Dependence of nanoparticle-cell recognition efficiency on the surface orientation of scFv targeting ligands
Serena
Mazzucchelli
a,
Silvia
Sommaruga
a,
Maureen
O'Donnell
b,
Patrizia
Galeffi
c,
Paolo
Tortora
b,
Davide
Prosperi
*bd and
Miriam
Colombo
*ab
aDipartimento di Scienze Biomediche e Cliniche “Luigi Sacco”, Università di Milano, Ospedale L. Sacco, Via G. B. Grassi 74, 20157 Milano, Italy
bNanoBioLab, Dipartimento di Biotecnologie e Bioscienze, Università di Milano-Bicocca, Piazza della Scienza 2, 20126 Milano, Italy. E-mail: davide.prosperi@unimib.it; m.colombo140@campus.unimib.it
cUTAGRI-GEN, ENEA, R. C. Casaccia, Via Anguillarese 301, 00123 Roma, Italy
dFondazione Don Carlo Gnocchi Onlus, via Capecelatro 66, 20148 Milano, Italy
First published on 16th April 2013
Abstract
The surface activation of multifunctional nanoparticles (MNPs) with peptide ligands directing their targeting to cancer cells is an emerging research area in nanobiotechnology. In this paper, water-soluble MNPs have been synthesized and functionalized with an scFv antibody variant specific toward the HER2 receptor overexpressed in several breast cancer cell lines. The scFv was genetically engineered to introduce a cysteine residue inside the loop sequence bridging the VH and VL lobes of the molecule and a histidine tag at the C-terminus in the VL fragment. The Cys and 6 × His functionalities were exploited as orthogonal reactive groups driving the scFv conjugation to MNPs. In this way, scFv positioning on the MNP surface was forced into two different orientations depending on the molecular binding site used for conjugation. The resulting scFv-functionalized MNP1 and MNP2, respectively, were assessed as to their labeling efficiency and selectivity to HER2-positive MCF7 cells. We demonstrate that, while both MNP1 and MNP2 were selective for HER2, there is a remarkable preference for scFv presentation with VH and VL lobes concurrently available for receptor recognition (MNP1) in terms of cell binding efficiency, suggesting that ligand orientation may strongly affect cell binding from MNPs.
Introduction
The advent of nanotechnology in bioscience has triggered great advances in the exploration of novel frontiers in biomedical research, providing previously unconceivable new tools for the diagnosis and treatment of several human pathologies, including cancer, inflammatory disorders and neurodegenerative diseases.1–4 In particular, the use of multifunctional nanoparticles (MNPs) is attracting broad interest due to their intrinsic propensity to combine optical and magnetic properties useful for bimodal imaging with targeting aptitudes suitable for localized delivery of therapeutics.5–7 While in the first generation of nanoscale systems most efforts have been mainly focused on governing the morpho-structural features of nanoparticles, including the size and shape, the second era of multifunctional nanomaterials is opening up new avenues toward the prediction and fine-tuning of their physical and (bio)chemical properties at the molecular and atomic level.Controversial evidence has been provided on the concrete relevance of molecular targeting to enhance the effectiveness of localized therapies based on such nanomaterials.8–10 When using MNPs for targeted cancer diagnosis, however, one of the main issues actually concerns the design and optimization of effective functionalization strategies to achieve an efficient targeting of specific cell receptors.11–13 For this reason, reliable methods that allow researchers to finely control the positioning and orientation of targeting molecules on the surface of MNPs are urgently needed to contribute to shedding light on this debate.14 Hence, many efforts have been devoted to develop new strategies for surface bioengineering of MNPs in order to maximize their targeting efficiency. So far, covalent conjugation has been the most reliable approach to afford stable nanoconstructs for application in a biological environment.15–17
Among the diverse classes of targeting ligands exploited for MNP modification, monoclonal antibodies (mAbs) have shown great potential in diagnosis and treatment of several cancer types.18 However, the use of entire mAbs for nanoparticle functionalization often results in an increase of immunogenicity of the nanoconstruct associated with a reduction of circulation life combined with a low tissue penetration.19 Recombinant antibodies with small sizes (i.e., 20–30 kDa) have been proposed to improve tissue penetration and biodistribution.20 In particular, single-chain fragment variable antibodies (scFv) obtained through genetic engineering, consisting of the variable VH and VL regions connected through a synthetic loop, hold great promise.21,22 We have recently demonstrated that the decreased affinity and specificity toward receptors caused by monovalent binding can be strongly improved by multimerization on the surface of nanoparticles.23 In general, an obvious drawback using peptides for MNP activation is that targeting selectivity may be appreciably dependent on the way in which the targeting molecule is presented on the surface of MNPs.24–27 Thus, a combination of stable positioning and controlled orientation of the immobilized scFv on MNP is mandatory to optimize the recognition efficiency with the specific biological counterpart.28 Further efforts are needed to design novel strategies toward proper ligand orientation on the MNP surface.29–33 For this reason, we aim at estimating the importance of presenting scFv molecules in nanoconjugates with optimized orientation to maximize the labeling efficiency of specific molecular receptors on malignant cells. In the present work, we have produced a newly designed variant of the previously reported scFv800E6,34 mutagenized to include a single cysteine residue in the loop bridging VH and VL fragments and a 6 × histidine tag at the C-terminal end of VL (CscFv), which exhibited a strong affinity for the breast carcinoma marker HER2 with a Ka of the order of 108 M−1. By taking advantage of the presence of two orthogonal functionalities on the polypeptide sequence, i.e. Cys residue and His-tag, scFv was immobilized in two different orientations on the surface of magnetofluorescent nanoparticles (MNP1 and MNP2, respectively). We speculated that CscFv molecules could expose both VH and VL lobes upon conjugation via Cys thiol, while only VH should be available for receptor binding when using His-tag for Ni-NTA immobilization due to hindrance constraints, as schematically illustrated in Fig. 1. The aim of this study was to assess whether the different modes of scFv immobilization on MNPs were capable of affecting the labeling efficiency of HER2-positive breast cancer cells.
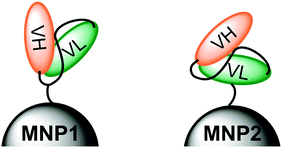 | ||
| Fig. 1 Schematic representation of the molecular positioning of an anti-HER2 scFv antibody via the Cys residue in the loop (MNP1, left) or via the 6 × His tag at the C-terminal end of VL (MNP2, right). | ||
Results and discussion
CscFv expression in P. pastoris
To produce the CscFv variant in P. pastoris, the nucleotide sequence of the original scFv800E6 was mutated from pPICZαA-scFv800E6 vector34 by site-directed mutagenesis. The resulting pPICZαA-CscFv vector included the tightly regulated, methanol-inducible alcohol-oxidase-1 (AOX1) promoter, along with the open reading frame of the first 90 residues of the prepro α-factor signal sequence, to drive the secretion of the induced polypeptides into the yeast medium. In addition, one myc-tag and a 6 × His-tag were appended to the C terminus of CscFv to detect and purify the secreted protein, respectively. A schematic representation of the expression construct used in this work is reported in Fig. 2A. The linearized plasmid was used to electroporate the KM71H P. pastoris host strains. To directly compare the secretion levels of CscFv, sixteen independent zeocine-resistant transformants were induced for 48 h in a small-scale culture experiment and their conditioned media analyzed by immunoblotting with anti-myc-HRP antibody. Four KM71H clones secreted CscFv in good amounts, while only three of them did not secrete the protein at all (Fig. 2B). The KM71H-pPICZαA-CscFv-14 clone was selected as the better producer and used for the next studies.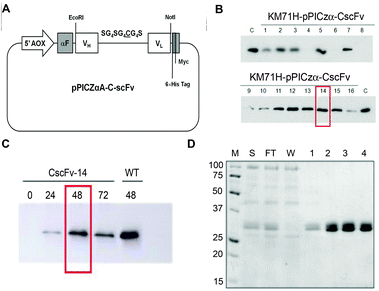 | ||
| Fig. 2 (A) Schematic diagram of the pPICZαA-CscFv expression vector. The recombinant gene encoding CscFv was inserted into the pPICZαA vector under the control of the alcoholoxidase-1 (AOX1) promoter, in frame with the prepro α-factor signal sequence (αF) and with myc and 6 × His tags. (B) Small-scale screening of P. pastoris best producing clones. Equal amounts (10 μL) of the culture supernatants of KM71H-pPICZαA-CscFv clones were analyzed by SDS-PAGE and immunoblotted with the anti-myc-HRP antibody. As a control (C), the culture supernatant of wt KM71H-pPICZαA-scFv800E6 was loaded. (C) Medium scale experiment. The clone KM71H-pPICZαA-CscFv-14 was induced with 0.5% methanol and 0.8% glycerol. Cell culture supernatants were collected at 24 h time intervals and analyzed by SDS-PAGE and immunoblotting with the anti-myc-HRP antibody. (D) Purification of CscFv. SDS-PAGE analysis was performed on samples obtained by purification on Ni-NTA agarose resin: the supernatant (S), flow through (FT), wash (W) and eluted fractions (1, 2, 3 and 4). Proteins were visualized by Coomassie staining. M: molecular weight markers (kDa). | ||
In order to set up the optimal induction conditions, the KM71H-pPICZαA-CscFv-14 clone was induced in a medium-scale culture experiment under the induction conditions previously described for wild type (wt) scFv800E6 in the presence of 0.5% methanol and 0.8% glycerol for up to 72 h.34 Media were collected at different times after methanol addition and analyzed by immunoblotting. As shown in Fig. 2C, the maximum yield in secreted protein was obtained with 2% methanol and 0.8% glycerol after a 48 h induction, whereas no significant increase in CscFv secretion levels could be observed at longer times. After a 24 h induction, only a slight amount of the protein accumulated in the medium. In contrast, a marked increase in CscFv expression was recovered after 48 h, most probably upon depletion of glycerol carbon source (Fig. 2C).
