Biomaterial approaches to gene therapies for neurodegenerative disorders of the CNS
Ben
Newland
a,
Eilís
Dowd
b and
Abhay
Pandit
*a
aNetwork of Excellence for Functional Biomaterials (NFB), National University of Ireland, Galway, Ireland. E-mail: abhay.pandit@nuigalway.ie; Fax: +353 91 495585; Tel: +353 91 492758
bPharmacology and Therapeutics, National University of Ireland, Galway, Ireland
First published on 5th April 2013
Abstract
Neurodegeneration gives rise to a wide range of disorders which represent a growing health burden to both western societies and developing countries. Whilst for many disorders such as Alzheimer's and Parkinson's disease the cause is unknown, gene therapy is becoming the forefront of novel potential therapies described in the literature and has entered clinical trials. Furthermore, although in somewhat an earlier stage, biomaterials offer means of enhancing gene therapy strategies either through new delivery methods or provision of support for genetically manipulated cells. This review outlines recent uses of biomaterials in the CNS and captures recent advances in non-viral gene delivery to the brain. Three dimensional scaffolding systems for ex vivo gene delivery to the brain are also discussed highlighting the progress of hydrogel mediated cell delivery. This review also addresses the difficulties and safety considerations of these approaches; illustrating the ability of biomaterial strategies to significantly improve outcomes of gene therapies for neurodegenerative disorders.
 Ben Newland | Ben Newland is currently pursuing a PhD in Professor Pandit's laboratory at the National University of Ireland, Galway. He graduated with a BSc in Natural Sciences (chemistry/biology) from Durham University (UK). He later graduated with an MRes in Nanomaterials from Imperial College London (UK) before beginning his PhD research that focuses on the use of biomaterials to assist gene therapies for Parkinson's disease. |
 Eilís Dowd | Dr Eilís Dowd is currently a Lecturer in Pharmacology at National University of Ireland, Galway (Ireland). She earned her PhD at Edinburgh University (UK) after which she was awarded a Wellcome Trust Travelling Postdoctoral Fellowship which enabled her to complete post-doctoral research at McGill University (Canada) and Cardiff University (UK). In 2006 she joined the faculty at National University of Ireland, Galway. Her current research is focused on developing and validating novel pharmacological, cell and gene therapies for neurodegenerative diseases. |
 Abhay Pandit | Prof. Abhay Pandit is the Director of a Science Foundation Ireland funded Strategic Research Cluster “Network of Excellence for Functional Biomaterials” (NFB) at the National University of Ireland, Galway. Prof Pandit's postgraduate work focused on the modification of a fibrin scaffold to deliver a therapeutic biomolecule that resulted in a clinical trial at the Burn Centre at the University of Alabama at Birmingham (USA). He worked in the medical device industry in the USA for seven years. He has been at the National University of Ireland, Galway for the last ten years. His current research interests include developing responsive extracellular matrix-based systems for biomolecular delivery for a range of clinical targets. |
1 Introduction
Neurodegenerative disorders such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and multiple sclerosis (MS) are all characterised by the loss of neuron or glial cells in the brain or spinal cord. These conditions are progressive in nature, unlike other forms of neurodegeneration such as cerebrovascular accidents (stroke), or those caused by external factors, i.e. spinal cord injury (SCI) and traumatic brain injury (TBI). Dementia, of which AD is the leading cause, is the most common of the neurodegenerative disorders, with an estimated 24 million people being affected worldwide.1 The second most common neurodegenerative disorder, PD, has an incidence of 8.6 to 19.0 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inhabitants.2 In contrast to AD, where 60% of sufferers live in developing countries, PD is most prevalent in the USA and Europe.1,3 Whilst others are less common, the majority of neurodegenerative disorders are age related diseases, so through the general rise in ageing demographics, these disorders are likely to become more prevalent (e.g. a predicted 80 million will suffer from AD globally by 2040).1
000 inhabitants.2 In contrast to AD, where 60% of sufferers live in developing countries, PD is most prevalent in the USA and Europe.1,3 Whilst others are less common, the majority of neurodegenerative disorders are age related diseases, so through the general rise in ageing demographics, these disorders are likely to become more prevalent (e.g. a predicted 80 million will suffer from AD globally by 2040).1
Whilst the mechanisms involved in the pathogenesis of these diseases are in various states of elucidation, the aetiology for the majority of progressive cases remains unknown (with the exception of those caused by genetic inheritance e.g. familial PD4–6 or HD7). Another problem for patients of such diseases is that there are very few disease modifying therapies (such that exist are solely for the relapsing-remitting form of MS8). Thus, although therapies such as oral levadopa for PD patients offer a substantial rise in the quality of life, these therapies do not retard neuronal cell death. Whilst this may paint a bleak picture of the current state of therapies for the treatment of neurodegenerative diseases, one must also consider the inherent nature of the diseases that give a positive outlook for future interventions. The first aspect of these degenerative states is that the progression is often slow (with the most notable exceptions being SCI, TBI, stroke and to a lesser extent ALS), which gives a window of opportunity for intervention. The second is that neurons are influenced by growth factors such as nerve growth factor (NGF),9 brain-derived neurotrophic factor (BDNF)10 and glial-derived neurotrophic factor (GDNF),11 to name a few. The neuroprotective properties of these trophic factors is well documented,12–15 and they offer an exciting potential for early intervention with the more progressive diseases coupled with the possibility of tissue repair in the acute disorders.
The first growth factor to be identified, NGF,9 was found to protect cholinergic neurons in adult rats following axotomy,13–15 and the potential for therapeutic benefit in AD was noted that same year.16 However, the size of proteins and their inherent charge renders them incapable of crossing the blood–brain-barrier (BBB). Therefore, as with all protein infusion trials, the protein must be delivered intracerebrally. Clinical trials based on the direct injection/infusion of growth factors into the brains of patients with PD or AD, have resulted in varying outcomes. By a brief review of these trials one can highlight some potential problems inherent with direct protein delivery. Amidst rising concerns over the safety of intracerebroventricular injections of NGF,17,18 a clinical trial involving three patients with AD was undertaken, whereby the patients received continuous intracerebroventricular infusions for a period of three months.19 Due to the short half-life of growth factors, a continuous infusion directly into the lateral ventricle was required. However, this trial showed no improvement in cognitive tests and had to be discontinued due to patient weight loss and pain. Whilst these side effects had been predicted in animal models, the lack of patient amelioration may be contributed to the lack of labelled NGF localising in basal forebrain neurons of primates following similar administrations.20 These studies therefore suggest that direct administration to the target tissue was required. Nevertheless, it must be noted that one major advantage of growth factor administrations is that the negative side effects reversed when delivery ceased.19
A more targeted approach to growth factor delivery was reported in 2003 for five patients with PD who participated in a Phase I trial. Intraputaminal administration of GDNF, that acts upon dopaminergic neurons (neurons lost during PD), for the course of a year produced an overall improvement in the Unified Parkinson's Disease Rating Scale (UPDRS) score for all five patients.21 Another Phase I trial, involving ten patients receiving intraputaminal administration of GDNF, led to similar conclusions; that an overall benefit was observed during the 12 months, with only minor side effects.22 However the trial was halted by the sponsor Amgen following blood sample analysis detecting GDNF binding antibodies in seven of the ten patients. A follow up study of the patients one year post-treatment showed no overt adverse effect from this, but showed that the melioration due to GDNF was reversed without the continuation of treatment.23 In contrast to these trials, where improvements in UPDRS scores were observed, a randomised, multicentre, double blind and placebo controlled trial involving thirty four PD patients showed no significant improvement in UPDRS score by intraputaminal administration of GDNF.24 However, much criticism of the study has arisen especially over delivery-specific issues such as the type of cannula used (differing from the multiport one used in the first study) and dose used (lower than both the previous studies).25 Though the protective effects of neurotrophic factors have been well reported, it is likely that issues surrounding optimised delivery remain a cause for a lack of overall efficacy of these therapies.26
In light of these findings, the potential benefits that gene therapy based approaches to neurodegenerative diseases may offer must be assessed. By grouping gene therapies for brain applications into two major subcategories – direct in vivo gene delivery and indirect ex-vivo gene therapy – it is noted that both approaches have the potential to mediate therapeutic effects with a single, one-time administration. In the case of using these approaches to manipulate over-expression of neurotrophic proteins either in host cells (in vivo) or implanted cells (ex vivo), single administrations could be sufficient, provided that suitable efficacy can be achieved. Whilst reaching such levels of efficacy remains a target (to which the use of biomaterials can be a useful aide – discussed later) the single surgical procedure will obviate the implantation of a catheter into the patient's brain. In doing so, catheter related problems such as local excoriation,23 infection,21 migration from the desired position24 and the need for surgical revision for catheter re-positioning21 can be eliminated. Another drawback of the direct infusion process needed for growth factor therapies also drives a positive rationale to turn focus towards gene therapies. Although this effect is less obvious, it arises in applications where a large spread of the neurotrophic factor is required (i.e. in the relatively large structure of the striatum for PD) and may incur serious implications. In order to increase this diffusion distance of the neurotrophic factor from the cannula port, convection enhanced delivery (CED) can be used.27 The process involves the infusion of a relatively large liquid volume (normally in pulses) that relies on the local pressure to drive the liquid through the brain interstitium. However, analysis of the brains of adult rhesus monkeys ten weeks post CED of GDNF showed gross tissue loss of up to approximately 2 mm in diameter around the catheter tip.28 Although the authors attribute this tissue damage to local flow rates from the single port catheter, potential methods of countering this negative side effect are not discussed. It has been found that a single four micro litre injection of phosphate buffered saline into the striatum of adult Sprague Dawley rats does not result in such gross tissue loss.29 Thus, provided that acute toxicity of the in vivo or ex vivo approach can be avoided, the single injection for a gene therapy strategy is less detrimental to the surrounding tissue. This review will therefore focus on an injectable, single dose therapeutic intervention mediated through the use of biomaterials.
2 Neurological disorder specific biomaterials toolbox
Using biomaterials to mediate a one-off intervention strategy has the foreseeable benefits as outlined in the introduction. Biomaterials can be used in several gene based strategies for neurological therapies. Potentially, such materials can therefore be used to replace existing viral gene vectors, as controlled delivery devices to the CNS, for neuron regeneration via functionalised scaffolds and as a method of enhancing ex vivo cell delivery. It is also highly feasible for a combination of these features to be used together in a complementary or combinatorial role, as will be described later on. Fig. 1 shows a schematic representation of these possibilities which are in relative infancy for neural applications. However, much progress has been made in areas such as non-viral gene delivery as a general shift can be noted from early non-targeted vectors (see Tables 1 and 2) to new nanoparticle formations (Table 3) and targeted vectors that to deliver genetic cargo to specific tissue or cell types (Table 4).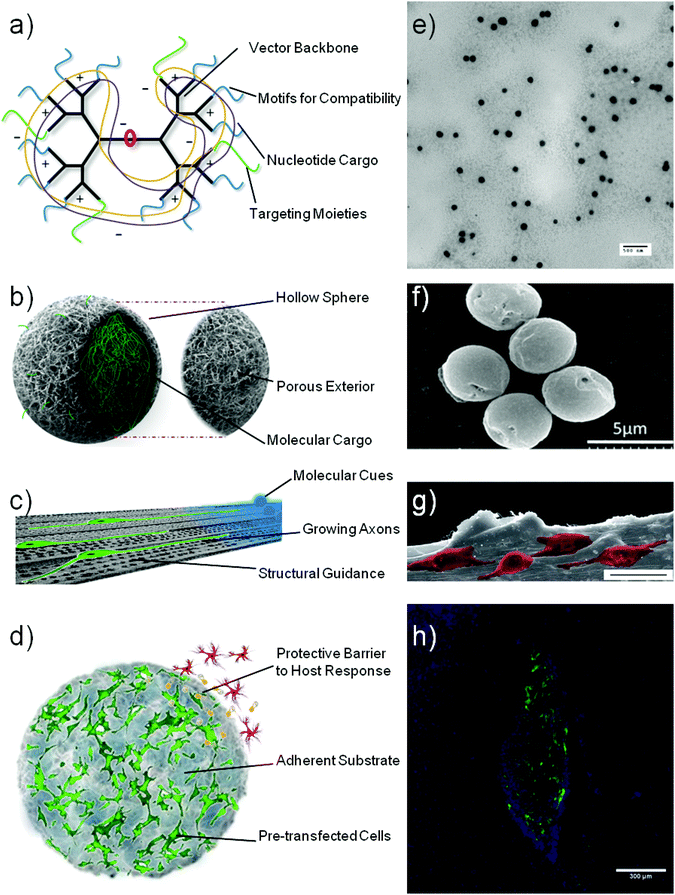 | ||
| Fig. 1 Schematic representation of various biomaterial constructs that can aid gene therapy applications for neurodegenerative disorders: nucleic acid vectors (a), sustained delivery devices (b), neural conduits (c) and cell support constructs (d). Subsequent microscopy images show: transmission electron micrograph of polyplexes formed by condensation of plasmid DNA with a polymeric vector (e), scanning electron micrograph of collagen hollow spheres prior to polyplex loading (f), scanning electron micrograph of PC12 cells (pseudocoloured) growing on an aligned collagen scaffold (scale bar = 25 μm) (g) and fluorescent micrograph of GFP transgenic stem cells growing within a collagen hydrogel (h) taken with permissions29,35,36 (e–h). | ||
| Vector | Gene | Methods | Outcomes | Researcher/Year |
|---|---|---|---|---|
| a Intracerebroventricular. b Dioctadecylamido glycylspermine. c Dioleoylphosphatidyl ethanolamine. d Aromatic L-amino acid decarboxylase. e 6-Hydroxydopamine. f Myristoyl lauroyl rosenthal inhibitor. g Green fluorescent protein. h Heat shock protein. | ||||
| Lipofectin® | β-Galactosidase or chloramphenicol acetyltransferase | Direct injection into the neonatal mouse brain, unspecified conditions, analysed up to 9 days post injection. | Qualitative X-gal positive staining or chloramphenicol acetyltransferase immunopositive cells in several brain regions. | Ono et al., 1990130 |
| Lipofectin® | β-Galactosidase or cholecystokinin octapeptide | Stereotactic ICVa injection into P77PMC rat model of audiogenic epileptic seizures, analysed up to 14 days post injection. | ∼3 fold reduction in the vulnerability of seizure score 4 days after injection. | Li-Xin et al., 1992131 |
| Lipofectin® | β-Galactosidase | Stereotactic injection into the caudate putamen of the adult mouse. Heterozygous deficient gusmps/+ mouse used to control for false positive X-gal staining. Analysis up to 21 days post injection. | Qualitative analysis of X-gal positive staining, found largely along needle tract, no obvious difference between promoters used. | Rossler et al., 1994132 |
| DOGSb/DOPEc | Luciferase or β-galactosidase | Direct injection approximately into the striatum of the newborn mouse. Analysis conducted 1, 2 and 3 days post injection. | Quantitative analysis show highest luciferase expression with low charge ratios (0.8 and 1.8), 1 μg of DNA and 24 h post injection. | Schwartz et al., 1995133 |
| Lipofectin® | β-Galactosidase or cholecystokinin | Stereotactic ICVa injection into P77PMC rat model of audiogenic epileptic seizures. Also injection into the hippocampus for visualisation of transfection. 1–21 day analysis. | Successful suppression of audiogenic seizures 4 days post injection (10 μg/20 μl). Qualitative X-gal positive staining of ventricular ependymal cells. | Zhang et al., 1997135 |
| Lipofectin® | β-Galactosidase | Either single stereotactic injection or continual infusion with Alzet® osmotic minipumps in the adult rat septum. Animals were sacrificed 4 and 6 days post surgery. | Qualitative analysis. X-gal staining proved non-specific, β-gal immunohistochemistry revealed only low intensity staining, concluding low efficiency of transfection. | Kofler et al., 1998136 |
| DOGSb | β-Galactosidase | Continual infusion with Alzet® osmotic minipumps in the adult rat caudate putamen. Analysis 1, 7 and 14 days post injection. | Quantification of transfected cells following β-gal immunohistochemistry. Highest transfected cell number after 7 days of continual infusion or highest dose (50 μg). | Imaoka et al., 1998137 |
| DOGSb | Tyrosine hydroxylase (TH) or L-AADCd | 7 day continual infusion into the striatum of the 6-OHDAe rat model of PD. Functional recovery assessed by apomorphine induced rotations. | Functional improvement through TH transfection up to 6 weeks post infusion. Improved further by dual infusion of both TH and L-AADC. | Imaoka et al., 1998138 |
| Lipofectin® | TH | Stereotactic injection into two sites of the striatum of the 6-OHDAe rat model of PD. | Functional improvement for up to 3 weeks as assessed by apomorphine induced rotations. Astrocyte specific expression of TH. | Segovia et al., 1998144 |
| MLRIf-DOPEc | Luciferase, GFPg or Hsp 70h | Stereotactic ICVa injection of vectors into the adult rat ventricle. Analysis 1 day or 44 h post injection. | Qualitative immunohistochemical analysis show cells stained positive for Hsp70. GFP expression detected by immunofluorescence. | Hecker et al., 2001153 |
| JetSI™/DOPEc | Luciferase siRNA | Direct injection of siRNA formulations along with plasmid formulations into approximately the ventricular area of the newborn mouse. Analysis up to 60 h post injection. | Maximal downregulation of the polymer induced upregulation of luciferase expression achieved at a dose of 0.05 pico moles of siRNA. | Hassani et al., 2005150 |
| Vector | Gene | Methods | Outcomes | Researcher/Year |
|---|---|---|---|---|
| a Polyethyleneimine. b Dopamine transporter. c B-cell lymphoma-extra-large. d Polyaminoethyl propylene phosphate. e Tyrosine kinase deleted FGF receptor-1. f L-Cysteinyl-poly-L-lysine 30-mer conjugated with PEG. g Arginine ester of PAMAM. h High mobility group box-1. i 6-Hydroxydopamine. | ||||
| PEIa | Luciferase | Direct injection into approximately the striatum of the newborn mouse. Transfection analysed 1 day post injection. | Quantitative analysis of luciferase activity showed maximal transfection with an N:P ratio of 9:1. |
Bousiff et al., 199540 |
| PEI | Luciferase | Stereotactic injection into the mouse cerebral cortex, hippocampus and hypothalamus. Analysis up to 90 days post injection. | A molecular weight of 25 kDa, an N:P ratio of 6, and a 3 day time point resulted in highest transfection. |
Abdallah et al., 1996152 |
| PEI | DATb | Stereotactic injection of sense and antisense plasmid into the adult rat substantia nigra. Analysed 3 and 7 days post injection. | Immunoautoradiographic labelling revealed DAT levels increased and decreased via sense and antisense plasmid respectively. | Martres et al., 1998157 |
| PEI | β-Galactosidase | Stereotactic injection into the adult mouse ventricle analysed 1 and 2 days post injection. | Qualitative analysis; transfection in neurons and glia throughout the ventricle. | Goula et al., 1998154 |
| PEI | β-Galactosidase, luciferase or Bcl-Xlc | Direct injection of marker genes, or dual injection of marker genes with Bcl-Xl into the newborn mouse ventricle. Analysis 1 and 7 days post injection. | 100 ng of DNA produced largest luciferase activity which was greater than 1 μg of DNA after both 1 and 7 days. Anti-apoptotic Bcl-X1 increased luciferase activity. | Lemkine et al., 1999155 |
| PEI | β-Galactosidase, luciferase or Bcl-Xlc | Stereotactic injection into the mouse ventricle. Analysed 4, 7 and 3 months post injection. | Neural stem cells transfected and expression passed to immediate progeny. Bcl-X1 aided transfection process. | Lemkine et al., 2002156 |
| PEI-PEG | Luciferase | Intrathecal injection into the adult rat subarachnoid space. Analysed 1–28 days post injection. | Addition of PEG increases transfection capability which is maximal 1 day post injection. | Tang et al., 2003158 |
| PPE-EAd or PEI | Luciferase | Injection into the mouse cerebellomedullary cistern. Analysed 1–28 days post injection. | Luciferase activity higher than naked DNA in the cerebellum, diencephalon and cortex. | Li et al., 2004159 |
| PEI | β-Galactosidase or FGFR1(TK-)e | Stereotactic injection into the rat substantia nigra, β-gal used as control. Analysed 7 or 28 days post injection. | Viral transfection with FGFR1(TK-) reduced TH positive cells but PEI group was statistically no different to control. | Corso et al., 2005160 |
| β-Cyclodextrin-PEI | Luciferase | Luciferase activity analysed 2 days post injection into the rat subarachnoid space. | Cyclodextrin conjugation of 600 kDa PEIs matched transfection via 25 kDa PEI. | Tang et al., 2006161 |
| PEI | Luciferase | Direct injection at 5 sites into the cerebral parenchyma of 5 dogs. Analysed after 3 days. | No increased luciferase activity over naked DNA injections. | Oh et al., 2007162 |
| CK30PEG10kf | Luciferase, GFP or GDNF | Stereotactic injection into the adult rat striatum. Luciferase activity, GFP or GDNF expression analysed at time points between 1 and 56 days. | Persistent luciferase activity achieved for 56 days. Neurons and glia transfected and increased GDNF levels post transfection. | Yurek et al., 200961 |
| e-PAM-Rg | HMGB1h siRNA | Stereotactic injection into the post ischemic rat brain (occlusion model of stroke). Analysed 2 days post occlusion. | Injection reduced HMGB1 expression in normal brain, and reduced infarct volume caused by middle cerebral artery occlusion. | Kim et al., 2010163 |
| CK30PEG10kf | GDNF | Stereotactic injection into the healthy adult rat striatum or 6-OHDAi model of PD. Analysed up to 98 days or 6 months. | GDNF levels elevated for 6 months in healthy brain, but elevated more in lesioned brain, indicating astrocyte transfection. | Fletcher et al., 2011164 |
| CK30PEG10kf | Luciferase | Stereotactic injection into the rat cortex, striatum or substantia nigra, or mouse striatum. Bioluminescence imaging (BLU) used to detect transgene expression up to 1 year. | Live BLU imaging and ex vivo BLU imaging revealed transfection in the target regions. Stable luciferase expression for 1 year in the mouse striatum. | Yurek et al., 2011165 |
| e-PAM-Rg | HMGB1h siRNA | Intranasal delivery the post ischemic rat brain (occlusion model of stroke). Analysed 2 days post occlusion. | Knockdown in the cortex and striatum. Reduction in the infarct volume and functional improvement in the rota rod test. | Kim et al., 2012166 |
| Vector | Gene | Methods | Outcomes | Researcher/Year |
|---|---|---|---|---|
| a Organically modified silica. b Enhanced green fluorescent protein. c FGF receptor-1. d PolyQ peptides with 20 (normal) or 127 (expanded) glutamine repeats. e Glyceraldehyde 3-phosphate dehydrogenase. f Functionalised carbon nanotubes. g Protease for N-terminal cleavage of the amyloid precursor protein. | ||||
| Calcium phosphate | β-Galactosidase | Stereotactic injection into the adult rat substantia nigra. Analysed after 7 and 28 days. | Qualitative analysis showed transfection 28 days post injection, but not after 7 days. | Corso et al., 2005170 |
| ORMOSILa nanoparticles | EGFPb or FGFR1c | Stereotactic injection in the mouse substantia nigra or lateral ventricle. Analysis after 7 and 10 days. | Fiber-optic Cell-viZio probe monitoring allows qualitative analysis and shows transfected cells surrounding the lateral ventricle. | Bharali et al., 2005175 |
| ORMOSILa | EGFP, polyQ(20) or polyQ(127)d | Two stereotactic injections into the mouse or rat lateral ventricle 2 weeks apart, and intrastriatal injections analysed up to 4 weeks post injection. | Immunohistochemical analysis shows expression of peptides and astrocyte reactivity. Mice with polyQ(127) showed functional impairment in fine movements test. | Klejbor et al., 2007176 |
| ORMOSILa | EGFPb, FGF-2 or FGFR1c | Stereotactic injection into the adult mouse lateral ventricle. Analysed up to 18 days post transfection. | Qualitative EGFP, FGF-2 and FGFR1 expression in sites surrounding the lateral ventricle. Neuronal differentiation of cells transfected with FGF-2 or FGFR1. | Stachowiak et al., 2009177 |
| Gold nanorods | GAPDHe siRNA | Stereotactic injection into the rat hippocampus. Uptake and knockdown measured up to 10 days post injection. | Specific uptake in neurons (qualitative) and 70% knockdown of GAPDH achieved in the CA1 region of the hippocampus. | Bonoiu et al., 2011180 |
| f-CNTf | Caspase-3 siRNA | Stereotactic injection into the mouse and rat prior to, or post endothelin-1 (ET-1) administration for a model of stroke. Apoptotic cell number analysed (1 day post administration) or skilled reaching ability (9–13 days post treatment). | f-CNT mediated delivery prior to ET-1 resulted in reduction in apoptotic cell number, but not post ET-1 treatment. Functional analysis showed an improvement in skilled reaching ability compared to controls. | Al-Jamal et al., 2011179 |
| Targeted exosomes | GAPDH siRNA or BACE1g siRNA | Systemic delivery (tail vein), mice euthanized 3 days post last treatment. | Specific knockdown (<50%) of GAPDH in striatum, midbrain and cortex, achieved only with targeted vectors. Reduction in BACE1 expression and protein levels. | Alvarez-Erviti et al. 2011181 |
| Vector | Targeting moiety | Gene | Summary | Senior Author/Year(s) |
|---|---|---|---|---|
| a Poly-L-lysine. b Glial cell line-derived neurotrophic factor. c 6-Hydroxydopamine. d 1,2-Dioleoyl-3-trimethylammonium-propane. e Enhanced green fluorescent protein. f Polyethyleneimine. g Poly(amido amine). h Green fluorescent protein. i 6-Hydroxydopamine. j 2-Dimethylaminoethyl methacrylate. k Vascular endothelial growth factor. l Dioleoylphosphatidyl ethanolamine. | ||||
| PEGylated immunoliposomes (PIL's) | Human insulin or transferrin (Tf) monoclonal antibodies (mAb) | Multiple | Systemic injection of PEGylated immunoliposomes (also termed Trojan horse liposomes) into mice, rats and primates results in transgene expression in the brain. Off-target expression can be reduced by use of specific promoters. Therapeutic genes such as tyrosine hydroxylase resulted in functional improvement in PD models. | Pardridge et al., 2000–200850,59,185–195 |
| PLLa | Neurotensin (NT) | Multiple | Stereotactic injection of polyplexes into the striatum results in transfection of dopaminergic neurons (via the neurotensin high affinity receptor). GDNFb encoding gene transfection 1 week after 6-OHDAc lesion resulted in partial recovery at an atomical and motor level. | Martinez-Fong et al., 2001–2006202–204 |
| DOTAPd-Cholesterol | Tf | Multiple | Stereotactic intrastriatal injection of NGF encoding gene loaded liposomes resulted in large reduction of kainic acid induced infarct volume in rats. Luciferase positive mouse used to prove concept of siRNA mediated knockdown, and c-Jun siRNA delivery resulted in increased neuronal survival post excitotoxic damage. | Pedroso de Lima et al., 2005–2009197–199 |
| PLLa | Monoclonal antibody MC192 | EGFPe, GDNFb | Intramuscular administration into the hind limb of newborn or adult rats results in retrograde transport and expression in the CNS with cell rescue from axotomy induced death. | Rush et al., 2006207 |
| Self-assembled PEIf | Nerve growth factor (NGF) | Luciferase | Intrathecal injection into the lumbar spinal cord in rats resulted in transgene activity in the spinal cord, which was more specific to dorsal root ganglia than non-targeted vector controls. | Wang et al., 2007206 |
| PEG-PAMAMg | Lactoferrin (Lf) or Tf | Multiple | Widespread GFPh expression in the mouse brain with Tf targeted vectors after intravenous administration. Quantitative analysis showed higher luciferase activity in mouse brain when Lf is used. GDNFb gene delivered to 6-OHDAi and rotenone rat models of PD resulted in improved locomotor activity. | Pei et al., 2007–201058,208,209,243 |
| PEIf in liposome | 8D3 mAb | VCAM-1 decoy oligo-deoxynucleotides | Encapsulating polyplexes in PEG stabilised liposomes resulted in extended circulation times and 8D3 mAb significantly improved brain uptake post systemic injection into mice. | Bickel et al., 2009201 |
| PEG-PDMAEMAj | Peptide TGN (TGNYKALHPHNG), Angiopep or rabies viral glycoprotein (RVG) | Luciferase, EGFPe | Targeted polyplexes resulted in higher brain uptake following systemic delivery than non-targeted polyplexes, significant luciferase activity was observed, widespread EGFPe expression particularly around the lateral ventricle. | Jiang et al., 2009–2013215,244,245 |
| PEG-PEIf | Tet1 | β-Galactosidase, Luciferase | Stereotactic injection of targeted polyplexes into the mouse lateral ventricle mediated luciferase expression that extended to the hindbrain and persisted up to 10 days. Targeted polyplexes transfected neural stem cells in the subventricular zone. | Pun et al., 2010246 |
| PEG-Liposome | Tf | β-Galactosidase , VEGFk | 2 days after middle artery occlusion (rat stroke model) femoral vein injection of Tf targeted liposomes resulted in higher VEGF mRNA, protein levels, microvessel density and higher percentage of residual cerebral blood flow. | Kong et al., 2011200 |
| DOTAPd/DOPEl | Peptide P (K16GACLPHKSMPCG) | GFPh | Lipid–peptide–DNA complexes injected by stereotactic surgery into the rat corpus callosum or striatum could be imaged by MRI due to gadolinium labelling. Microglia transfected near injection site. | Hart et al., 2012247 |
2.1 Non-viral gene vectors
An important research focus for materials design in gene based therapies for neurological disorders is in replacement of the existing viral vectors themselves. Whilst a number of safety concerns have arisen through the use of viral vectors,30–33 several recent trials have shown the safety of non-genome integrating viral vectors.34 However, for therapies to enter widespread use, they must be economically viable in terms of product manufacture, preparation procedures and administration techniques. Non-viral gene vectors often offer the possibility of facile upscale and safe handling procedures, which, along with the ability to incorporate large plasmids, promise substantial benefits over their viral counterpart. The use of polymeric or liposomal gene vectors in the brain has therefore been the focus of much research.The majority of non-viral gene vectors use a charge–charge interaction to condense nucleic acids (e.g. DNA, siRNA, mRNA etc.) through the negative charge of the nucleic acids arising through the phosphate groups, and cationic moieties on the vector. This interaction, resulting in a particle commonly termed with the suffix “-plex”, for example polyplex (polymer formed complex), often condenses the DNA to such an extent that intercalating dyes can no longer interpolate with the DNA base pairs.37 These charge based non-viral nucleic acid vectors, reviewed elsewhere,38 have been prepared in a variety of forms outlined in Fig. 2, ranging from the conventional polymer or liposomal vectors39 to the more recent nanoparticle or nanotube vectors. Of these vectors, polyethylenimine (PEI) based vectors40 and liposomal vectors41 have been the focus of much research attention and are discussed later in the context of use in the mammalian brain.
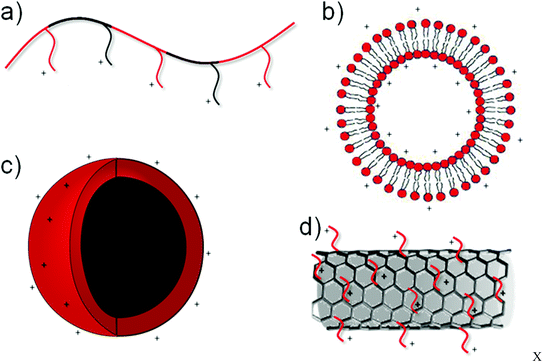 | ||
| Fig. 2 Schematic representation of various non-viral transfection agents depicting cationic polymers (a), liposomes (b), nanoparticles (c) and functionalised carbon nanotubes (d). | ||
The majority of research in the field of non-viral vectors aims at increasing vector efficiency whilst reducing its toxicity. Increasing efficiency may be sought through: changing vector composition,42 varying the molecular weight of the vector,43 altering vector structure,44,45 mediating selective intracellular degradation,46 increasing circulation time (for systemically administered vectors),47,48 membrane disruption49 or increasing the specificity of targeting,50–52 to name a few. Whilst there is often a clear relationship between the vector to nucleic acid ratio and the toxicity of the system as a whole (e.g. polyplex/lipoplex etc.)53 the majority of work aimed at reducing toxicity sought to reduce the toxicity of the vector itself. Polyethylene glycol (PEG), a neutral polyether, has been used extensively to alter the properties of biomedical materials through its intrinsic properties of good solubility with a lack of toxic effects.54 Modification of gene vectors with PEG has, to a large extent, focused upon reducing the vector toxicity,55 which can shield surface charge56 and reduce protein/blood component aggregation.57 Another common use of PEG is as a spacer between vector and targeting moieties. However, when considering the commercial feasibility of up-scaling non-viral vectors for applications in neurological disorders, one must consider that the majority of PEGylation strategies involve post modification of the original vector,58–61i.e. an additional synthesis/purification step. An attractive alternative lies in chemistries that incorporate PEG during the original synthesis producing a PEGylated vector after a single purification step,62 allowing low cost possibilities for the scale-up of safe vectors that would be required for such intervention strategies.
2.2 Controlled delivery devices
In order to overcome the problem of the short half-life of growth factors, controlled delivery devices have been designed to function as a sustainable protein release platform, thus allowing an increased action time of the growth factor.63,64 Synthetic polymers such as poly(D,L-lactide-co-glycolide) (PLGA) have been used to fabricate biodegradable microspheres that do not augment the host response to implantation in the rat brain.65 Furthermore, microspheres based on such polymers, can be loaded with growth factors such as NGF,66,67 GDNF68 and NT-369 for sustained delivery applications. Other synthetic materials such as PEG have been used to create microspheres for the fabrication of protein gradients, whereby different densities of microspheres are graded during centrifugation.70Attractive alternative materials for the manufacture of controlled delivery devices are natural biopolymers such as fibrin, chitosan, collagen, elastin and hyaluronan. Recently, attention has been drawn towards the use of carriers based on these materials to deliver plasmid DNA, often in a complexed form (e.g. polyplex/lipoplex etc.).71–76 Whilst these studies were not focused around their application in neurological disorders, a recent study showed that polyplex mediated toxicity could be ameliorated in brain tissue by use of collagen hollow spheres as a sustained delivery mechanism.35 For therapeutic applications in neurological disorders, such devices hold the promise not only to reduce the toxicity of gene vectors, but also to prolong the therapeutic time period.
As mentioned earlier, discontinuing GDNF protein infusion during a clinical trial reversed the positive effects first observed,19 indicating that for gene therapies to become a one-off intervention, a long window of transgene expression is required. By controlling the release of gene vectors or bound nucleic acids such as siRNA or plasmid DNA, it could be envisioned that the transient expression associated with non-viral gene delivery may be extended. It has also been shown previously that the use of a different promoter sequence on plasmids encoding luciferase can alter the transgene expression levels and duration in the rat brain.77 In the same study, a GDNF encoding plasmid containing the polyubiquitin C promoter produced transgene expression in the rat brain which for lasted for the full study period of 21 days. It should also be noted here that the use of minicircle vector DNA technology devoid of a bacterial backbone provides another means of extending transgene expression time.78 Therefore, via careful plasmid design, the use of an efficient vector and the packaging of the system within a sustained delivery device such as the aforementioned spheres, transgene expression in the brain perhaps could be extended to several months and possibly up to one year. Although the goal remains a single intervention strategy, and most of the progressive neurological disorders extend beyond a one year time period, controlled delivery devices at the very least offer an increased transgene expression window. It is therefore likely that sustained delivery devices will play an important future role in the assistance of non-viral gene delivery; however, further development to allow temporal release of the transfecting agents over longer periods is likely to be required. If repeat administrations do become a necessity, less invasive administration should be sought such as targeted systems for systemic delivery (see Section 3.4).
2.3 Functionalised scaffolds
With the exception of SCI, where surgical intervention is a current option, therapeutic strategies must be injectable and cause low tissue damage/toxicity/host response. However, brain implantation of materials has been performed at sites where surgical intervention is theoretically possible, such as at the periphery (cortex).79,80 Implantation of scaffolds inevitably requires the removal of brain tissue of the size scale proportional to that of the scaffold to be implanted. The depth then also determines how much tissue is removed, for example a study implanting synthetic channelled scaffolds required 1 mm × 4 mm × 1 mm for cortical implantation and a depth of 6 mm to be taken for implantation near the subventricular zone.81 For this reason, functionalised scaffolds may be better suited for applications in the spinal cord where, although invasive, implantation does not necessarily entail surrounding tissue removal or damage.Within the biomaterials toolbox, hydrogels, scaffolds or hollow conduits may be used for guidance of nerve growth following neuronal damage.82,83 Much of the current research into material design and preparation has centred upon physical guidance cues to direct axonal regeneration36,84–86 or the release of neurotrophins to provide chemical cues for neuronal regeneration.87–89 In terms of using scaffolds for genetic therapies, functionalisation of the scaffold has been performed with either gene vectors90–93 or genetically modified cells.94,95 Scaffolds functionalised to sustain the release of gene vectors, for example those coding NT-3, a protein involved in the path finding of axons,96 could serve a dual purpose of providing both physical and molecular cues to regenerating axons.97 It could be argued here that the transient nature of transgene expression following non-viral gene transfer may be advantageous as the growth factor would likely be most valuable during the initial regeneration phase.98
2.4 Hydrogels
Hydrogels are polymer networks which contain water as the liquid phase dispersed amongst the polymer chains. Whilst in many cases their inability to flow in a steady-state renders them with properties of a solid, they typically can absorb many times their dry mass in water.99 The interaction of the chains either through charge interaction or chemical crosslinking, alters the physical properties of the gels allowing their use for a variety of biomedical and tissue engineering applications.99,100 Hydrogels can be formed from natural materials101 or synthetic materials102 and can be prepared in different ways depending upon the size required, administration method and application. For example, preformed macroscopic gels have been produced through casting methods for applications in SCI103 and biocompatibility studies in the brain.104 Such preformed macrostructures necessitate surgical implantation, an undesired characteristic for applications in the brain, and one which can be bypassed by the use of materials that undergo gelation in situ, post injection. These hydrogels still form microscopic/macroscopic networks however they have the benefit of injectability by being capable of having all components of the hydrogel prepared in solution pre-injection, with gel formation only occurring post injection.105Natural biopolymers, such as collagen, form hydrogels in physiological conditions (through warmth, neutral pH and the presence of salt) by the change in structure of the elongated triple helices (in the secondary structure), to that of compact coils.106 Environmental responsive hydrogels107 have been prepared based on a range of natural biopolymers such as chitosan,108 hyaluronan,109 collagen110 and polysaccharides111 to name a few. Hydrogels composed of synthetic materials have also been designed to be responsive to a variety of stimuli such as heat,112,113 light,114,115 pH changes,116 salt content117 and enzymes118,119 or via multiple stimuli.120,121 Hydrogels with filaments of sizes in the order of nanometers have been termed nanogels or nanofiber gels which can also gel in situ, and are thus injectable.122–126 Whilst a large proportion of hydrogel mediated delivery of therapeutic agents such as growth factors has focused on use in the context of SCI,127–129 the final part of this review will offer a perspective on the potential for hydrogels to improve ex vivo gene therapies for neurological disorders.
3 Strategies for in vivo therapies
3.1 Liposomal gene vectors
The earliest examples of non-viral transfection to the mammalian brain were published in 1990. The liposomal vector termed Lipofectin® was then subsequently used for a number of studies in the rat brain (see Table 1 for a summary of liposomal transfection). Likewise the reporter gene β-galactosidase, detectable by X-gal staining, was commonly used to detect whether transgene expression was occurring. Although β-galactosidase expression can be quantified, the early studies using liposomal vectors were largely qualitative in nature130–132 until the plasmid encoding Firefly luciferase was used.133 This study also used a different transfection vector which was a combination of a cationic lipid dioctadecylamido glycylspermine (DOGS) with a neutral lipid dioleoylphosphatidyl ethanolamine (DOPE). By the use of the luciferase plasmid the brains of the newborn mice could be removed, the cerebral hemispheres homogenised, and a luciferase assay kit used to detect transfection. A luminescence read out in terms of relative light units could be obtained, which the authors found was highest 24 hours post transfection and dropped off over the next two days.In a breed of rat with congenital audiogenic seizures (P77PMC rats) administration of exogenous cholecystokinin-8 (CCK-8) was shown to reduce these seizures.134 For a measure of functional outcome of transfection via the Lipofectin® vector, a plasmid encoding CCK was loaded to Lipofectin® and administered to the rat ventricle.135 Four days post administration, a reduction in the vulnerability of seizure score by approximately three-fold was observed compared to that of the control of an empty plasmid.
It was not until 1998 that it was first reported that in fact low numbers of cells were observed to be transfected in the rat septum when the Lipofectin® transfection vector was used.136 In fact that same year, two studies were published that reported cannulation of animals, and the use of mini-pumps to continuously administer a DOGS vector.137,138 Continual infusion gave greater transfection results by day seven, which still reduced at day 14.137 However, when this transfection method was used in the 6-hydroxydopamine (6-OHDA) rat model of PD, with plasmids encoding either tyrosine hydroxylase (TH) or aromatic L-amino acid decarboxylase (AADC), an improvement in the apomorphine induced rotations scores was observed for 5 weeks.138 In the normal healthy brain, TH is a predominant enzyme involved in conversion of dietary tyrosine to the dopamine precursor L-3,4-dihydroxyphenylalanine (L-DOPA).139L-DOPA is then decarboxylated via the enzyme AADC to dopamine,140 the neurotransmitter that is diminished in PD.141 This process occurs in dopaminergic neurons142 (and serotonergic neurons) which are lost during the progression of PD; thus replacement of these key enzymes could become a strategy to replace the existing method of administering L-DOPA, first discovered in 1967.143 Another study showed that by dual administration (same time point) of Lipofectin® carrying the TH encoding plasmid, apomorphine induced rotations in the 6-OHDA rat model could be ameliorated, compared to the control plasmid group.144 It should be noted here that such strategies are purely symptomatic in nature and, whilst they have the potential to improve the quality of a PD patient's life substantially, they will not halt the pathological progression of the disease.
A study using the reporter gene encoding for green fluorescent protein was also able to show liposome mediated expression in three 30 μm thick coronal sections of the rat brain. Although no quantification of the transfection efficiency was performed, immunostaining using anti heat shock protein 70 (Hsp 70) antibodies revealed cellular staining when plasmid Hsp 70 was administered with the modified DOPE liposome. Hsp 70 holds critical cellular roles in protein folding and rescue of misfolded proteins.145 Therefore when misfolded proteins play an important role in pathogenesis,146 inducement of over-expression of Hsp 70 may provide potential therapeutic strategies to reduce protein aggregates such as α-synuclein in PD, hyperphosphorylated tau in AD and fronto-temporal dementia, prion in Prion disease, and huntingtin in HD.147
While early studies of nucleic acid delivery to the brain were largely based on over-expression of a protein by transfection using plasmid DNA, the delivery of silencing constructions such as small-interfering RNA (siRNA) to the brain is an emerging field of interest.148,149 However, one such early proof of concept study used a polymeric vector based on PEI to deliver a luciferase encoding plasmid whilst co-administering a luciferase silencing construct delivered by a liposomal formulation.150 The study showed that the up-regulation of luciferase expression can be reduced when the siRNA was co administered. In this study there was a discrepancy between the effectiveness of the delivery vehicles depending on the nucleic acids delivered. The polyethylenimine based polymeric vector produced greater over-expression of luciferase while the liposome formulation showed more effective gene silencing.
3.2 Polymeric gene vectors
The most studied polymer for delivery of nucleic acids to the brain is the cationic polymer PEI. With every third atom a protonizable amine group, it has a high charge density, allowing the ability to condense nucleic acids into polyplexes. Linear and branched versions of PEI have been developed for the purpose of gene delivery, with linear PEI containing only secondary amines, as opposed to branched PEI that contains primary, secondary and tertiary amines.151 The first study that used PEI to deliver plasmid DNA into the mammalian brain was published in 1995.40 Branched PEI was used to deliver a plasmid encoding luciferase into newborn mouse brain at an approximate site of the striatum. One day post injection, the animals were sacrificed and, via an enzymatic reaction, were shown to have higher transgene expression compared to those treated with the naked plasmid (no complexing agent). This study was followed up by the same group using a lower molecular weight PEI (Mw = 25 kDa) in the adult mouse brain,152 then linear PEI (Mw = 22 kDa) delivered via intraventricular injection into the newborn and adult mouse brain.154–156 These studies showed that the polyplex characteristics vary dramatically depending whether a salt solution is used (large polyplex aggregates) or a 5% glucose formulation is used (dispersed polyplexes)154 and that intraventricular injection results predominantly in the transfection of neural stem cells and their immediate progeny.156 Interestingly, lowering the amount of DNA delivered,155 or co-expressing a pro-survival molecule B-cell lymphoma-extra-large (Bcl-xL) increased the transgene expression,155,156 indicating that perhaps the toxicity of the vector is limiting transfection performance.The first study delivering a functional gene (as opposed to marker genes) via the PEI vector delivered a plasmid encoding the dopamine transporter (DAT) to the rat striatum.157 DAT regulates the synaptic concentration of dopamine in a spatial and temporal manner, thus having possible implications in PD and/or the therapeutics of schizophrenia.157 Using plasmids encoding for either over-expression or down-regulation (antisense sequence), the levels of DAT could be increased or decreased respectively when delivered via the linear PEI vector.
The modification of non-viral vectors for use in the CNS will largely be dealt with in Section 3.4 with the addition of targeting moieties for improved efficacy in the target tissue. However, branched 25 kDa PEI has been shown to be more efficacious at CNS gene transfer by the addition of α-malemidyl-N-hydroxysuccinimidyl functionalised 2 kDa PEG strands directly to the amine groups of the PEI molecule.158 The PEGylated PEI showed reduced toxicity in vitro, and although no comparison was made with naked DNA, PEGylated PEI showed greater luciferase activity over the non modified PEI in many parts of the CNS following lumbar injection into the rat subarachnoid space. The same group performed a similar surgical procedure but with a transfection vector consisting of low molecular weight (600 Da) PEI conjugated to a β-cyclodextrin core.161 The vector showed a similar transfection profile as the 25 kDa branched PEI control in vivo, but with much less toxicity in vitro. Interestingly, this research group had also developed a biodegradable polyaminoethyl propylene phosphate (PPE-AE) polymer which they used to deliver a luciferase plasmid to the mouse cerebellomedullary cistern.159 In these studies, PPE-EA was compared to 25 kDa branched PEI and naked plasmid, revealing that after 28 days the PPE-EA was able to mediate higher luciferase activity than the comparison injections. In more recent years, the shift of focus has been towards the development of biodegradable transfection agents as a means for safer and less toxic agents,46 the same rationale behind the developments of the PPE-AE agent.
To compare the efficacy of luciferase either administered as a naked plasmid, via PEI or via an adenovirus vector, a study was conducted in the brain of adult dogs.162 Bearing in mind the small repetition number, it was clear that transfection via the PEI vector was approximately 1.5% that of adenoviral transfection activity. However a lower host response was observed with PEI mediated gene delivery in terms of white blood cell count in the cerebrospinal fluid and observation of inflammatory cells at the site of injection.
A significant advancement in the ability to reduce the size of the polyplex came through the use of polylysine conjugated to 10 kDa PEG moieties (later L-Cysteinyl-poly-L-lysine (CK30PEG10k)). Initial studies showed polyplex sizes as low as 25 nm and the ability to transfect post-mitotic human neuroblastoma cells in vitro.167 The size of these compacted DNA nanoparticles was further reduced to 9–11 nm, deemed small enough to cross the nuclear pore,168 and later they were used for gene delivery applications in the brain.61,164,165,169 A large increase of luciferase activity over naked DNA controls was observed, the duration of which can be vastly extended through the use of the polyubiquitin C promoter instead of the cytomegalovirus promoter.61 Transgene expressions of up to 56 days (luciferase, maximum period assessed61,165) or 98 days (GDNF164) was observed in the rat striatum. Live in vivo bioluminescence and ex vivo bioluminescence studies showed large portions of the striatum exhibiting luciferase activity.165 Whilst the bulk of the studies that have used polymeric vectors have not assessed functional outcomes in pre-clinical models, the recent ability to vastly extend transgene expression time is a significant advancement for non-viral transfection in the context of future therapies for neurological disorders.
3.2 Other gene vectors
Whilst the majority of non-viral transfection studies in the brain have used vectors that fall under the category of liposomal or polymeric agents, other vectors have been used for transgene expression analysis. Nanoparticles comprised of calcium phosphate, using the positive charge of the calcium ion to condense DNA, have been used to deliver β-galactosidase to the rat substantia nigra (SN).170 The cell bodies of dopaminergic neurons lost during PD reside in the substantia nigra pars compacta and terminate in the striatum, thus the SN provides a site of interest for both modelling and therapeutic applications in PD.171–174 However, no β-galactosidase positive staining was observed after seven days, and expression was observed in an insufficient number of cells after 28 days to warrant continuation of studies (the authors used PEI and the herpes simplex virus type-1 (HSV) instead for further study).Amine functionalised nanoparticles prepared from triethoxyvinylsilane and 3-aminopropyltriethoxysilane using organic micelles as a sacrificial template have been used as a highly effective gene vector in the mouse brain.175–177 Like the compacted DNA nanoparticles,61,164,165 these organically modified silica nanoparticles (ORMOSIL), are able to condense DNA to diameters of the order of 20 nm,178 much smaller than conventional polyplex/lipoplexes. Although no quantification was performed, ORMOSIL mediated delivery of the plasmid encoding enhanced GFP (EGFP) was observed in the substantia nigra, or in regions surrounding the lateral ventricles, depending on the injection site. Furthermore, delivery of the plasmid encoding the nucleus targeting fibroblast growth factor receptor type 1 (FGFR1) to the lateral ventricle affected the proliferation of the stem progenitor cells in the sub ventricular zone, as analysed by a reduction in DNA incorporation of bromodeoxyuridine. A follow up study showed that this withdrawal of the stem progenitor cells from the cell cycle, via FGFR1 transfection with ORMOSIL, caused their differentiation into double cortin expressing migratory neuroblasts and neurons, followed by subsequent migration.177
Although less arduous than cell transplantation therapies, generating neurons via non-viral modification of the host's own stem cells perhaps offers less control over the placement of these neurons, as this work showed migration as far as the cortex and olfactory bulbs. However, if properly harnessed, such cell migration distances will allow for cell replacement applications in the striatum (HD and PD), motor cortex (ALS) and cerebral cortex (AD).
High aspect ratio materials have also been used to deliver siRNA to the mammalian brain in the form of amine functionalised multiwalled carbon nanotubes (f-CNT).179 These were used to condense siRNA to knockdown caspase-3, a regulator of cell death, in the mouse or rat motor cortex. Endothelin-1 was administered to model stroke by causing vasoconstriction and ischemic injury either prior to, or post the siRNA treatment. Whilst no quantitative PCR was performed to analyse the caspase-3 expression, f-CNT mediated delivery prior to injury reduced the number of apoptosis stained cells in mice (via TUNEL staining), and maintained the rat skilled reaching score of the prior-to-injury training period. Treatment one hour post injury gave a strong trend towards a lower number of apoptotic cells; however this was not statistically different from control groups. Nevertheless these studies, and others using the cerebral artery occlusion model,163,166 prove the concept of non-viral siRNA mediated improvement in cell survival.
Exosomes, cell secreted vesicles, have recently been used to deliver either GAPDH siRNA or BACE1 siRNA to the mouse brain after systemic delivery.181 BACE1 is a protease that is responsible for N-terminal cleavage of the amyloid precursor protein, thus has implications for AD as they form β-amyloid peptides, a major component of insoluble plaques found in AD patients.182,183 Only when the rabies virus glycoprotein was incorporated into the exosome membrane could specific knockdown be observed in the brain, and not in off-site organs.181 These targeted exosomes mediated over 50% BACE1 knockdown in the gene expression and the associated protein concentration when non-targeted administration showed no change. For the true realisation of systemically delivered gene vectors for neurological disease therapies, the above mentioned specificity of targeting must be accomplished to avoid off-site side effects.
3.4 Targeted vectors
In 1984 an important discovery was published that would have implications for subsequent research targeting the brain. The study showed that when monoclonal antibodies (mAb) against transferrin (Tf) receptors were administered systemically they labelled the capillaries in the brain, but not other tissues.184 Research groups optimising non-viral gene therapy to the brain have since extensively used either mAb against Tf receptors or used the Tf glycoprotein itself in the construct preparation. Perhaps the most reported has been the systemic use of PEGylated liposomes for brain applications by way of having a mAb (usually against the Tf receptor or human insulin receptor) incorporated into their structure.50,59,185–195 These Trojan horse liposomes or PEGylated immunoliposomes (PILs) have delivered complementary DNA, plasmid DNA and siRNA across the blood brain barrier of mice, rats and non-human primates for a variety of applications. Reducing off-site effects has been achieved through selection of specific plasmid promoters and has been reviewed extensively elsewhere.196 Other groups that have used Tf for targeting non-viral vectors have also used PEGylated liposomes as the basic vector,197–200 or as in one case, PEI that was delivered within a liposome to increase circulation time.201A range of other vectors and targeting moieties have been developed for the delivery of nucleic acids to the brain, however, of particular note is neurotensin (NT) targeted poly-L-lysine (PLL).202–204 By adding the NT peptide to the PLL construct the authors were able to specifically target the dopaminergic neurons. Furthermore, intranigral injection of this vector carrying the GDNF encoding gene was able to ameliorate the behavioural effects of the 6-OHDA lesion,204 suggesting a potential future role in PD therapeutics.205 However, it is not only the brain that has been the focus of targeting gene delivery in the context of neurological disorder applications. Peripheral neurons have also been the site of proof-of-concept studies206 or GDNF over-expression via neurotrophic receptor targeting (p75NTR).207
A study that compared Tf with another targeting ligand, lactoferrin (Lf), argued that although Tf receptors are only found on the capillaries of the brain, the binding sites were often occupied by high plasma levels of Tf and that perhaps Lf may allow for better brain uptake.58 Tf and Lf were covalently added to PEGylated PAMAM dendrimers, and compared to PEGylated or unmodified PAMAM controls for bio-distribution and luciferase expression. Whilst Lf modification mediated the highest brain uptake/transgene expression, with Tf also above controls, a large amount of off target uptake and expression was also occurring. Approximately 0.15% injected dose per gram uptake was observed in the brain in comparison to 4% in the kidney. Further studies also demonstrated the ability of Lf modified PAMAM to deliver GDNF to the brain in PD models.208,209
Much of the research on polymeric vectors has focused on PEI, PAMAM and PLL, however another monomer often used for the preparation of gene vectors is 2-(dimethylamino)ethyl methacrylate (DMAEMA).56,210,211 Altering the polymer structure of polyDMAEMA can alter the transfection capability of the vector45 and with carefully controlled chemistries212,213 a knot structured polyDMAEMA can be formed that allows neuronal cell transfection.214 An alternative method has been to use PEGylated DMAEMA coupled with a different blood brain barrier targeting peptide (termed TGN).215 Whilst the non-targeted vectors largely accumulated in the kidneys, TGN conjugated vectors mediated greater transfection in the brain, showing that systemic therapies may provide a route to future therapeutic intervention once the problem of off-site expression is fully resolved.
4 Ex vivo strategies
4.1 Current limitations
The use of donor cells to deliver therapeutic transgene molecules to the host tissue is an attractive alternative to in vivo gene delivery.216 By removal of the vector system required for transgene expression, toxic effects of the vector itself can be bypassed. In addition, if stable transfection has occurred (gene inserted into the chromosomal DNA of the cell) the desired protein will continue to be produced during the cells’ lifetime. Stem cells such as mesenchymal stem cells are typically hard to transfect, but delivery via poly(amidoamine) dendrimers can be improved by altering hydrophobicity of the vector or adding peptide molecules such as RGD.217–219 Aside from the inherent factors such as cell sourcing, transfection and care during expansion,220 a major limitation of current ex vivo gene therapy for neurodegenerative therapies is the poor cell survival that follows transplantation into the host brain.221,222 Studies have shown that the volume of the rat mesenchymal stem cells (rMSCs) grafted into the adult brain is reduced by 35% in as few as four days post transplantation.223 Another recent study that observed the graft behaviour of different stem cell grafts reported that all cell types showed survival rates of approximately 3% at two weeks post transplantation into the CNS.224 It should also be noted that extended culture time prior to transplantation also has a detrimental effect on cell survival post transplantation.225,226 Whilst previous studies have shown the ability of GDNF producing rMSCs to produce local striatal neuro-protective effects against a 6-OHDA lesion, the area of secreted GDNF decreased in accordance with the reduction in graft volume.227 Examination of the host response to cell transplantation reveals that grafts typically cause a microglial reaction throughout the graft site and an astrocyte/gliotic scar that surrounds the transplant.223,224,228 Although it has been suggested that hypoxia and host graft site disruption may be factors contributing to transplant death, it has also been suggested that the innate host reaction is largely responsible.2294.2 Use of biomaterials
As a means of protecting transplanted cells from the host glial cell response, polymers such as polyethersulfone have been used for cell encapsulation in long thin fibres implanted into the brain.230 Brain implantable devices for growth factor delivery,231,232 rely on the design of inert/non inflammatory biomaterials.233 Methods such as these allow the option of device retraction,234 and allow enough GDNF release in the striatum for behavioural recovery in 6-OHDA Parkinsonian rats.230 Furthermore, devices that were explanted after 13 weeks showed numerous surviving cells and GDNF production was at the level of that pre-transplantation. However, such ability to promote cell survival through a physical barrier (that only allows small molecule diffusion), comes at the cost of a large region of tissue disruption by the 500 μm diameter fibre 5 mm in length. Furthermore, as the device extends through the cortex, GDNF release would presumably also occur in off-site regions.Recent clinical trials in patients with AD have shown that NGF releasing implants were well tolerated; however, functional benefits were only seen in two out of six patients.235 Prior mixing of cells with injectable materials that form soft hydrogels upon injection may attempt to overcome some of the above shortcomings whilst still providing a physical substrate to adhere to and a barrier to the host immune environment.236–238
Hydrogels have been used as a regenerative platform for axonal re-growth both with239 and without transplanted cells.80,240,241 In terms of providing a barrier, when a hydrogel consisting of heparin covalently crosslinked with star-PEG was injected into the rat striatum, minimal penetration of the hydrogel by host cells was observed.242 To assess the cell attachment properties in vitro, functionalisation of the hydrogel with RGD peptides and basic fibroblast growth factor was performed. The addition of both showed far greater cell numbers attached to the hydrogel and viable than the non-functionalised hydrogel or poly-D-lysine control. A study that transplanted induced pluripotent stem-neural progenitor cells with or without a surrounding synthetic hydrogel noted that the hydrogel impeded graft infiltration by astrocytes, but not by microglia.248 As the hydrogel mediated 1.2% cell survival compared to 0% survival in PBS (perhaps a transplantation media would be more supportive), no graft comparison could be made. However, recent studies have shown this to be the case even for grafted cells without a supportive matrix.223 Interestingly another study using a hyaluronan–heparin–collagen hydrogel promoted embryonic stem–neural progenitor cell survival in the infarct cavity of focal ischemia stroke.249 The authors reported a reduction of microglial infiltration, but no reduction is angiogenesis (linked to activated microglia250).
In vitro studies have shown that viability of cells grown within a hydrogel can be improved via BDNF functionalisation251 and that the presence of extracellular matrix proteins provides a more permissive environment for re-growth.252 Indeed, hydrogels often require the incorporation of natural peptides such as RGD239 or polypeptides such as poly-D-lysine253,254 to improve cell adhesion. An extracellular matrix based hydrogel termed Matrigel™, a hydrogel composed of extracellular matrix proteins from mouse sarcomas, has been also used to improve stem cell survival post transplantation.255 Matrigel™ caused a reduction in CD45 positive (microglia, macrophages and lymphocytes) in the graft at the early time-point (24 hours), whilst another study observed less graft infiltration of GFAP positive cells (astrocytes).256 Future strategies to promote cell survival post-transplantation could arise through the provision of a stable environment for the cells in vitro prior to injection, which is transplanted together during the grafting process. To facilitate this, the inherent shear stresses encountered during injection must be harnessed.257 Cell containing hydrogels of gelatinous texture prior to transplantation can be tailored to shear-thin upon injection, and re-gel at the transplant site.258
Another means of providing physical support for transplanted stem cells has been provided by the co-injection of poly(lactic-co-glycolic acid) (PLGA) microparticles.259 In addition, functionalising synthetic microparticles so as to release growth factors provides a synthetic stimulatory microenvironment.260 PLL coated (PLGA) microparticles engineered to release NGF were able to control stem cell survival post transplantation. Lastly, another non-gelatinous and growth factor mediated means of enhancing cell survival was provided through the injection of compacted DNA nanoparticles carrying the GDNF encoding gene one week prior to transplantation.169 The 6-OHDA lesion model of PD was induced one week prior to nanoparticle injection then fetal dopaminergic neurons were transplanted a week later. A four-fold increase in graft volume was observed compared to saline controls.
5 Host response considerations
Studies have specifically investigated the toxicity and host response to polymeric and liposomal vectors in the rat striatum in comparison to an adeno associated virus control.29 Often, small regions of tissue damage were observed at the injection site, that were not present for PBS or naked DNA controls, or when the vectors were packaged in an extracellular matrix sustained delivery device. This, therefore, provides further justification for peripheral administration (intranasal,261 or intramuscular262) for subsequent transfer to the CNS. The toxicity of nanomaterials in the brain has been reviewed elsewhere;263 however it is important to note that the safety concerns associated with high aspect ratio materials such as carbon nanotubes have been elevated after the publication of an asbestos type response in the mouse peritoneal cavity.264 The study showed that when a dose of 100 μg ml−1 was administered, the elevated inflammatory response was only seen due to the long/untangled materials. Functionalisation of carbon nanotubes requires modification of the pristine graphitic sheets, thus in effect this introduces defects and sites for enzymatic degradation.265 Aside from increasing dispersion and excretion rates,266 a specific study of the toxicity and short-term fate of (250 μg ml−1) f-CNT in the (mouse) cortex showed that indeed structural deformation and partial degradation of the functionalised materials had occurred as early as 2 days.267 Gold nanorods have also been used for proof-of-concept knockdown of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the rat hippocampus by siRNA delivery.180 However, these nanorods are of lower aspect ratio and complex DNA into complexes typically less than 100 nm in length.Whilst for many in vivo applications, a biodegradable material is favourable to allow eventual clearance; this must also be viewed in context. Recent studies have shown that biodegradable hydrogels implanted into the rat brain can cause a more elevated astrocyte response than a non-degradable counterpart.104 The effect of a continually changing environment along with increasing number of degradation products necessitates a more active host response.268 Macroscale implants, for instance probes for deep brain stimulation (DBS), composed of inert materials are well tolerated by the host immune system and are regularly used in clinical practice. The biggest problem of adverse side effects of DBS is thought to arise through either incorrect implantation or hardware failure.269 Therefore non-degradable implants may have chronic issues. The acute issues associated with degradable materials can be addressed through chemical modifications or functionalisation. Thus biomaterial design must always be tailored specifically to the requirements of the application.
6 Summary
As the prevalence and healthcare costs of neurodegenerative disorders rises, so too does the requirement for new therapeutic strategies. Through the design of new materials, different and combinatorial approaches may be sought that could mediate disease modifying gene therapies. Targeted non-viral vectors are becoming increasingly efficacious, whilst methods of delivery are improving, for example; transferrin mediated transport across the BBB. The reduction of off-target effects such as transgene expressing in the liver must remain a high priority to increase the safety profile of such targeted/systemic techniques. Sustained delivery offers the potential to lengthen the therapeutic effect of gene therapy strategies, which could be beneficial for the more progressive disorders. Both synthetic and natural materials (often a combination of both) have been designed using structural and chemical guidance cues to improve recovery after spinal cord injury in preclinical models. The later have also been mediated through gene therapy functionalisation within the guiding scaffold or conduit. Soft hydrogels hold the potential to assist gene therapies for neurological disorders through a range of avenues such as better control of gene vector delivery, enhancing vector placement, improving ex vivo transplant survival or a combination of the above. Biomaterial strategies for neurodegenerative diseases are in their relative infancy, implying that the full potential has not yet been explored, but also that safety profiles must be thoroughly characterised in the target tissue. Long-term effects of implants must be analysed including host reactions to degradation products. However, careful rational design and feedback driven optimisation will allow the production of highly effective biomaterials for use in neurodegenerative disorder therapies in the future.Acknowledgements
The authors wish to thank Science Foundation of Ireland, Strategic Research Cluster (SRC) (grant no. 07/SRC/B1163) for financial support.Notes and references
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer and M. Scazufca, Lancet, 2005, 366, 2112–2117 CrossRef.
- D. Twelves, K. S. M. Perkins and C. Counsell, Mov. Disord., 2003, 18, 19–31 CrossRef.
- G. Alves, E. Forsaa, K. Pedersen, M. Dreetz Gjerstad and J. Larsen, J. Neurol., 2008, 255, 18–32 CrossRef.
- M. H. Polymeropoulos, C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. Di Iorio, L. I. Golbe and R. L. Nussbaum, Science, 1997, 276, 2045–2047 CrossRef CAS.
- C. Paisán-Ruíz, S. Jain, E. W. Evans, W. P. Gilks, J. Simón, M. Van Der Brug, A. L. De Munain, S. Aparicio, A. M. Gil, N. Khan, J. Johnson, J. R. Martinez, D. Nicholl, I. M. Carrera, A. S. Peňa, R. De Silva, A. Lees, J. F. Martí-Massó, J. Pérez-Tur, N. W. Wood and A. B. Singleton, Neuron, 2004, 44, 595–600 CrossRef.
- A. Zimprich, S. Biskup, P. Leitner, P. Lichtner, M. Farrer, S. Lincoln, J. Kachergus, M. Hulihan, R. J. Uitti, D. B. Calne, A. J. Stoessl, R. F. Pfeiffer, N. Patenge, I. C. Carbajal, P. Vieregge, F. Asmus, B. Müller-Myhsok, D. W. Dickson, T. Meitinger, T. M. Strom, Z. K. Wszolek and T. Gasser, Neuron, 2004, 44, 601–607 CrossRef CAS.
- M. E. MacDonald, C. M. Ambrose, M. P. Duyao, R. H. Myers, C. Lin, L. Srinidhi, G. Barnes, S. A. Taylor, M. James, N. Groot, H. MacFarlane, B. Jenkins, M. A. Anderson, N. S. Wexler, J. F. Gusella, G. P. Bates, S. Baxendale, H. Hummerich, S. Kirby, M. North, S. Youngman, R. Mott, G. Zehetner, Z. Sedlacek, A. Poustka, A.-M. Frischauf, H. Lehrach, A. J. Buckler, D. Church, L. Doucette-Stamm, M. C. O'Donovan, L. Riba-Ramirez, M. Shah, V. P. Stanton, S. A. Strobel, K. M. Draths, J. L. Wales, P. Dervan, D. E. Housman, M. Altherr, R. Shiang, L. Thompson, T. Fielder, J. J. Wasmuth, D. Tagle, J. Valdes, L. Elmer, M. Allard, L. Castilla, M. Swaroop, K. Blanchard, F. S. Collins, R. Snell, T. Holloway, K. Gillespie, N. Datson, D. Shaw and P. S. Harper, Cell, 1993, 72, 971–983 CrossRef.
- C. Humphries, Nature, 2012, 484, S10–S10 CrossRef.
- R. Levi-Montalcini and P. U. Angeletti, Physiol. Rev., 1968, 48, 534–569 CAS.
- J. Leibrock, F. Lottspeich, A. Hohn, M. Hofer, B. Hengerer, P. Masiakowski, H. Thoenen and Y.-A. Barde, Nature, 1989, 341, 149–152 CrossRef CAS.
- L. Lin, D. Doherty, J. Lile, S. Bektesh and F. Collins, Science, 1993, 260, 1130–1132 CAS.
- Y.-A. Barde, Neuron, 1989, 2, 1525–1534 CrossRef CAS.
- F. Hefti, J. Neurosci., 1986, 6, 2155–2162 CAS.
- L. R. Williams, S. Varon, G. M. Peterson, K. Wictorin, W. Fischer, A. Bjorklund and F. H. Gage, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 9231–9235 CrossRef CAS.
- L. Kromer, Science, 1987, 235, 214–216 CAS.
- F. Hefti and W. J. Weiner, Ann. Neurol., 1986, 20, 275–281 CrossRef CAS.
- L. R. Williams, Exp. Neurol., 1991, 113, 31–37 CrossRef CAS.
- J. Winkler, G. A. Ramirez, H. G. Kuhn, D. A. Peterson, P. A. Day-Lollini, G. R. Stewart, M. H. Tuszynski, F. H. Gage and L. J. Thal, Ann. Neurol., 1997, 41, 82–93 CrossRef CAS.
- M. Eriksdotter Jonhagen, A. Nordberg, K. Amberla, L. Bäckman, T. Ebendal, B. Meyerson, L. Olson, Seiger, M. Shigeta, E. Theodorsson, M. Viitanen, B. Winblad and L. O. Wahlund, Dement. Geriatr. Cogn. Disord., 1998, 9, 246–257 CrossRef CAS.
- C. J. Emmett, G. R. Stewart, R. M. Johnson, S. P. Aswani, R. L. Chan and L. B. Jakeman, Exp. Neurol., 1996, 140, 151–160 CrossRef CAS.
- S. S. Gill, N. K. Patel, G. R. Hotton, K. O'Sullivan, R. McCarter, M. Bunnage, D. J. Brooks, C. N. Svendsen and P. Heywood, Nat. Med., 2003, 9, 589–595 CrossRef CAS.
- J. T. Slevin, G. A. Gerhardt, C. D. Smith, D. M. Gash, R. Kryscio and B. Young, J. Neurosurg., 2005, 102, 216–222 CrossRef CAS.
- J. T. Slevin, D. M. Gash, C. D. Smith, G. A. Gerhardt, R. Kryscio, H. Chebrolu, A. Walton, R. Wagner and A. B. Young, Neurosurg. Focus, 2006, 20, 1–7 CrossRef.
- A. E. Lang, S. Gill, N. K. Patel, A. Lozano, J. G. Nutt, R. Penn, D. J. Brooks, G. Hotton, E. Moro, P. Heywood, M. A. Brodsky, K. Burchiel, P. Kelly, A. Dalvi, B. Scott, M. Stacy, D. Turner, V. G. F. Wooten, W. J. Elias, E. R. Laws, V. Dhawan, A. J. Stoessl, J. Matcham, R. J. Coffey and M. Traub, Ann. Neurol., 2006, 59, 459–466 CrossRef CAS.
- T. B. Sherer, B. K. Fiske, C. N. Svendsen, A. E. Lang and J. W. Langston, Mov. Disord., 2006, 21, 136–141 CrossRef.
- P. Aebischer and J.-L. Ridet, Trends Neurosci., 2001, 24, 533–540 CrossRef CAS.
- R. H. Bobo, D. W. Laske, A. Akbasak, P. F. Morrison, R. L. Dedrick and E. H. Oldfield, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2076–2080 CrossRef CAS.
- D. M. Gash, Z. Zhang, Y. Ai, R. Grondin, R. Coffey and G. A. Gerhardt, Ann. Neurol., 2005, 58, 224–233 CrossRef CAS.
- B. Newland, T. C. Moloney, G. Fontana, S. Browne, M. T. Abu-Rub, E. Dowd and A. S. Pandit, Biomaterials, 2013, 34, 2130–2141 CrossRef CAS.
- E. Check, Nature, 2002, 420, 116–118 CrossRef CAS.
- S. Hacein-Bey-Abina, C. von Kalle, M. Schmidt, F. Le Deist, N. Wulffraat, E. McIntyre, I. Radford, J.-L. Villeval, C. C. Fraser, M. Cavazzana-Calvo and A. Fischer, N. Engl. J. Med., 2003, 348, 255–256 CrossRef.
- S. Hacein-Bey-Abina, C. Von Kalle, M. Schmidt, M. P. McCormack, N. Wulffraat, P. Leboulch, A. Lim, C. S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P. Fraser, J. I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A. Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E. Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L. E. Leiva, M. Wissler, C. Prinz, T. H. Rabbitts, F. Le Deist, A. Fischer and M. Cavazzana-Calvo, Science, 2003, 302, 415–419 CrossRef CAS.
- R. Deyner and M. R. Douglas, Parkinson's Dis., 2012, 2012, 1–13 Search PubMed.
- W. J. Marks Jr., J. L. Ostrem, L. Verhagen, P. A. Starr, P. S. Larson, R. A. E. Bakay, R. Taylor, D. A. Cahn-Weiner, A. J. Stoessl, C. W. Olanow and R. T. Bartus, Lancet Neurol., 2008, 7, 400–408 CrossRef.
- S. Browne, G. Fontana, B. J. Rodriguez and A. Pandit, Mol. Pharm., 2012, 9, 3099–3106 CrossRef CAS.
- M. T. Abu-Rub, K. L. Billiar, M. H. van Es, A. Knight, B. J. Rodriguez, D. I. Zeugolis, S. McMahon, A. J. Windebank and A. Pandit, Soft Matter, 2011, 7, 2770–2781 RSC.
- C. Holladay, M. Keeney, B. Newland, A. Mathew, W. Wang and A. Pandit, Nanoscale, 2010, 2, 2718–2723 RSC.
- M. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS.
- C. Tros de Ilarduya, Y. Sun and N. Düzgüneş, Eur. J. Pharm. Sci., 2010, 40, 159–170 CrossRef CAS.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS.
- J. J. Green, R. Langer and D. G. Anderson, Acc. Chem. Res., 2008, 41, 749–759 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 1999, 45, 268–275 CrossRef CAS.
- B. Newland, H. Tai, Y. Zheng, D. Velasco, A. Di Luca, S. M. Howdle, C. Alexander, W. Wang and A. Pandit, Chem. Commun., 2010, 46, 4698–4700 RSC.
- C. V. Synatschke, A. Schallon, V. R. Jérôme, R. Freitag and A. H. E. Müller, Biomacromolecules, 2011, 12, 4247–4255 CrossRef CAS.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS.
- M. Ogris, S. Brunner, S. Schüller, R. Kircheis and E. Wagner, Gene Ther., 1999, 6, 595–605 CrossRef CAS.
- M. Neu, O. Germershaus, M. Behe and T. Kissel, J. Controlled Release, 2007, 124, 69–80 CrossRef CAS.
- J. G. Schellinger, J. A. Pahang, R. N. Johnson, D. S. H. Chu, D. L. Sellers, D. O. Maris, A. J. Convertine, P. S. Stayton, P. J. Horner and S. H. Pun, Biomaterials, 2013, 34, 2318–2326 CrossRef CAS.
- N. Shi, Y. Zhang, C. Zhu, R. J. Boado and W. M. Pardridge, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12754–12759 CrossRef CAS.
- H. Wei, J. G. Schellinger, D. S. H. Chu and S. H. Pun, J. Am. Chem. Soc., 2012, 134, 16554–16557 CrossRef CAS.
- R. M. Levine, C. M. Scott and E. Kokkoli, Soft Matter, 2012, 9, 985–1004 RSC.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- M. Lee and S. W. Kim, Pharmacol. Res., 2005, 22, 1–10 CrossRef CAS.
- C. Tu, N. Li, L. Zhu, L. Zhou, Y. Su, P. Li and X. Zhu, Polym. Chem., 2013, 4, 393–401 RSC.
- S. Kolli, S.-P. Wong, R. Harbottle, B. Johnston, M. Thanou and A. D. Miller, Bioconjugate Chem., 2013, 24, 314–332 CrossRef CAS.
- R. Huang, W. Ke, Y. Liu, C. Jiang and Y. Pei, Biomaterials, 2008, 29, 238–246 CrossRef CAS.
- C.-F. Xia, R. J. Boado, Y. Zhang, C. Chu and W. M. Pardridge, J. Gene Med., 2008, 10, 306–315 CrossRef CAS.
- A. M. O'Mahony, B. M. D. C. Godinho, J. Ogier, M. Devocelle, R. Darcy, J. F. Cryan and C. M. O'Driscoll, ACS Chem. Neurosci., 2012, 3, 744–752 CrossRef CAS.
- D. M. Yurek, A. M. Fletcher, G. M. Smith, K. B. Seroogy, A. G. Ziady, J. Molter, T. H. Kowalczyk, L. Padegimas and M. J. Cooper, Mol. Ther., 2009, 17, 641–650 CrossRef CAS.
- B. Newland, Y. Zheng, Y. Jin, M. Abu-Rub, H. Cao, W. Wang and A. Pandit, J. Am. Chem. Soc., 2012, 134, 4782–4789 CrossRef CAS.
- J.-P. Benoit, N. Faisant, M.-C. Venier-Julienne and P. Menei, J. Controlled Release, 2000, 65, 285–296 CrossRef CAS.
- B. Song, J. Song, S. Zhang, M. A. Anderson, Y. Ao, C.-Y. Yang, T. J. Deming and M. V. Sofroniew, Biomaterials, 2012, 33, 9105–9116 CrossRef CAS.
- P. Menei, V. Daniel, C. Montero-Menei, M. Brouillard, A. Pouplard-Barthelaix and J. P. Benoit, Biomaterials, 1993, 14, 470–478 CrossRef CAS.
- P. J. Camarata, R. Suryanarayanan, D. A. Turner, R. G. Parker and T. J. Ebner, Neurosurgery, 1992, 30, 313–319 CrossRef CAS.
- P. Menei, J. M. Pean, V. Nerrière-Daguin, C. Jollivet, P. Brachet and J. P. Benoit, Exp. Neurol., 2000, 161, 259–272 CrossRef CAS.
- M. S. Ward, A. Khoobehi, E. B. Lavik, R. Langer and M. J. Young, J. Pharm. Sci., 2007, 96, 558–568 CrossRef CAS.
- G. J. R. Delcroix, E. Garbayo, L. Sindji, O. Thomas, C. Vanpouille-Box, P. C. Schiller and C. N. Montero-Menei, Biomaterials, 2011, 32, 1560–1573 CrossRef CAS.
- J. L. Roam, H. Xu, P. K. Nguyen and D. L. Elbert, Biomaterials, 2010, 31, 8642–8650 CrossRef CAS.
- S. Özbas-Turan, C. Aral, L. Kabasakal, M. Keyer-Uysal and J. Akbuga, J. Pharm. Sci., 2003, 6, 27–32 Search PubMed.
- C. Helary, S. Browne, A. Mathew, W. Wang and A. Pandit, Acta Biomater., 2012, 8, 4208–4214 CrossRef CAS.
- B. C. Dash, S. Mahor, O. Carroll, A. Mathew, W. Wang, K. A. Woodhouse and A. Pandit, J. Controlled Release, 2011, 152, 382–392 CrossRef CAS.
- G. Réthoré, A. Mathew, H. Naik and A. Pandit, Tissue Eng., Part C, 2009, 15, 605–613 CrossRef.
- S. Mahor, E. Collin, B. C. Dash and A. Pandit, Curr. Drug Delivery, 2011, 8, 354–362 CrossRef CAS.
- M. M. Kulkarni, U. Greiser, T. O'Brien and A. Pandit, Mol. Pharm., 2010, 8, 439–446 Search PubMed.
- D. M. Yurek, A. M. Fletcher, G. M. Smith, K. B. Seroogy, A. G. Ziady, J. Molter, T. H. Kowalczyk, L. Padegimas and M. J. Cooper, Mol. Ther., 2009, 17, 641–650 CrossRef CAS.
- Z.-Y. Chen, C.-Y. He, A. Ehrhardt and M. A. Kay, Mol. Ther., 2003, 8, 495–500 CrossRef CAS.
- F. Cui, W. Tian, S. Hou, Q. Xu and I. S. Lee, J. Mater. Sci.: Mater. Med., 2006, 17, 1393–1401 CrossRef CAS.
- S. Woerly, P. Petrov, E. Sykova, T. Roitbak, Z. Simonova and A. R. Harvey, Tissue Eng., Part A, 1999, 5, 467–488 CrossRef CAS.
- C. Martínez-Ramos, A. Vallés-Lluch, J. M. G. Verdugo, J. L. G. Ribelles, J. A. Barcia Albacar, A. B. Orts, J. M. Soria López and M. M. Pradas, J. Biomed. Mater. Res. Part A, 2012, 100, 3276–3286 CrossRef.
- N. N. Madigan, S. McMahon, T. O'Brien, M. J. Yaszemski and A. J. Windebank, Respir. Physiol. Neurobiol., 2009, 169, 183–199 CrossRef CAS.
- W. T. Daly, L. Yao, M. T. Abu-rub, C. O'Connell, D. I. Zeugolis, A. J. Windebank and A. S. Pandit, Biomaterials, 2012, 33, 6660–6671 CrossRef CAS.
- F. Yang, R. Murugan, S. Wang and S. Ramakrishna, Biomaterials, 2005, 26, 2603–2610 CrossRef CAS.
- S. Stokols and M. H. Tuszynski, Biomaterials, 2006, 27, 443–451 CrossRef CAS.
- B. Lanfer, A. Hermann, M. Kirsch, U. Freudenberg, U. Reuner, C. Werner and A. Storch, Tissue Eng., Part A, 2010, 16, 1103–1113 CrossRef CAS.
- K. Moore, M. MacSween and M. Shoichet, Tissue Eng., Part A, 2006, 12, 267–278 CrossRef CAS.
- C. E. Kang, M. D. Baumann, C. H. Tator and M. S. Shoichet, Cells Tissues Organs, 2013, 197, 55–63 CrossRef CAS.
- M. D. Wood, A. M. Moore, D. A. Hunter, S. Tuffaha, G. H. Borschel, S. E. Mackinnon and S. E. Sakiyama-Elbert, Acta Biomater., 2009, 5, 959–968 CrossRef CAS.
- L. De Laporte, A. Lei Yan and L. D. Shea, Biomaterials, 2009, 30, 2361–2368 CrossRef CAS.
- L. Yao, S. Yao, W. Daly, W. Hendry, A. Windebank and A. Pandit, Drug Discovery Today, 2012, 17, 998–1005 CrossRef CAS.
- L. De Laporte, A. Huang, M. M. Ducommun, M. L. Zelivyanska, M. O. Aviles, A. F. Adler and L. D. Shea, Acta Biomater., 2010, 6, 2889–2897 CrossRef CAS.
- U. Mittnacht, H. Hartmann, S. Hein, H. Oliveira, M. Dong, A. P. Pêgo, J. Kjems, K. A. Howard and B. Schlosshauer, Nano Lett., 2010, 10, 3933–3939 CrossRef CAS.
- A. Hurtado, L. D. F. Moon, V. Maquet, B. Blits, R. Jérôme and M. Oudega, Biomaterials, 2006, 27, 430–442 CrossRef CAS.
- S. Stokols, J. Sakamoto, C. Breckon, T. Holt, J. Weiss and M. H. Tuszynski, Tissue Eng., 2006, 12, 2777–2787 CrossRef CAS.
- L. Schnell, R. Schneider, R. Kolbeck, Y.-A. Barde and M. E. Schwab, Nature, 1994, 367, 170–173 CrossRef CAS.
- L. Yao, S. Yao, W. Daly, W. Hendry, A. Windebank and A. Pandit, Drug Discovery Today, 2012, 17, 998–1005 CrossRef CAS.
- K. W. Lu, Z. Y. Chen, D. D. Jin, T. S. Hou, L. Cao and Q. Fu, J. Neurotrauma, 2002, 19, 1081–1090 CrossRef.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 43, 3–12 CrossRef.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- Z. Z. Khaing and C. E. Schmidt, Neurosci. Lett., 2012, 519, 103–114 CrossRef CAS.
- S. Woerly, Biomaterials, 1993, 14, 1056–1058 CrossRef CAS.
- T. A. Kapur and M. S. Shoichet, J. Biomed. Mater. Res., Part A, 2004, 68, 235–243 Search PubMed.
- K. B. Bjugstad, K. Lampe, D. S. Kern and M. Mahoney, J. Biomed. Mater. Res., Part A, 2010, 95, 79–91 CrossRef CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- C. Xu and J. Kopeček, Polym. Bull., 2007, 58, 53–63 CrossRef CAS.
- L. Klouda and A. G. Mikos, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS.
- N. Bhattarai, H. R. Ramay, J. Gunn, F. A. Matsen and M. Zhang, J. Controlled Release, 2005, 103, 609–624 CrossRef CAS.
- H. Tan, C. M. Ramirez, N. Miljkovic, H. Li, J. P. Rubin and K. G. Marra, Biomaterials, 2009, 30, 6844–6853 CrossRef CAS.
- S. Doleski, L. Yao, A. Pandit and C. Elvira, J. Biomed. Mater. Res., Part A, 2010, 94, 457–465 CrossRef.
- A. V. Reis, M. R. Guilherme, O. A. Cavalcanti, A. F. Rubira and E. C. Muniz, Polymer, 2006, 47, 2023–2029 CrossRef CAS.
- S. E. Stabenfeldt, A. J. García and M. C. LaPlaca, J. Biomed. Mater. Res., Part A, 2006, 77, 718–725 CrossRef.
- C. Gong, S. Shi, P. Dong, B. Kan, M. Gou, X. Wang, X. Li, F. Luo, X. Zhao, Y. Wei and Z. Qian, Int. J. Pharm., 2009, 365, 89–99 CrossRef CAS.
- K. A. Smeds and M. W. Grinstaff, J. Biomed. Mater. Res., 2001, 54, 115–121 CrossRef CAS.
- R. H. Schmedlen, K. S. Masters and J. L. West, Biomaterials, 2002, 23, 4325–4332 CrossRef CAS.
- D.-Q. Wu, Y.-X. Sun, X.-D. Xu, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Biomacromolecules, 2008, 9, 1155–1162 CrossRef CAS.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- J. Zhang, R. Xie, S.-B. Zhang, C.-J. Cheng, X.-J. Ju and L.-Y. Chu, Polymer, 2009, 50, 2516–2525 CrossRef CAS.
- Y. Dong, P. Gunning, H. Cao, A. Mathew, B. Newland, A. O. Saeed, J. P. Magnusson, C. Alexander, H. Tai, A. Pandit and W. Wang, Polym. Chem., 2010, 1, 827–830 RSC.
- A. O. Saeed, B. Newland, A. Pandit and W. Wang, Chem. Commun., 2012, 48, 585–587 RSC.
- J. Tan, H. Kang, R. Liu, D. Wang, X. Jin, Q. Li and Y. Huang, Polym. Chem., 2010, 2, 672–678 RSC.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- B. Palmgren, Y. Jiao, E. Novozhilova, S. I. Stupp and P. Olivius, Exp. Neurol., 2012, 235, 599–609 CrossRef CAS.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS.
- J. Piantino, J. A. Burdick, D. Goldberg, R. Langer and L. I. Benowitz, Exp. Neurol., 2006, 201, 359–367 CrossRef CAS.
- A. Jain, Y.-T. Kim, R. J. McKeon and R. V. Bellamkonda, Biomaterials, 2006, 27, 497–504 CrossRef CAS.
- J. A. Burdick, M. Ward, E. Liang, M. J. Young and R. Langer, Biomaterials, 2006, 27, 452–459 CrossRef CAS.
- T. Ono, Y. Fujino, T. Tsuchiya and M. Tsuda, Neurosci. Lett., 1990, 117, 259–263 CrossRef CAS.
- Z. Li-Xin, W. Min and H. Ji-Sheng, NeuroReport, 1992, 3, 700–702 CrossRef.
- B. J. Roessler and B. L. Davidson, Neurosci. Lett., 1994, 167, 5–10 CrossRef CAS.
- B. Schwartz, C. Benoist, B. Abdallah, D. Scherman, J. P. Behr and B. A. Demeneix, Hum. Gene Ther., 1995, 6, 1515–1524 CrossRef CAS.
- L. X. Zhang, Y. Zhou, Y. Du and J. S. Han, Neuropeptides, 1993, 25, 73–76 CrossRef CAS.
- L. X. Zhang, X. L. Li, M. A. Smith, R. M. Post and J. S. Han, Neuroscience, 1997, 77, 15–22 CrossRef CAS.
- P. Kofler, B. Wiesenhofer, C. Rehrl, G. Baier, G. N. Stockhammer and C. Humpel, Cell Transplant., 1998, 7, 175–185 CrossRef CAS.
- T. Imaoka, I. Date, T. Ohmoto, T. Yasuda and M. Tsuda, Brain Res., 1998, 780, 119–128 CrossRef CAS.
- T. Imaoka, I. Date, T. Ohmoto and T. Nagatsu, Hum. Gene Ther., 1998, 9, 1093–1102 CrossRef CAS.
- T. Nagatsu, M. Levitt and S. Udenfriend, J. Biol. Chem., 1964, 239, 2910–2917 CAS.
- W. Lovenberg, H. Weissbach and S. Udenfriend, J. Biol. Chem., 1962, 237, 89–93 CAS.
- H. Bernheimer, W. Birkmayer, O. Hornykiewicz, K. Jellinger and F. Seitelberger, J. Neurol. Sci., 1973, 20, 415–455 CrossRef CAS.
- T. Hökfelt, K. Fuxe and M. Goldstein, Brain Res., 1973, 53, 175–180 CrossRef.
- G. C. Cotzias, M. H. Van Woert and L. M. Schiffer, N. Engl. J. Med., 1967, 276, 374–379 CrossRef CAS.
- J. Segovia, P. Vergara and M. Brenner, Gene Ther., 1998, 5, 1650–1655 CAS.
- F. U. Hartl, Nature, 1996, 381, 571–580 CrossRef CAS.
- T. Nakamura and S. Lipton, Apoptosis, 2009, 14, 455–468 CrossRef CAS.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef.
- S. P. Mathupala, Expert Opin. Ther. Pat., 2009, 19, 137–140 CrossRef CAS.
- L. Flintoft, Nat. Rev. Genet., 2011, 12, 296–296 CrossRef CAS.
- Z. Hassani, G. F. Lemkine, P. Erbacher, K. Palmier, G. Alfama, C. Giovannangeli, J.-P. Behr and B. A. Demeneix, J. Gene Med., 2005, 7, 198–207 CrossRef CAS.
- U. Lungwitz, M. Breunig, T. Blunk and A. Göpferich, Eur. J. Pharm. Biopharm., 2005, 60, 247–266 CrossRef CAS.
- B. Abdallah, A. Hassan, C. Benoist, D. Goula, J. P. Behr and B. A. Demeneix, Hum. Gene Ther., 1996, 7, 1947–1954 CrossRef CAS.
- J. G. Hecker, L. L. Hall and V. R. Irion, Mol. Ther., 2001, 3, 375–384 CrossRef CAS.
- D. Goula, J. S. Remy, P. Erbacher, M. Wasowicz, G. Levi, B. Abdallah and B. A. Demeneix, Gene Ther., 1998, 5, 712–717 CAS.
- G. F. Lemkine, D. Goula, N. Becker, L. Paleari, G. Levi and B. A. Demeneix, J. Drug Target., 1999, 7, 305–312 CrossRef CAS.
- G. F. Lemkine, S. Mantero, C. Migné, A. Raji, D. Goula, P. Normandie, G. Levi and B. A. Demeneix, Mol. Cell. Neurosci., 2002, 19, 165–174 CrossRef CAS.
- M.-P. Martres, B. Demeneix, N. Hanoun, M. Hamon and B. Giros, Eur. J. Neurosci., 1998, 10, 3607–3616 CrossRef CAS.
- G. P. Tang, J. M. Zeng, S. J. Gao, Y. X. Ma, L. Shi, Y. Li, H. P. Too and S. Wang, Biomaterials, 2003, 24, 2351–2362 CrossRef CAS.
- Y. Li, J. Wang, C. G. L. Lee, C. Y. Wang, S. J. Gao, G. P. Tang, Y. X. Ma, H. Yu, H. Q. Mao, K. W. Leong and S. Wang, Gene Ther., 2004, 11, 109–114 CrossRef CAS.
- T. D. Corso, G. Torres, C. Goulah, I. Roy, A. S. Gambino, J. Nayda, T. Buckley, E. K. Stachowiak, E. J. Bergey, H. Pudavar, P. Dutta, D. C. Bloom, W. J. Bowers and M. K. Stachowiak, Mol. Brain Res., 2005, 139, 361–366 CrossRef CAS.
- G. P. Tang, H. Y. Guo, F. Alexis, X. Wang, S. Zeng, T. M. Lim, J. Ding, Y. Y. Yang and S. Wang, J. Gene Med., 2006, 8, 736–744 CrossRef CAS.
- S. Oh, G. E. Pluhar, E. A. McNeil, K. M. Kroeger, C. Liu, M. G. Castro, P. R. Lowenstein, A. Freese and J. R. Ohlfest, J. Neurosurg, 2007, 107, 136–144 CrossRef CAS.
- I.-D. Kim, C.-M. Lim, J.-B. Kim, H. Y. Nam, K. Nam, S.-W. Kim, J.-S. Park and J.-K. Lee, J. Controlled Release, 2010, 142, 422–430 CrossRef CAS.
- A. M. Fletcher, T. H. Kowalczyk, L. Padegimas, M. J. Cooper and D. M. Yurek, Neuroscience, 2011, 194, 220–226 CrossRef CAS.
- D. M. Yurek, A. M. Fletcher, M. McShane, T. H. Kowalczyk, L. Padegimas, M. R. Weatherspoon, M. D. Kaytor, M. J. Cooper and A. G. Ziady, Mol. Imaging, 2011, 10, 327–339 CAS.
- I.-D. Kim, J.-H. Shin, S.-W. Kim, S. Choi, J. Ahn, P.-L. Han, J.-S. Park and J.-K. Lee, Mol. Ther., 2012, 20, 829–839 CrossRef CAS.
- G. Liu, D. Li, M. K. Pasumarthy, T. H. Kowalczyk, C. R. Gedeon, S. L. Hyatt, J. M. Payne, T. J. Miller, P. Brunovskis, T. L. Fink, O. Muhammad, R. C. Moen, R. W. Hanson and M. J. Cooper, J. Biol. Chem., 2003, 278, 32578–32586 CrossRef CAS.
- T. L. Fink, P. J. Klepcyk, S. M. Oette, C. R. Gedeon, S. L. Hyatt, T. H. Kowalczyk, R. C. Moen and M. J. Cooper, Gene Ther., 2006, 13, 1048–1051 CrossRef CAS.
- D. M. Yurek, A. M. Flectcher, T. H. Kowalczyk, L. Padegimas and M. J. Cooper, Cell Transplant, 2009, 18, 1183–1196 CrossRef.
- T. D. Corso, G. Torres, C. Goulah, I. Roy, A. S. Gambino, J. Nayda, T. Buckley, E. K. Stachowiak, E. J. Bergey, H. Pudavar, P. Dutta, D. C. Bloom, W. J. Bowers and M. K. Stachowiak, Folia Morphol., 2005, 64, 1130–1144 Search PubMed.
- S. R. Sinclair, J. W. Fawcett and S. B. Dunnett, Eur. J. Neurosci., 1999, 11, 4341–4348 CrossRef CAS.
- D. Kirik, L. E. Annett, C. Burger, N. Muzyczka, R. J. Mandel and A. Björklund, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2884–2889 CrossRef CAS.
- P. Mulcahy, A. O'Doherty, A. Paucard, T. O'Brien, D. Kirik and E. Dowd, Neuroscience, 2012, 203, 170–179 CrossRef CAS.
- D. B. Hoban, E. Connaughton, C. Connaughton, G. Hogan, C. Thornton, P. Mulcahy, T. C. Moloney and E. Dowd, Brain Behav. Immun., 2013, 27, 91–100 CrossRef CAS.
- D. J. Bharali, I. Klejbor, E. K. Stachowiak, P. Dutta, I. Roy, N. Kaur, E. J. Bergey, P. N. Prasad and M. K. Stachowiak, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11539–11544 CrossRef CAS.
- I. Klejbor, E. K. Stachowiak, D. J. Bharali, I. Roy, I. Spodnik, J. Morys, E. J. Bergey, P. N. Prasad and M. K. Stachowiak, J. Neurosci. Methods, 2007, 165, 230–243 CrossRef CAS.
- E. K. Stachowiak, I. Roy, Y.-W. Lee, M. Capacchietti, J. M. Aletta, P. N. Prasad and M. K. Stachowiak, Integr. Biol., 2009, 1, 394–403 RSC.
- I. Roy, T. Y. Ohulchanskyy, D. J. Bharali, H. E. Pudavar, R. A. Mistretta, N. Kaur and P. N. Prasad, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 279–284 CrossRef CAS.
- K. T. Al-Jamal, L. Gherardini, G. Bardi, A. Nunes, C. Guo, C. Bussy, M. A. Herrero, A. Bianco, M. Prato, K. Kostarelos and T. Pizzorusso, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10952–10957 CrossRef CAS.
- A. C. Bonoiu, E. J. Bergey, H. Ding, R. Hu, R. Kumar, K.-T. Yong, P. N. Prasad, S. Mahajan, K. E. Picchione, A. Bhattacharjee and T. A. Ignatowski, Nanomed., 2011, 6, 617–630 CrossRef CAS.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. A. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS.
- S. Sinha, J. Anderson, V. John, L. McConlogue, G. Basi, E. Thorsett and D. Schenk, Ann. N. Y. Acad. Sci., 2000, 920, 206–208 CrossRef CAS.
- F. M. Laird, H. Cai, A. V. Savonenko, M. H. Farah, K. He, T. Melnikova, H. Wen, H.-C. Chiang, G. Xu, V. E. Koliatsos, D. R. Borchelt, D. L. Price, H.-K. Lee and P. C. Wong, J. Neurosci., 2005, 25, 11693–11709 CrossRef CAS.
- W. A. Jefferies, M. R. Brandon, S. V. Hunt, A. F. Williams, K. C. Gatter and D. Y. Mason, Nature, 1984, 312, 162–163 CrossRef CAS.
- N. Shi and W. M. Pardridge, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7567–7572 CrossRef CAS.
- N. Shi, R. J. Boado and W. M. Pardridge, Pharm. Res., 2001, 18, 1091–1095 CrossRef CAS.
- Y. Zhang, F. Calon, C. Zhu, R. J. Boado and W. M. Pardridge, Hum. Gene Ther., 2003, 14, 1–12 CrossRef CAS.
- Y. Zhang, F. Schlachetzki and W. M. Pardridge, Mol. Ther., 2003, 7, 11 CrossRef CAS.
- Y. Zhang, F. Schlachetzki, J. Y. Li, R. J. Boado and W. M. Pardridge, Mol. Vis., 2003, 9, 465–472 CAS.
- Y. Zhang, F. Schlachetzki, Y. F. Zhang, R. J. Boado and W. M. Pardridge, Hum. Gene Ther., 2004, 15, 339–350 CrossRef CAS.
- Y. Zhang, J. Bryant, A. Charles, R. J. Boado and W. M. Pardridge, Clin. Cancer Res., 2004, 10, 3667–3677 CrossRef CAS.
- C. Zhu, Y. Zhang, J. Yi Li, R. J. Boado and W. M. Pardridge, J. Gene Med., 2004, 6, 906–912 CrossRef CAS.
- C. Chu, Y. Zhang, R. J. Boado and W. M. Pardridge, Pharm. Res., 2006, 23, 1586–1590 CrossRef CAS.
- C. F. Xia, C. Chu, J. Li, Y. Wang, Y. Zhang, R. J. Boado and W. M. Pardridge, J. Gene Med., 2007, 9, 605–612 CrossRef CAS.
- Y. Zhang, Y. Wang, R. J. Boado and W. M. Pardridge, Pharm. Res., 2008, 25, 400–406 CrossRef CAS.
- R. J. Boado and W. M. Pardridge, J. Drug Delivery, 2011, 2011, 296151 Search PubMed.
- M. T. G. da Cruz, A. L. C. Cardoso, L. P. de Almeida, S. Simoes and M. C. Pedroso de Lima, Gene Ther., 2005, 12, 1242–1252 CrossRef.
- A. L. C. Cardoso, S. Simoes, L. P. de Almeida, N. Plesnila, M. C. Pedroso de Lima, E. Wagner and C. Culmsee, J. Controlled Release, 2008, 132, 113–123 CrossRef CAS.
- A. L. C. Cardoso, P. Costa, L. P. de Almeida, S. Simões, N. Plesnila, C. Culmsee, E. Wagner and M. C. Pedroso de Lima, J. Controlled Release, 2010, 142, 392 CrossRef CAS.
- H. Zhao, X. Bao, R. Wang, G. Li, J. Gao, S. Ma, J. Wei, M. Feng, Y. Zhao, W. Ma, Y. Yang, Y.-N. Li and Y.-G. Kong, Hum. Gene Ther., 2011, 22, 207–215 CrossRef CAS.
- Y. T. Ko, R. Bhattacharya and U. Bickel, J. Controlled Release, 2009, 133, 230–237 CrossRef CAS.
- I. Alvarez-Maya, I. Navarro-Quiroga, M. A. Meraz-Ríos, J. Aceves and D. Martinez-Fong, Mol. Med., 2001, 7, 186–192 CAS.
- I. Navarro-Quiroga, J. Antonio González-Barrios, F. Barron-Moreno, V. Ì. c. González-Bernal, D. B. Martinez-Arguelles and D. Martinez-Fong, Mol. Brain Res., 2002, 105, 86–97 CrossRef CAS.
- J. A. Gonzalez-Barrios, M. Lindahl, M. J. Bannon, V. Anaya-Martínez, G. Flores, I. Navarro-Quiroga, L. E. Trudeau, J. Aceves, D. B. Martinez-Arguelles and R. Garcia-Villegas, Mol. Ther., 2006, 14, 857–865 CrossRef CAS.
- D. Martinez-Fong, M. J. Bannon, L.-E. Trudeau, J. A. Gonzalez-Barrios, M. L. Arango-Rodriguez, N. G. Hernandez-Chan, D. Reyes-Corona, J. Armendáriz-Borunda and I. Navarro-Quiroga, Nanomedicine, 2012, 8, 1052–1069 CrossRef CAS.
- J. Zeng, X. Wang and S. Wang, Biomaterials, 2007, 28, 1443–1451 CrossRef CAS.
- S. Barati, P. R. Hurtado, S. H. Zhang, R. Tinsley, I. A. Ferguson and R. A. Rush, Exp. Neurol., 2006, 202, 179–188 CrossRef CAS.
- R. Huang, L. Han, J. Li, F. Ren, W. Ke, C. Jiang and Y. Pei, J. Gene Med., 2009, 11, 754–763 CrossRef CAS.
- R. Huang, W. Ke, Y. Liu, D. Wu, L. Feng, C. Jiang and Y. Pei, J. Neurol. Sci., 2010, 290, 123–130 CrossRef CAS.
- A. Schallon, J. Valérie, A. Walther, C. V. Synatschke, A. H. E. Müller and R. Freitag, React. Funct. Polym., 2010, 70, 1–10 CrossRef CAS.
- A. Nouri, R. Castro, V. Kairys, J. L. Santos, J. Rodrigues, Y. Li and H. Tomás, J. Mater. Sci.: Mater. Med., 2012, 23, 2967–2980 CrossRef CAS.
- Y. Zheng, H. Cao, B. Newland, Y. Dong, A. Pandit and W. Wang, J. Am. Chem. Soc., 2011, 133, 13130–13137 CrossRef CAS.
- Y. Zheng, B. Newland, H. Tai, A. Pandit and W. Wang, Chem. Commun., 2012, 48, 3085–3087 RSC.
- B. Newland, M. Abu-Rub, M. Naughton, Y. Zheng, A. V. Pinoncely, E. Collin, W. Wang and A. Pandit, ACS Chem. Neurosci., 2013 DOI:10.1021/cn4000023.
- Y. Qian, Y. Zha, B. Feng, Z. Pang, B. Zhang, X. Sun, J. Ren, C. Zhang, X. Shao, Q. Zhang and X. Jiang, Biomaterials, 2013, 34, 2117–2129 CrossRef CAS.
- L. J. Fisher and J. Ray, Curr. Opin. Neurobiol., 1994, 4, 735–741 CrossRef CAS.
- J. L. Santos, D. Pandita, J. Rodrigues, A. P. Pêgo, P. L. Granja, G. Balian and H. Tomás, Mol. Pharm., 2010, 7, 763–774 CrossRef CAS.
- J. L. Santos, H. Oliveira, D. Pandita, J. Rodrigues, A. P. Pêgo, P. L. Granja and H. Tomás, J. Controlled Release, 2010, 144, 55–64 CrossRef CAS.
- D. Pandita, J. L. Santos, J. Rodrigues, A. P. Pêgo, P. L. Granja and H. Tomás, Biomacromolecules, 2011, 12, 472–481 CrossRef CAS.
- S. B. Dunnett and A. E. Rosser, Curr. Opin. Organ Transplant., 2011, 16, 632–639 CrossRef CAS.
- O. Sabate, P. Horellou, E. Vigne, P. Colin, M. Perricaudet, M. H. Buc-Caron and J. Mallet, Nat. Genet., 1995, 9, 256–260 CrossRef CAS.
- K. Reekmans, J. Praet, J. Daans, V. Reumers, P. Pauwels, A. Linden, Z. Berneman and P. Ponsaerts, Stem Cell Rev. Rep., 2012, 8, 262–278 CrossRef.
- T. C. Moloney, P. Dockery, A. J. Windebank, F. P. Barry, L. Howard and E. Dowd, Neurorehabil. Neural Repair, 2010, 24, 645–656 CrossRef.
- J. Praet, K. Reekmans, D. Lin, N. De Vocht, I. Bergwerf, B. Tambuyzer, J. Daans, N. Hens, H. Goossens, P. Pauwels, Z. Berneman, A. Van der Linden and P. Ponsaerts, Cell Transplant, 2012, 21, 1867–1881 CrossRef.
- R. Zietlow, V. Pekarik, R. J. E. Armstrong, P. Tyers, S. B. Dunnett and A. E. Rosser, J. Anat., 2005, 207, 227–240 CrossRef CAS.
- R. Zietlow, S. V. Precious, C. M. Kelly, S. B. Dunnett and A. E. Rosser, Exp. Neurol., 2012, 235, 563–573 CrossRef.
- T. C. Moloney, G. E. Rooney, F. P. Barry, L. Howard and E. Dowd, Brain Res., 2010, 1359, 33–43 CrossRef CAS.
- T. M. Coyne, A. J. Marcus, D. Woodbury and I. B. Black, Stem Cells, 2006, 24, 2483–2492 CrossRef.
- T. M. Coyne, A. J. Marcus, K. Reynolds, I. B. Black and D. Woodbury, Transplantation, 2007, 84, 1507–1516 CrossRef.
- A. Sajadi, J.-C. Bensadoun, B. L. Schneider, C. Lo Bianco and P. Aebischer, Neurobiol. Dis., 2006, 22, 119–129 CrossRef CAS.
- N. Törnqvist, L. Björklund, P. Almqvist, L. Wahlberg and I. Strömberg, Exp. Neurol., 2000, 164, 130–138 CrossRef.
- J.-C. Bensadoun, L. Pereira de Almeida, E. G. Fine, J. L. Tseng, N. Déglon and P. Aebischer, J. Controlled Release, 2003, 87, 107–115 CrossRef CAS.
- J. Voges, R. Lehrke, D. G. Kim, C. Lucas, R. Schröder, V. Sturm and H. Stricker, J. Exp. Ther. Oncol., 2002, 2, 70–76 CrossRef CAS.
- L. Fjord-Larsen, P. Kusk, J. Tornoe, B. Juliusson, M. Torp, C. R. Bjarkam, M. S. Nielsen, A. Handberg, J. C. H. Sorensen and L. U. Wahlberg, Mol. Ther., 2010, 18, 2164–2172 CrossRef CAS.
- M. Eriksdotter-Jönhagen, B. Linderoth, G. Lind, L. Aladellie, O. Almkvist, N. Andreasen, K. Blennow, N. Bogdanovic, V. Jelic, A. Kadir, A. Nordberg, E. Sundström, L. O. Wahlund, A. Wall, M. Wiberg, B. Winblad, Å. Seiger, P. Almqvist and L. Wahlberg, Dement. Geriatr. Cogn. Disord., 2012, 33, 18–28 CrossRef.
- D. R. Nisbet, K. E. Crompton, M. K. Horne, D. I. Finkelstein and J. S. Forsythe, J. Biomed. Mater. Res. Part B: Appl. Biomater., 2008, 87, 251–263 CrossRef.
- M. M. Pakulska, B. G. Ballios and M. S. Shoichet, Biomed. Mater., 2012, 7, 024101 CrossRef.
- M. J. Cooke, K. Vulic and M. S. Shoichet, Soft Matter, 2010, 6, 4988–4998 RSC.
- N. K. Loh, S. Woerly, S. M. Bunt, S. D. Wilton and A. R. Harvey, Exp. Neurol., 2001, 170, 72–84 CrossRef CAS.
- S. Woerly, V. D. Doan, F. Evans-Martin, C. G. Paramore and J. D. Peduzzi, J. Neurosci. Res., 2001, 66, 1187–1197 CrossRef CAS.
- T. Zhang, Y. Yan, X. Wang, Z. Xiong, F. Lin, R. Wu and R. Zhang, J. Bioact. Compat. Polym., 2007, 22, 19–29 CrossRef CAS.
- U. Freudenberg, A. Hermann, P. B. Welzel, K. Stirl, S. C. Schwarz, M. Grimmer, A. Zieris, W. Panyanuwat, S. Zschoche, D. Meinhold, A. Storch and C. Werner, Biomaterials, 2009, 30, 5049–5060 CrossRef CAS.
- R.-Q. Huang, Y.-H. Qu, W.-L. Ke, J.-H. Zhu, Y.-Y. Pei and C. Jiang, FASEB J., 2007, 21, 1117–1125 CrossRef CAS.
- W. Ke, K. Shao, R. Huang, L. Han, Y. Liu, J. Li, Y. Kuang, L. Ye, J. Lou and C. Jiang, Biomaterials, 2009, 30, 6976–6985 CrossRef CAS.
- Y. Liu, R. Huang, L. Han, W. Ke, K. Shao, L. Ye, J. Lou and C. Jiang, Biomaterials, 2009, 30, 4195–4202 CrossRef CAS.
- E. J. Kwon, J. Lasiene, B. E. Jacobson, I.-K. Park, P. J. Horner and S. H. Pun, Biomaterials, 2010, 31, 2417–2424 CrossRef CAS.
- M. J. Writer, P. G. Kyrtatos, A. S. Bienemann, J. A. Pugh, A. S. Lowe, C. Villegas-Llerena, G. D. Kenny, E. A. White, S. S. Gill, C. W. McLeod, M. F. Lythgoe and S. L. Hart, J. Controlled Release, 2012, 162, 340–348 CrossRef CAS.
- J. Zhang, T. Tokatlian, J. Zhong, Q. K. T. Ng, M. Patterson, W. E. Lowry, S. T. Carmichael and T. Segura, Adv. Mater., 2011, 23, 5098–5103 CrossRef CAS.
- J. Zhong, A. Chan, L. Morad, H. I. Kornblum, F. Guoping and S. T. Carmichael, Neurorehabili. Neural Repair, 2010, 24, 636–644 CrossRef.
- Y. Yamasaki, Y. Itoyama and K. Kogure, Keio J. Med., 1996, 45, 225–229 CrossRef CAS.
- T. Nakaji-Hirabayashi, K. Kato and H. Iwata, Biomaterials, 2009, 30, 4581–4589 CrossRef CAS.
- L. N. Novikova, A. Mosahebi, M. Wiberg, G. Terenghi, J.-O. Kellerth and L. N. Novikov, J. Biomed. Mater. Res., Part A, 2006, 77, 242–252 CrossRef.
- K. E. Crompton, J. D. Goud, R. V. Bellamkonda, T. R. Gengenbach, D. I. Finkelstein, M. K. Horne and J. S. Forsythe, Biomaterials, 2007, 28, 441–449 CrossRef CAS.
- W. M. Tian, S. P. Hou, J. Ma, C. L. Zhang, Q. Y. Xu, I. S. Lee, H. D. Li, M. Spector and F. Z. Cui, Tissue Eng., Part A, 2005, 11, 513–525 CrossRef CAS.
- M. Uemura, M. M. Refaat, M. Shinoyama, H. Hayashi, N. Hashimoto and J. Takahashi, J. Neurosci. Res., 2010, 88, 542–551 CAS.
- S.-S. Park, Y. J. Lee, S. H. Lee, D. Lee, K. Choi, W.-H. Kim, O.-K. Kweon and H. J. Han, Cytotherapy, 2012, 14, 584–597 CrossRef CAS.
- B. A. Aguado, W. Mulyasasmita, J. Su, K. J. Lampe and S. C. Heilshorn, Tissue Eng., Part A, 2011, 18, 806–815 CrossRef.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS.
- E. Bible, D. Y. S. Chau, M. R. Alexander, J. Price, K. M. Shakesheff and M. Modo, Biomaterials, 2009, 30, 2985–2994 CrossRef CAS.
- W. M. Saltzman, M. W. Mak, M. J. Mahoney, E. T. Duenas and J. L. Cleland, Pharm. Res., 1999, 16, 232–240 CrossRef CAS.
- A. P. Perez, C. Mundiña-Weilenmann, E. L. Romero and M. J. Morilla, Int. J. Nanomedicine, 2012, 7, 1373–1385 CAS.
- S. Wang, N. Ma, S. J. Gao, H. Yu and K. W. Leong, Mol. Ther., 2001, 3, 658–664 CrossRef CAS.
- A. Nunes, K. T. Al-Jamal and K. Kostarelos, J. Controlled Release, 2012, 161, 290–306 CrossRef CAS.
- C. A. Poland, R. Duffin, I. Kinloch, A. Maynard, W. A. H. Wallace, A. Seaton, V. Stone, S. Brown, W. MacNee and K. Donaldson, Nat. Nanotechnol., 2008, 3, 423–428 CrossRef CAS.
- G. P. Kotchey, S. A. Hasan, A. A. Kapralov, S. H. Ha, K. Kim, A. A. Shvedova, V. E. Kagan and A. Star, Acc. Chem. Res., 2012, 45, 1770–1781 CrossRef CAS.
- L. Lacerda, A. Soundararajan, R. Singh, G. Pastorin, K. T. Al-Jamal, J. Turton, P. Frederik, M. A. Herrero, S. Li, A. Bao, D. Emfietzoglou, S. Mather, W. T. Phillips, M. Prato, A. Bianco, B. Goins and K. Kostarelos, Adv. Mater., 2008, 20, 225–230 CrossRef CAS.
- A. Nunes, C. Bussy, L. Gherardini, M. Meneghetti, M. A. Herrero, A. Bianco, M. Prato, T. Pizzorusso, K. T. Al-Jamal and K. Kostarelos, Nanomedicine, 2012, 7, 1485–1494 CrossRef CAS.
- D. Y. Lewitus, K. L. Smith, W. Shain, D. Bolikal and J. Kohn, Biomaterials, 2011, 32, 5543–5550 CrossRef CAS.
- A. L. Benabid, S. Chabardes, J. Mitrofanis and P. Pollak, Lancet Neurol., 2009, 8, 67–81 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |