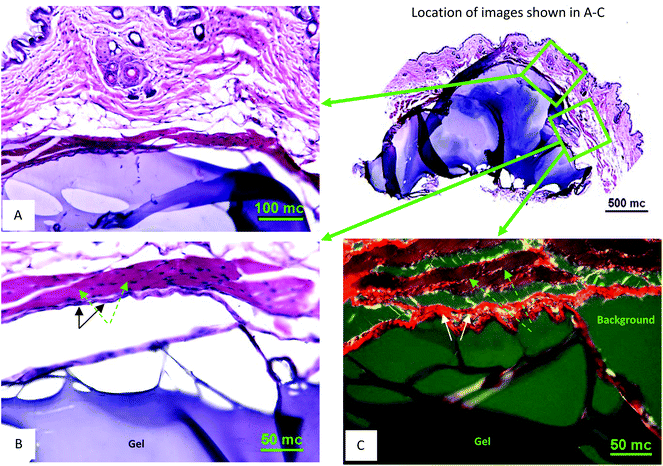 | ||
| Fig. 7 In vivo subcutaneous characterization of OMNCL hydrogel. (A) H&E stained tissue section at 20× magnification with gel associated with the outer skin. The gel is stained blue and surrounding tissue stained blue and red (an overview of the skin-gel injection area is shown on the right at 4× magnification). (B) H&E stained tissue section at 40× magnification from sequentially obtained tissue sections, showing the gel (blue body at the bottom), subtle fibrous capsule (marked with black arrows) and supra-capsular muscle layer (purple-red, marked with green arrows). (C) Picro-Sirius Red stained section (40× magnification) obtained from the same area as B. Hydrogel is at the bottom; the capsule surrounding the hydrogel is a bright-red fibrous structure (marked with white arrows) and muscle mass shown in brown-red (marked with green arrows). The scale bars indicate length in micrometers (mc). | ||
Discussion
In this report we have detailed the synthesis, reaction mechanism and characterization of a two-component hydrogel formulation that sets rapidly by OMNCL at physiologic pH and ionic strength. The OMNCL reaction involves covalent capture of the thiol side chain of N-terminal Cys to initially form a thioester bond. The proximity and orientation of the primary amine in relation to the thioester leads to a S-to-N acyl rearrangement to form the more stable amide bond. This mechanism of amide bond formation via a rearrangement is similar to native chemical ligation (NCL),24 with the exception that in OMNCL a oxo-ester is employed as opposed to a thioester (Scheme 1).18Implementation of NCL in a biological context can be problematic due to the adverse biological effects of the buffers and reducing agents typically employed in NCL reactions. Furthermore, we have shown that the small molecule thiol leaving group liberated during NCL can be toxic to cells.13 OMNCL, on the other hand, is often practiced in solutions containing concentrated guanidine (5–6 M),18–20 which would have adverse effects in a biological system because of its strong denaturing potential. Our results show that hydrogel formation by OMNCL proceeds well in phosphate buffer, achieving rapid gel formation without the use of catalysts or additives. In the pH range of our experiments (6–7.3), the rate of gel formation by OMNCL was adjustable through pH control and ranged from several seconds to less than a minute. This is in contrast to gels formed by NCL chemistry,7,12 which have slower gelation rates at physiological pH.
We elucidated the mechanism of cross-linking by following the reaction of P8NHS and L-Cys by 1H NMR (Fig. 2, 3A,B), taking advantage of chemical shift differences to reveal the temporal evolution of reactants, intermediates and products. Several lines of evidence from these studies point to cross-link formation via OMNCL. First, the rapid increase in thioester cross-link in parallel with the rapid decrease in P8NHS at the beginning of the reaction is indicative of thiol capture. The thioester intermediate reaches as high as >50% relative abundance within the first few minutes but then decreases until it is no longer detectable. This decrease cannot be explained by thioester hydrolysis, as the hydrolysis product (P8G) was never present at greater than ∼10% relative abundance at pH 6 or 7. Second, the kinetics of thioester cross-link disappearance was roughly matched with the kinetics of amide cross-link emergence, which is a strong indicator of S-to-N rearrangement. It should be noted that at neutral and acidic pH the thiol should be considerably more reactive than the primary amino group due to the high pKa of the terminal amine of L-Cys (pKa ∼ 10.7823). As evidence of this, in the reaction between P8NHS and L-Cys at neutral pH, conversion to P8G-AM-Cys and P8G-TE-Cys was 83% within the first minute. However, reaction of P8NHS with S-methyl-L-Cys yielded only 20% conversion under the same conditions, despite the significantly lower pKa of the S-methyl-L-Cys amino group (pKa ∼ 8.75)23 compared to L-Cys. Finally, further support for OMNCL pathway was provided by a competitive reaction between P8NHS, L-Cys and L-Gly, where 80% of the reaction proceeded with L-Cys and only 15% with L-Gly despite the lower amino pKa of L-Gly (pKa ∼ 9.623).
Taking these findings into consideration, we therefore propose the OMNCL reaction pathway shown in Scheme 2 for the gel-forming reaction between P8NHS and P8Cys. For reasons elaborated above, we consider the hydrolysis of P8NHS and the thioester intermediate as minor competing reactions under our conditions. Both of these reactions would produce acid terminated polymer endgroups that would not be capable of contributing to gel formation. It is impossible to exclude some contribution from direct amide bond formation between the terminal amine of P8Cys and P8NHS. However, it should be noted that the pKa value of the N-terminal amino of Cys is 10.78 whereas that of the thiol side chain is 8.33.23 Thus, at the pH values of 6–7.3 employed in gel formation, most N-terminal amino groups of Cys would be rendered inactive toward reaction with the NHS activated ester by protonation. In support of this, mixtures of P8NHS and 8-arm PEG-amine form gels more than 10 times slower in PBS buffer.
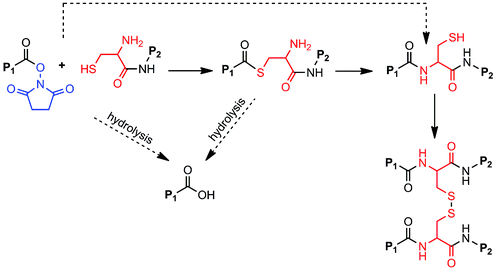 | ||
| Scheme 2 OMNCL cross-linking of P8Cys and P8NHS. Fast reaction pathways are indicated by solid arrows, slow pathways by dashed arrows. Thiol capture followed by S-to-N acyl rearrangement results in polymer cross-linking. Secondary cross-links arise through the formation of disulfide bonds among network-bound Cys residues. P1 = P8NHS; P2 = P8Cys. | ||
Most polymer hydrogels typically absorb water under physiologic conditions, leading to a significant increase in volume. The results of our gel swelling experiments were notable for two reasons. First, the increase in relative wet weight (0%) for OMNCL hydrogels is considerably lower than many experimental and clinically approved PEG-based hydrogels. For example, studies of PEG based hydrogels cross-linked via amide or thioester bonds report increasing relative wet weight values of 50%25 to 400%26 within the first 24 hours. In some cases, swelling of implanted materials may lead to complications such as nerve compression27–31 or other serious problems requiring intervention.32 Secondly, the swelling experiments provided important evidence of the latent formation of disulfide bonds, and insight into the mechanical consequences of this secondary cross-linking mechanism. Exposure of OMNCL gels to reducing agent resulted in an increase in swelling that was reversible upon removal of reducing agent, implying the formation of disulfide bonds within the gel network. This can be understood to be a result of the S-to-N acyl rearrangement step that releases the thiol side chain of Cys, which then becomes available for disulfide bond formation with other network-bound thiols. Through comparison of the kinetics of the OMNCL reaction (Fig. 3A,B) and the swelling results (Fig. 5), we surmised that the reduction in swelling observed after ∼5 h results from disulfide bond formation. Thus, we conclude that the hydrogels form initially by OMNCL and are later further cross-linked through the formation of disulfide bonds.
Our in vitro cell experiments demonstrated low cytotoxicity of OMNCL hydrogel extracts and high viability of encapsulated cells (Fig. 6). This led us to undertake an initial in vivo evaluation of OMNCL hydrogels in a subcutaneous implant model. Gels were formed by injection of precursor solutions and then evaluated at 6 weeks. Histological sections of explants showed low-level acute inflammatory response to implanted gels and deposition of a thin fibrous capsule surrounding the implant (Fig. 7). These findings confirm the potential of OMNCL hydrogels for in vivo applications.
Activated ester PEG polymers are currently approved by the FDA in the form of the medical sealants DuraSeal™ and COSEAL™. An important distinction between these existing materials and our OMNCL hydrogels relates to the pH range of the cross-linking reaction. Both DuraSeal™ and COSEAL™ are deployed at highly alkaline pH (typically pH 9–10),33 whereas the OMNCL hydrogels reported here are capable of rapid gel formation at neutral pH. In our studies, PEG was chosen as the backbone for both components because it is non-cytotoxic and has demonstrated favorable results in previous in vivo studies. PEG is also attractive as a simple platform for quantitative NMR studies and is amenable towards chemical modification. However, it is important to note that the OMNCL hydrogel chemistry described here can be easily adapted for use with other suitable polymer platforms.
In summary, we have detailed the synthesis and characterization of a two-component hydrogel formulation that sets rapidly at physiologic pH and ionic strength in the absence of catalysts or other additives. The cross-linking mechanism was revealed by NMR studies to be primarily OMNCL, proceeding by thiol capture to form a thioester intermediate followed by a S-to-N acyl rearrangement to generate amide cross-links. OMNCL hydrogels exhibit attractive mechanical properties that include high compressive moduli and good adhesion to tissue, and low equilibrium swelling due to the latent formation of secondary disulfide cross-links. The biological performance of OMNCL hydrogels was assessed, showing high in vitro cytocompatibility and low acute inflammatory response in vivo. OMNCL hydrogels represent attractive candidates for in vivo applications such as wound repair and sealing, drug delivery, and tissue engineering.
Acknowledgements
The authors would like to thank the NIH for funding this project through grants R01 DE021215 and R01 DE021104. This research utilized the facilities of the Central Laboratory for Materials Mechanical Properties (CLAMMP) and the Integrated Molecular Structure Education and Research Center (IMSERC) at Northwestern University (Evanston, IL). These studies were also supported by a grant from the Commonwealth of Pennsylvania and an NCI Cancer Center Grant (P30 CA10815) to the Wistar Institute as well as supported by the Wistar Animal Core Facility.References
- M. Schnolzer and S. B. Kent, Science, 1992, 256, 221 CAS.
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338 RSC.
- D. Bang and S. B. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534 CrossRef CAS.
- A. G. Komarov, K. M. Linn, J. J. Devereaux and F. I. Valiyaveetil, ACS Chem. Biol., 2009, 4, 1029 CrossRef CAS.
- Y. Kajihara, N. Yamamoto, R. Okamoto, K. Hirano and T. Murase, Chem. Rec., 2010, 10, 80 CrossRef CAS.
- E. Byun, J. Kim, S. M. Kang, H. Lee, D. Bang and H. Lee, Bioconjugate Chem., 2010, 22, 4 Search PubMed.
- B. H. Hu, J. Su and P. B. Messersmith, Biomacromolecules, 2009, 10, 2194 CrossRef CAS.
- J. P. Jung, J. L. Jones, S. A. Cronier and J. H. Collier, Biomaterials, 2008, 29, 2143 CrossRef CAS.
- S. E. Paramonov, V. Gauba and J. D. Hartgerink, Macromolecules, 2005, 38, 7555 CrossRef CAS.
- I. R. Ruttekolk, A. Chakrabarti, M. Richter, F. Duchardt, H. Glauner, W. P. R. Verdurmen, J. R. Rademann and R. Brock, Mol. Pharmacol., 2011, 79, 692 Search PubMed.
- I. R. Ruttekolk, F. Duchardt, R. Fischer, K. H. Wiesmüller, J. Rademann and R. Brock, Bioconjugate Chem., 2008, 19, 2081 CrossRef CAS.
- J. Su, B. H. Hu, W. L. Lowe Jr., D. B. Kaufman and P. B. Messersmith, Biomaterials, 2010, 31, 308 CrossRef CAS.
- P. B. Messersmith, J. Su and B. H. Hu, Pat. US20110262492A1, Catalyst and byproduct-free native chemical ligation using cyclic thioester precursors, 2011, 27 Search PubMed.
- J. Offer, Biopolymers, 2010, 94, 530 CrossRef CAS.
- Y. Yuan, J. Chen, Q. Wan, R. M. Wilson and S. J. Danishefsky, Pept. Sci., 2010, 94, 373 Search PubMed.
- L. E. Canne, S. J. Bark and S. B. H. Kent, J. Am. Chem. Soc., 1996, 118, 5891 CrossRef CAS.
- J. D. Warren, J. S. Miller, S. J. Keding and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6576 CrossRef CAS.
- Q. Wan, J. Chen, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 15814 CrossRef CAS.
- G. M. Fang, H. K. Cui, J. S. Zheng and L. Liu, ChemBioChem, 2010, 11, 1061 CrossRef CAS.
- M. J. Weissenborn, R. Castangia, J. W. Wehner, R. Sardzik, T. K. Lindhorst and S. L. Flitsch, Chem. Commun., 2012, 48, 4444 RSC.
- B. H. Hu, Xiaoyang and T. Hu, in Faming Zhuanli Shenqing, Guangzhou Shengyu Pharmaceutical Science and Technology Co., Ltd, China, 2011, p. 20 Search PubMed.
- I. Strehin, Z. Nahas, K. Arora, T. Nguyen and J. Elisseeff, Biomaterials, 2010, 31, 2788 CrossRef CAS.
- W. P. Jencks and J. Regenstein, in Handbook of Biochemistry and Molecular Biology, ed. R. L. Lundblad, CRC Press, 4th edn, 2010, p. 595 Search PubMed.
- P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776 CrossRef CAS.
- N. E. Epstein, Spine J., 2010, 10, 1065 Search PubMed.
- M. M. Saunders, Z. C. Baxter, A. Abou-Elella, A. R. Kunselman and J. C. Trussell, Fertil. Steril., 2009, 91, 560 Search PubMed.
- G. Lee, C. K. Lee and M. Bynevelt, Spine, 2010, 35, E1522 Search PubMed.
- M. Mulder, J. Crosier and R. Dunn, Spine, 2009, 34, E144 Search PubMed.
- B. J. Neuman, K. Radcliff and J. Rihn, Clin. Orthop. Relat. Res., 2011, 470, 1640 Search PubMed.
- D. Thavarajah, P. De Lacy, R. Hussain and R. M. Redfern, Spine, 2010, 35, E25 Search PubMed.
- S. R. Parker, P. Harris, T. J. Cummings, T. George, H. Fuchs and G. Grant, J. Neurosurg.-Pediatr., 2011, 8, 177 Search PubMed.
- C. Pace Napoleone, A. Valori, G. Crupi, S. Ocello, F. Santoro, P. Vouhe, N. Weerasena and G. Gargiulo, Interact. Cardiovasc. Thorac. Surg., 2009, 9, 978 Search PubMed.
- D. G. Wallace, G. M. Cruise, W. M. Rhee, J. A. Schroeder, J. J. Prior, J. Ju, M. Maroney, J. Duronio, M. H. Ngo, T. Estridge and G. C. Coker, J. Biomed. Mater. Res., 2001, 58, 545 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm00201b |
| This journal is © The Royal Society of Chemistry 2013 |