CscFv protein purification
CscFv was purified from the medium of KM71H-pPICZαA-CscFv-14 clone induced for 48 h under optimized conditions by a single-step purification protocol on an Ni-NTA agarose column by taking advantage of the C-terminal His tag. The eluted fractions were quantified by the Bradford assay and analyzed by SDS-PAGE (Fig. 2D). Two closely migrating bands around 29–30 kDa were co-purified and detected in each of the eluted fractions. These bands were probably different isoforms of the secreted scFv, the slower migrating one being possibly due to inefficient cleavage of Glu–Ala repeats by Ste13 aminopeptidase during the processing of the propeptide in the Golgi complex, as previously suggested.34 Under these induction conditions, we estimated a yield in purified CscFv of approximately 5.4 mg L−1.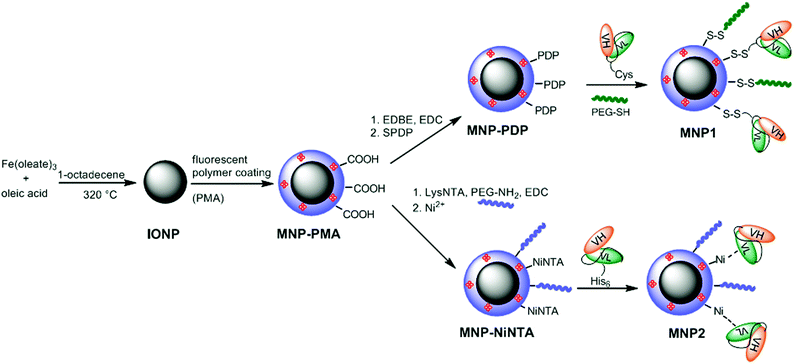 | ||
| Scheme 1 Synthesis of MNP1 and MNP2. | ||
Synthesis of MNP1
Highly uniform and monodisperse 8 nm hydrophobic iron oxide nanoparticles (IONP) were synthesized by solvothermal decomposition from iron oleate in a solution of octadecene in the presence of oleic acid as a capping agent.35 IONP were suspended in chloroform and transferred to water phase by mixing with a 0.5 M solution of a fluorescent amphiphilic polymer (PMA) in sodium borate buffer (SBB, pH 12). PMA was obtained by condensation of poly(isobutylene-alt-maleic anhydride) and dodecylamine and labeled with fluoresceinamine.31 The resulting highly soluble MNP-PMA were modified by condensation with 2,20-(ethylenedioxy)bis(ethylamine) (EDBE) and further functionalized with N-succinimidyl-3-[2-pyridyldithio]propionate (SPDP) (MNP-PDP). The thiol-reactive MNP-PDP were reacted with CscFv. To improve the conjugation efficiency, CscFv was pretreated with 1 mM dithiothreitol (DTT) in order to prevent CscFv dimerization. The residual PDP functionalities were saturated with excess PEG500-SH (Scheme 1). The excess of chemicals and biological reagents was removed by dialysis and MNP-scFv were collected. We estimated an average of 5–6 CscFv molecules per nanoparticle measuring the released pyridine-2-thione by UV absorption at λabs = 343 nm. MNP1 were 53.6 ± 4.4 nm in size, as measured by dynamic light scattering (DLS) in phosphate buffer saline (PBS, pH 7.4) at a concentration of 5 μg mL−1, with a ζ potential of −51.12 ± 2.36 mV. MNP-PMA exhibited a maximal fluorescence emission at 512 nm. To exclude the event of CscFv nonspecific adsorption, an SDS-PAGE experiment was performed in the presence or not of 2-mercaptoethanol. Only under reducing conditions, CscFv was released, which confirmed that CscFv was indeed bound only via the Cys site (data not shown).Synthesis of MNP2
The carboxylate functionalities on MNP-PMA were reacted with the synthesized chelating agent lysine-NTA (LysNTA) and the residual COOH groups saturated with PEG-NH2. The purified nanoparticles were brought to pH 8.4 with NaOH, and then NiCl2 was added resulting in fully armed Ni2+-functionalized nanoparticles (MNP-NiNTA). CscFv were incubated for 10 min at room temperature (RT) in the presence of MNP-NiNTA (Scheme 1). The resulting MNP2 were washed three times with PBS. The hydrodynamic size of MNP2 in PBS (pH 7.4) was 55.4 ± 5.3 nm, as determined by DLS. The ζ potential of the nanoparticles was measured before and after the nickel addition: NTA-conjugated MNP were negatively charged (−55.27 ± 1.22 mV) due to the presence of NTA carboxylic groups. The capture of Ni2+ ions by the external carboxylic groups of NTA resulted in a decrease in the negative charge of MNP-NiNTA (−42.21 ± 3.38 mV).The synthetic conditions were optimized in order to have basically the same amount of CscFv on MNP1 and MNP2. CscFv bound to MNP2 was quantified by a protein assay of supernatants at 280 nm using a calculated ε280 nm of 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1, obtaining the conjugation of 6.0 × 1015 CscFv molecules per mg nanoparticle, corresponding approximately to 5–6 CscFvs per particle. As a control, no traces of CscFv were recovered bound to MNPs in the absence of Ni2+ ions, suggesting that the immobilization of CscFv was indeed mediated by His-tag interaction with NiNTA groups on nanoparticles.
600 M−1, obtaining the conjugation of 6.0 × 1015 CscFv molecules per mg nanoparticle, corresponding approximately to 5–6 CscFvs per particle. As a control, no traces of CscFv were recovered bound to MNPs in the absence of Ni2+ ions, suggesting that the immobilization of CscFv was indeed mediated by His-tag interaction with NiNTA groups on nanoparticles.
In Fig. 3, no apparent change in core size and morphology was observed after surface modification in MNP1 and MNP2. Also the average hydrodynamic size of MNP1 and MNP2 was basically the same, as determined by dynamic light scattering (DLS), although the size distribution of MNP1 was quite narrower than MNP2, suggesting a better colloidal stability in buffered solution. This result indicates that the size, shape and extent of aggregation would not affect significantly the comparison between MNP1 and MNP2 in terms of HER2-binding efficiency.
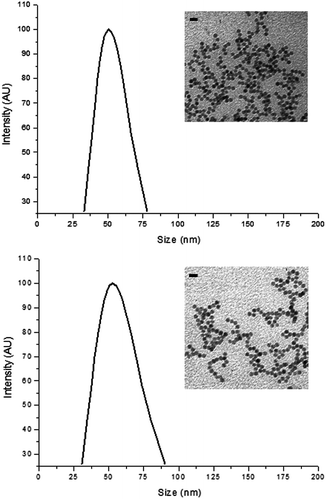 | ||
| Fig. 3 Characterization of MNP1 (A) and MNP2 (B). Size distribution was determined by dynamic light scattering in PBS, pH 7.4, at a colloidal concentration of 5 μg mL−1. Insets: TEM graphs of MNP1 (A) and MNP2 (B) in water, scale bars = 20 nm. | ||
Assessment of the MNP1 and MNP2 binding capability of HER2-positive cells
The binding capability of MNP1 and MNP2 towards the breast cancer biomarker HER2 was assayed using HER2-positive MCF7 cells. As a specificity control, an HER2− MDA-MB-468 cell line and MCF7 cells previously saturated with trastuzumab (TZ), a humanized monoclonal antibody with a high affinity toward HER2, were used.33 HER2+ MCF7 cells and HER2− MDA cells were incubated for 1 h at 4 °C (to minimize spontaneous endocytosis) in the presence of MNP1 or MNP2 (0.02 mg mL−1). VHVL-oriented conjugation in MNP1 showed a fourfold higher percentage of cells in the positive region as compared to MNP2. Moreover, the MNP1 conjugation strategy showed a remarkably higher specificity than MNP2, as demonstrated by a comparison between the results obtained with HER2+ and HER2− cell lines in both cases (Fig. 4). This observation is in accordance with previous evidence that site-specific conjugation of scFv on spherical nanoparticles exploiting the C-terminal His-tag is not able to significantly improve the binding efficiency compared with randomly oriented VHVL-conjugation.23 To confirm that nanoparticle binding to cancer cells was mediated by specific receptor recognition, HER2 was first saturated with free TZ.36 In this case, both MNP1 and MNP2 no longer exhibited a significant binding capability toward MCF7 cells compared to the negative control.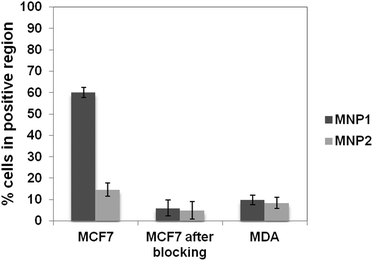 | ||
| Fig. 4 Assessment of HER2 binding capability of MNP1 and MNP2. The fraction of cells in the positive region is reported in the graph after 1 h incubation of MCF7 (HER2+) and MDA cells (HER2−, negative control) at 4 °C with 0.02 mg mL−1 of MNP1 or MNP2 previously saturated or not with TZ (5 mg, MCF7 after blocking). These values were normalized using MCF7 and MDA untreated cells, respectively, as controls. Data were expressed as means ± SE (standard error) of three individual experiments. | ||
MNP1 were further assessed by immunofluorescence experiments to confirm the selective recognition of HER2 (Fig. 5). After 1 h incubation at 37 °C with cells, MNP1 accumulated in the plasma membrane of MCF7 cells and inside the cell cytoplasm, showing a localization similar to that observed for free CscFv (CTRL+, Fig. 5). In contrast, no fluorescence signal was revealed when HER2− MDA cells were incubated with MNP1 (CTRL−), supporting the high binding specificity observed in the flow cytometry assay.
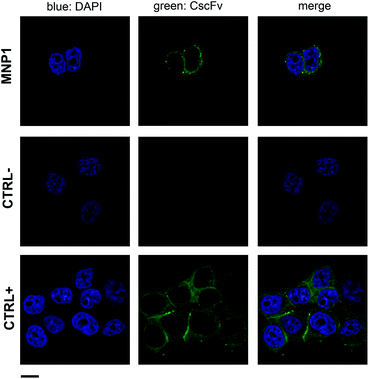 | ||
| Fig. 5 MNP1 capability of labeling HER2-positive cell membrane. HER2-positive MCF7 cells were incubated for 1 h at 37 °C with FITC-labeled MNP1 (0.1 mg mL−1). CscFv incubation with MCF7 cells was used as a positive control, while MNP1 incubation with HER2-negative MDA cells was used as a negative control. Free CscFv was revealed by a FITC-conjugated secondary antibody (green). Nuclei were stained with DAPI (blue). Scale bar = 10 μm. | ||
Experimental section
Materials and methods
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Water was deionized and ultrafiltered by a MilliQ apparatus from Millipore Corporation (Billerica, MA) before use. DLS measurements were performed at 90° with a 90Plus Particle Size Analyzer from Brookhaven Instruments Corporation (Holtsville, NY), working at 15 mW of a solid-state laser (λ = 661 nm). Zeta-potential measurements were performed on the same instrument, equipped with a couple of AQ-809 electrodes, and analyses were processed by ZetaPlus software. Viscosity and refractive index of pure water were used to characterize the solvent. Nanoparticles were dispersed in the solvent and sonicated in an S15H Elmasonic apparatus (Elma, Singen, Germany) before analysis. The final sample concentration used for measurements was typically 5 μg mL−1. Cell culture medium and chemicals were purchased from EuroClone. Confocal microscopy analysis was performed with a Leica SP2 AOBs microscope confocal system. Images were acquired with 63× magnification oil immersion lenses at 1024 × 1024 pixel resolution.Synthesis of (Nα,Nα-bis[carboxymethyl]-Nε-[benzyloxycarbonyl]-L-lysine (Nε-Z-NTA)
Bromoacetic acid (2.5 g, 18 mmol) was dissolved in 1.5 M NaOH (9 mL). Nε-Benzyloxycarbonyl-L-lysine (1.2 g, 4.2 mmol), dissolved in 1.5 M NaOH (15 mL), was added dropwise to the bromoacetic solution in ice under stirring. After 2 h, ice was removed and the reaction mixture was kept under stirring overnight at RT. After heating at 50 °C for 2 h, 1 M HCl (24 mL) was added dropwise and the formation of a white precipitate was observed. Stirring was continued for 5 min. The precipitate was filtered off in vacuo, washed with 0.1 M HCl (12 mL) and with water (2 × 12 mL), and dried in vacuo for 30 min. The precipitate was exsiccated in an oven at 90 °C for 1 h.Synthesis of Nα,Nα-bis[carboxymethyl]-L-lysine (LysNTA)
N ε-Z-NTA (1.1 g, 2.72 mmol) was dissolved in a mixture of methanol (27.5 mL) and deionized water (1.4 mL) and, after the addition of a spatula tip of Pd/C, hydrogenated at RT. After 3 h, total hydrogenation was achieved as confirmed by TLC in CH3CN–H2O (4:1). After removing the solvent in vacuo, water (20 mL) was added to dissolve the resulting precipitate, whereas the excess catalyst could not be redissolved, so it could be filtered off. Ethanol (90 mL) was added and the product crystallized at 4 °C within 2 days. The crystals were collected by filtration and dried in vacuo. NMR characterization was in accordance with previously published data.37
Synthesis of PMA-coated Fe3O4 nanoparticles (MNP-PMA)
Oleate-coated magnetite nanoparticles (IONP) were synthesized according to a previously published protocol.35 Briefly, an iron–oleate complex was firstly prepared by reacting metal chlorides and sodium oleate in a solvent mixture containing ethanol, distilled water and hexane in a 4:3:7 ratio and the resulting solution was heated to 70 °C for 4 h and then cooled at RT. The organic phase containing the iron–oleate complex was washed three times with distilled water in a separatory funnel and the hexane was removed under vacuum resulting in a waxy solid form. Iron–oleate (36 g, 40 mmol) and oleic acid (5.7 g, 20 mmol) were dissolved in 1-octadecene (200 g) at RT. The reaction mixture was heated to 320 °C and kept at this temperature for 30 min to promote nanocrystal formation. The solution was then cooled to RT and centrifuged after the addition of acetone–hexane 4:1 (150 mL). The precipitate was washed several times with ethanol and IONP were collected and dispersed in chloroform. FITC-labeled PMA (0.5 M, 63 μL),31 obtained reacting overnight at RT 5 mL of 0.5 M PMA in CHCl3 with 1.0 M fluoresceinamine (0.5 mL) in ethanol, was added to IONP (4.6 mg). The organic solvent was evaporated and sodium borate buffer (SBB, pH 12, 10 mL) was added, yielding a stable nanoparticle dispersion, which was concentrated in Amicon tubes (100 kDa filter cutoff) by centrifuging at 3500 rpm for 20 min. Finally, the nanoparticles were washed twice with water (each centrifuge cycle was 5 min at 3500 rpm), yielding MNP-PMA.
MNP-PMA functionalization with SPDP (MNP-PDP)
0.1 M EDC (18 μL) was added to the MNP-PMA solution and 0.05 M 2,2-(ethylenedioxy)-bis-(ethylamine) (EDBE, 9 μL) was added after 2 min and stirred for 2 h. Next, the nanoparticle dispersion was concentrated and washed twice with water. Nanoparticles were then reacted for 4 h in the presence of N-succinimidyl-3-[2-pyridyldithio]-propionate (SPDP, 690 μL, 10 mg mL−1 in DMSO), concentrated and washed twice with water.Synthesis of MNP1
MNP-PDP (2.3 mg) were incubated in the presence of CscFv (0.46 mg) previously treated with 0.1 M DTT. The remaining PDP functional groups were saturated incubating with an excess of PEG500-SH (10 mg mL−1 in water) at RT for 1 h. The excess of chemicals and biological reagents was removed by dialysis and the MNP1 were collected.MNP-PMA functionalization with NiNTA (MNP-NiNTA)
MNP-PMA dispersion (2 mg) was reacted with 0.05 M EDC (3.2 μL) for 2 min, and then LysNTA (3.2 μL, 0.01 M in water) was added and stirred for 2 h. The remaining PMA functional groups were saturated with 1.5 μL of methoxypolyethylene glycol amine MW 2000 (0.01 M PEG-NH2) by incubating at RT for 1 h. The excess of chemicals and biological reagents was removed by centrifugation in an Amicon filter as described above and the nanoparticles were collected to a final volume of 2 mL. 1 M NaOH was added to the dispersion until the pH reached 8.4 and an NiCl2·6H2O solution (100 μL, 1 mg mL−1 in water) was added and shaken for 1.5 h. The nanoparticles were washed twice in water (MNP-NiNTA).Synthesis of MNP2
MNP-NiNTA (1.6 mg) were incubated with purified CscFv (0.8 mg) in phosphate buffer saline (PBS, pH 7.4) at a final volume of 0.5 mL and the mixture was stirred in an orbital shaker for 10 min at RT. MNP2 were isolated from unreacted CscFv by centrifugation at 11200 rpm for 5 min and the supernatant was discarded. Nanoparticles were washed three times with PBS (0.5 mL) and stored in PBS at 4 °C for further experiments. The amount of CscFv immobilized on MNP2 was determined by difference, measuring the absorbance of the supernatant and washes at 280 nm.
Strains and plasmids
E. coli DH5α was used as a host strain for the propagation of the plasmid vector. P. pastoris KM71H (arg4; aox1:ARG4) (his4) (Invitrogen) was used as hosts for expressing the CscFv gene. Plasmid pPICZαA (Invitrogen) was used for constructing the plasmid vector.Construction of the expression vector
The cDNA coding for scFv800E6 was amplified from pEmbl-scFv800E6 vector.38 Site-directed mutagenesis was performed to insert a Cys residue in the loop region between VH and VL domains using these mutagenic primers (S131C fw: 5′-CAGGCGGAGGTGGCT![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) TGGCGGTGGCGGATC-3′ and S131C rev: 5′-GATCCGCCACCGCCA
TGGCGGTGGCGGATC-3′ and S131C rev: 5′-GATCCGCCACCGCCA![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) AGCCACCTCCGCCTG-3′, substitution underlined) and pPICZαA-scFv800E6 as a template. The resulting PCR product was incubated for 1 h at 37 °C with DpnI to digest parental DNA. The resulting recombinant expression vector pPICZαA-CscFv was confirmed by restriction endonuclease digestion and DNA sequencing.
AGCCACCTCCGCCTG-3′, substitution underlined) and pPICZαA-scFv800E6 as a template. The resulting PCR product was incubated for 1 h at 37 °C with DpnI to digest parental DNA. The resulting recombinant expression vector pPICZαA-CscFv was confirmed by restriction endonuclease digestion and DNA sequencing.
Transformation in P. pastoris and screening of transformants
The recombinant expression vector pPICZαA-CscFv was linearized with HindIII and the digested product was transformed into the P. pastoris host strains KM71H by electroporation (1.5 kV, 400 Ω, 25 μF; Bio-Rad Gene Pulser). Transformants were selected on YPDS (1% yeast extract, 2% peptone, 2% dextrose, 1 M sorbitol) plates containing 100 μg mL−1 Zeocin. For small-scale screening of CscFv antibody fragment-productive clones, single clones from YPD agar plates were grown in 5 mL YPD medium at 30 °C overnight with shaking at 250 rpm. The cultures were centrifuged at 1500 rpm for 4 min and then the pellets were resuspended in 5 mL of BMMY (1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6.0), 1.34% YNB, 0.00004% biotin, 0.5% methanol) or BMDY (BMMY containing 2% dextrose instead of methanol). To maintain induction, methanol was added to the culture medium every 24 h at a final concentration of 0.5%. After 48 h induction, the cultures were centrifuged at 5000 rpm for 10 min and the supernatants analyzed by SDS-PAGE and Western blotting as described below.Optimization of induction conditions
In order to set up the optimal conditions for CscFv expression, medium-scale culture experiments were performed under induction conditions chosen for scFv800E6 wt.34 The best producing clone KM71H-pPICZαA-CscFv-14 was grown in 10 mL YPD medium at 30 °C overnight with shaking at 250 rpm. The cultures were centrifuged at 1500 rpm for 4 min and then the pellets were resuspended in 10 mL of BMMY supplemented with 0.5% methanol and 0.8% glycerol. The cultures, yielding an initial OD600 value of 10, were supplemented every 24 h with methanol to a final concentration to maintain the induction. Culture supernatants were sampled at different times to monitor CscFv production by Western blot analysis as described below.Purification of CscFv
For purification, 100 mL yeast culture in BMMY with 0.8% glycerol yielding an initial OD600 value of 10 was induced by daily addition of methanol to a final concentration of 0.5%. After 48 h of methanol treatment, the culture supernatant was filtered through 0.22 μm filters and dialyzed overnight in 50 mM sodium phosphate pH 8.0, 300 mM NaCl. The dialyzed medium was loaded at a flow rate of 0.5 mL min−1 onto a Ni-NTA agarose (Qiagen) column (bed volume 0.5 mL) pre-equilibrated with 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM imidazole. The column was washed with 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 20 mM imidazole and the protein eluted with a step-wise imidazole gradient, 100 mM to 200 mM, in the same buffer. Fractions were collected and analyzed by SDS-PAGE and Western blotting as described below. Protein content was determined using the Coomassie Plus Protein Assay Reagent from Pierce and bovine plasma immunoglobulin G as the standard protein.Analysis by SDS-PAGE and Western blotting
SDS-PAGE was performed according to Laemmli using 12% (v/v) polyacrylamide gels.39 The proteins were detected by Coomassie Brilliant Blue R-250 staining or transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore) for Western blotting analysis with horseradish peroxidase (HRP)-conjugated anti-myc antibody (Invitrogen). Target proteins were visualized using an enhanced chemiluminescence detection system (GE Healthcare).Cell cultures
MCF7 and MDA-MB-468 cell lines were used as HER2 positive and HER2 negative targets, respectively. Cells were cultured in 50% Dulbecco's Modified Eagle's Medium (DMEM) and 50% F12, supplemented with 10% fetal bovine serum, L-glutamine (2 mM), penicillin (50 UI mL−1) and streptomycin (50 mg mL−1) at 37 °C and 5% CO2 in a humidified atmosphere and subcultured prior to confluence using trypsin/EDTA.Flow cytometry
Cells were treated for FACS analysis with standard methods. Briefly, MCF7 and MDA-MB-468 cells were incubated for 1 h at 4 °C with MNP1 and with MNP2, and then were washed thrice with PBS. In blocking experiments, MCF7 cells were previously incubated with 5 μg of TZ for 1 h at 4 °C before nanoparticle incubation. Labeled cells were analyzed on a FACS Calibur flow cytometer (Becton Dickinson). 20000 events were acquired for each analysis, after gating on viable cells. Untreated cells were used to set the appropriate gates.
Confocal laser scanning microscopy
Cells were cultured on collagen (Sigma) pre-coated coverglass slides until sub-confluence. Cells were then incubated with MNP1 at 0.1 mg mL−1, or with an equal amount of free CscFv. Then cells were washed thrice with PBS, fixed for 10 min with 4% paraformaldehyde (Sigma) and treated for 10 min with 0.1 M glycine (Sigma) in PBS. A blocking step was performed for 1 h at RT with a solution containing 5% bovine serum albumin (Sigma), 2% goat serum in PBS. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) at 0.2 μg mL−1 in PBS with 0.1% saponin (Sigma) for 20 min at RT.Conclusions
In summary, we have designed and developed novel multifunctional nanoparticles based on a superparamagnetic iron oxide core coated with a fluorescent amphiphilic polymer suitable for nanoparticle dispersion in aqueous environment and for functionalization with a targeting scFv ligand selective for HER2 receptor overexpressed in breast cancer cells. In this paper, we have engineered an anti-HER2 scFv variant in order to introduce two orthogonal conjugation functionalities, namely a cysteine residue and a histidine tag, for the immobilization of the targeting ligand on the surface of nanoparticles. Cys was inserted into the bridging loop between VH and VL lobes, while His tag was placed at the C-terminus of VL. The different localizations of the orthogonal functionalities forced the molecule to assume a different orientation on the nanoparticle. We have assessed the resulting two scFv-functionalized nanoparticles (MNP1 and MNP2, respectively) as to their aptitude to bind selectively HER2-positive cancer cells by flow cytometry and found that the orientation of scFv on the nanoparticle surface strongly affected the cell labeling efficiency, with an optimal solution represented by MNP1 configuration. Finally, confocal microscopy confirmed the selectivity of HER2 targeting from MNP1 in two different breast cancer cell lines. These results suggest that the possibility to finely control the positioning and orientation of ligands in conjugated nanoparticles can play a fundamental role in biorecognition affinity and selectivity, highlighting the importance of developing efficient nanoconjugation strategies toward the optimal presentation of targeting ligands in multifunctional nanomaterials for biomedical applications.Acknowledgements
We thank R. Allevi from CMENA (UniMi) for TEM images. This work was supported by the “NanoBioSens” project (Sardegna-Lombardia), Fondazione Regionale per la Ricerca Biomedica (FRRB), NanoMeDia Project (A. O. “L. Sacco” and Regione Lombardia), and Fondazione “Romeo ed Enrica Invernizzi”. M. C. acknowledges a research fellowship from CMENA.Notes and references
- A. Schroeder, D. A. Heller, M. M. Winslow, J. E. Dahlman, G. W. Pratt, R. Langer, T. Jacks and D. G. Anderson, Nat. Rev. Cancer, 2012, 12, 39–50 CrossRef CAS.
- M. G. Harisinghani, J. Barentsz, P. F. Hahn, W. M. Deserno, S. Tabatabaei, C. H. van de Kaa, J. de la Rosette and R. Weissleder, N. Engl. J. Med., 2003, 348, 2491–2499 CrossRef.
- M. Piazza, M. Colombo, I. Zanoni, F. Granucci, P. Tortora, J. Weiss, T. Gioannini, D. Prosperi and F. Peri, Angew. Chem., Int. Ed., 2012, 50, 622–626 CrossRef.
- M. Srikanth and J. A. Kessler, Nat. Rev. Neurol., 2012, 8, 307–318 CrossRef CAS.
- S. A. McCarthy, G.-L. Davies and Y. K. Gun'ko, Nat. Protoc., 2012, 7, 1677–1693 CrossRef CAS.
- M. Colombo, M. Casula, S. Carregal-Romero, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306–4334 RSC.
- M. V. Yezhelyev, X. Gao, Y. Xing, A. Al-Hajj, S. Nie and R. M. O'Regan, Lancet Oncol., 2006, 7, 657–667 CrossRef CAS.
- E. Blanco, A. Hsiao, G. U. Ruiz-Esparza, M. G. Landry, F. Meric-Bernstam and M. Ferrari, Mol. Oncol., 2011, 5, 492–503 CrossRef CAS.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS.
- E. Tasciotti, X. Liu, R. Bhavane, K. Plant, A. D. Leonard, B. K. Price, M. M.-C. Cheng, P. Decuzzi, J. M. Tour, F. Robertson and M. Ferrari, Nat. Nanotechnol., 2008, 3, 251–157 CrossRef.
- D. Simberg, T. Duza, J. H. Park, M. Essler, J. Pilch, L. Zhang, A. M. Derfus, M. Yang, R. M. Hoffman, S. Bhatia, M. J. Sailor and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2010, 104, 932–936 CrossRef.
- M. Lewin, N. Carlesso, C.-H. Tung, X.-W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS.
- N. T. K. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- J.-M. Montenegro, V. Grazu, A. Sukhanova, S. Agarwal, J. M. de la Fuente, I. Nabiev, A. Greiner and W. J. Parak, Adv. Drug Deliv. Rev., 2013 DOI:10.1016/j.addr.2012.12.003.
- N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925–935 CrossRef CAS.
- C.-C. Yu, P.-C. Lin and C.-C. Lin, Chem. Commun., 2008, 1308–1310 RSC.
- L. Polito, D. Monti, E. Caneva, E. Delnevo, G. Russo and D. Prosperi, Chem. Commun., 2008, 621–623 RSC.
- L. Sanz, A. M. Cuesta, M. Compte and L. Alvarez-Vallina, Acta Pharmacol. Sin., 2005, 26, 641–648 CrossRef CAS.
- P. Holliger and P. J. Hudson, Nat. Biotechnol., 2005, 9, 1126–1136 CrossRef.
- A. Goel, S. Augustine, J. Baranowska-Kortylewicz, D. Colcher, B. J. M. Booth, G. Pavlinkova, M. Tempero and S. K. Batra, Clin. Cancer Res., 2001, 7, 175–184 CAS.
- L. Sanz, B. Blanco and L. Alvarez-Vallina, Trends Immunol., 2004, 25, 85–91 CrossRef CAS.
- A. M. Bayly, A. A. Kortt, P. J. Hudson and B. E. Power, J. Immunol. Methods, 2002, 262, 217–227 CrossRef CAS.
- S. Mazzucchelli, P. Verderio, S. Sommaruga, M. Colombo, A. Salvadè, F. Corsi, P. Galeffi, P. Tortora and D. Prosperi, Bioconjugate Chem., 2011, 22, 2296–2303 CrossRef CAS.
- S. Mazzucchelli, M. Colombo, P. Verderio, E. Rozek, F. Andreata, E. Galbiati, P. Tortora, F. Corsi and D. Prosperi, Angew. Chem., Int. Ed., 2013, 52, 3121–3125 CrossRef CAS.
- E. Occhipinti, P. Verderio, A. Natalello, E. Galbiati, M. Colombo, S. Mazzucchelli, A. Salvadè, P. Tortora, S. Maria Doglia and D. Prosperi, Nanoscale, 2011, 3, 387–390 RSC.
- M. E. Aubin-Tam and K. Hamad-Schifferli, Biomed. Mater., 2008, 3, 034001 CrossRef.
- K. Boeneman, J. R. Deschamps, S. Buckhout-White, D. E. Prasuhn, J. B. Blanco-Canosa, P. E. Dawson, M. H. Stewart, K. Susumu, E. R. Goldman, M. Ancona and I. L. Medintz, ACS Nano, 2010, 4, 7253–7266 CrossRef CAS.
- I. Medintz, Nat. Mater., 2006, 5, 842 CrossRef CAS.
- M. Colombo, S. Mazzucchelli, V. Collico, S. Avvakumova, L. Pandolfi, F. Corsi, F. Porta and D. Prosperi, Angew. Chem., Int. Ed., 2012, 51, 9272–9275 CrossRef CAS.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS.
- M. Colombo, S. Mazzucchelli, J. M. Montenegro, E. Galbiati, F. Corsi, W. J. Parak and D. Prosperi, Small, 2012, 8, 1492–1497 CrossRef CAS.
- M. Colombo, S. Sommaruga, S. Mazzucchelli, L. Polito, P. Verderio, P. Galeffi, F. Corsi, P. Tortora and D. Prosperi, Angew. Chem., Int. Ed., 2012, 51, 496–499 CrossRef CAS.
- S. Mazzucchelli, M. Colombo, C. De Palma, P. Verderio, M. D. Coghi, E. Clementi, P. Tortora, F. Corsi and D. Prosperi, ACS Nano, 2010, 4, 5693–5702 CrossRef CAS.
- S. Sommaruga, A. Lombardi, A. Salvadè, S. Mazzucchelli, F. Corsi, P. Galeffi, P. Tortora and D. Prosperi, Appl. Microbiol. Biotechnol., 2011, 91, 613–621 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS.
- M. Colombo, F. Corsi, D. Foschi, E. Mazzantini, S. Mazzucchelli, C. Morasso, E. Occhipinti, L. Polito, D. Prosperi, S. Ronchi and P. Verderio, Pharmacol. Res., 2010, 62, 150–165 CrossRef CAS.
- L. Schmitt, C. Dietrich and R. Tampe, J. Am. Chem. Soc., 1994, 116, 8485–8491 CrossRef CAS.
- P. Galeffi, A. Lombardi, M. D. Donato, A. Latini, M. Sperandei, C. Cantale and P. Giacomini, Vaccine, 2005, 23, 1823–1827 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |