DOI:
10.1039/C3BM60070J
(Paper)
Biomater. Sci., 2013,
1, 1055-1064
Nucleic acid-directed self-assembly of multifunctional gold nanoparticle imaging agents†‡
Received 13th March 2013, Accepted 4th June 2013
First published on 20th June 2013
Abstract
Gold nanoparticles have attracted much interest as a platform for development of multifunctional imaging and therapeutic agents. Multifunctionalized gold nanoparticles are generally constructed by covalent assembly of a gold core with thiolated ligands. In this study, we have assembled multifunctionalized gold nanoparticles in one step by nucleic acid hybridization of ODN (oligodeoxynucleotide)-derivatized gold nanoparticles with a library of pre-functionalized complementary PNAs (peptide nucleic acids). The PNAs were functionalized by conjugation with DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) for chelating 64Cu for PET imaging, PEG (polyethylene glycol) for conferring stealth properties, and Cy5 for fluorescent imaging. The resulting nanoparticles showed good stability in vitro by melting temperature studies, and in vivo by showing biodistribution behavior in a mouse that would be expected for a PEGylated gold nanoparticle rather than that for the radiolabelled PNA used in its assembly.
1. Introduction
Nanotechnology offers great potential for the development of highly sensitive diagnostic and targeted therapeutic agents. The particular size range of 10–100 nm gives nanoparticles extraordinary loading capacity for therapeutics and/or contrast agents as well as other functions such as targeting. Multifunctional and multimodal nanoparticles have been developed as magnetic resonance imaging (MRI), positron emission tomography (PET), photoacoustic, and optical imaging agents, as well as photodynamic, photothermal, and chemotherapeutic agents among others.1–6 Functionalization of nanoparticles is largely carried out by covalent bond formation using classical bioconjugation methods such as amide bond formation, addition or substitution reactions of thiols, hydrazone formation, and metal–thiol reactions, and more recently via various click reactions.7–10 These strategies generally suffer from difficulty in controlling the extent and reproducibility of the reactions and in characterizing the products, making it difficult to precisely control the composition and properties of the nanoparticles. In addition, the covalent nature of the conjugation reactions can inhibit biodegradation of the nanoparticles. Functionality has also been incorporated through weaker and less specific non-covalent self-assembly mechanisms such as electrostatic interactions, micelle or liposome formation, and host guest interactions.11–14 Nanoparticles produced in this way, however, have variable structure and stability, and are less well defined.A more promising approach to assembling nanoparticles with programmable composition and properties would be to use the higher specificity and tunable stability afforded by nucleic acid hybridization. Nucleic acid hybridization has been extensively used for constructing and assembling complex 2- and 3-dimensional materials, as well as for detecting nucleic acids by changes in the physical properties of the materials generated.15–19 It has also recently been used to construct nanoparticles for in vivo siRNA delivery20–23 and as reporters on nanoparticles for detecting the presence of complementary nucleic acids in cells (nano-flares).24–26 Nucleic acid hybridization has also been previously used in pre-targeting strategies to assemble targeting agents with imaging agents at the in vivo target site that are aimed at improving specificity.27–30 In most cases, nucleic acid hybridization has been used to attach only one functional entity to a nanoparticle. In principle, however, nucleic acid hybridization could also be used as a stable, but reversible, non-covalent click reaction to rapidly and combinatorially assemble much more highly functionalized nanoparticles containing any desired combination of targeting, imaging, therapeutic, endosome disrupting, and stealth functionalities (Fig. 1).
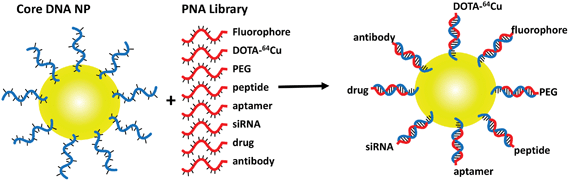 |
| | Fig. 1 Self-assembly of functionalized gold nanoparticles. Gold nanoparticles conjugated to thiolated ODNs are hybridized with any combination of functionalized PNAs to rapidly assemble a combinatorial library of functionalized gold NPs. | |
Gold nanoparticles are an ideal platform for nucleic acid directed self-assembly because they can be readily produced in a variety of sizes and shapes, and easily derivatized with thiolated oligodeoxynucleotides (ODNs).15,31–33 Instead of using a functionalized complementary ODN to hybridize to the ODN on the gold NP, we decided to make use of functionalized PNAs (peptide nucleic acid) because their unnatural polyamide backbone confers a number of advantages over ODNs.34,35 PNAs are highly resistant to degradation by biological systems and also protect the DNA or RNA to which they are bound from degradation. They also hybridize to DNA with higher affinity than does DNA making it possible to use shorter nucleic acid sequences for assembly. Most importantly, they can be conveniently synthesized by solid phase automated peptide synthesis, which allows the easy preparation of PNA-peptide hybrids and the incorporation of a wide variety of amino acid analogues and auxiliary agents. Herein, we report the preparation of functionalized gold nanoparticles through nucleic acid-directed assembly of PNA functionalized with PEG, the PET imaging agent 64Cu-DOTA, and fluorescent molecules (Fig. 2) with ODN-conjugated gold nanoparticles, that remain intact in vivo.
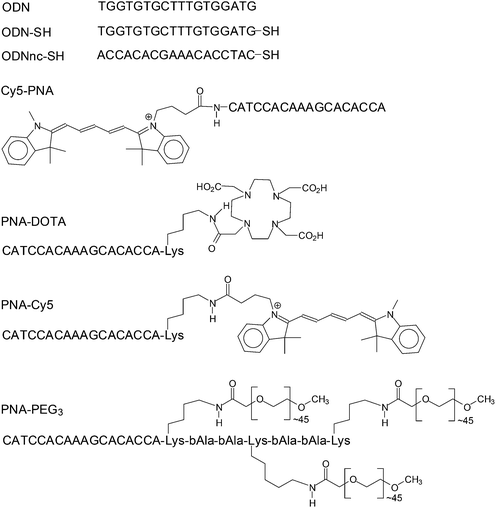 |
| | Fig. 2 Functionalized ODNs and PNAs used in this study. All ODNs are written in a 5′ → 3′ direction and all PNAs in an amino → carboxy direction. | |
2. Materials and methods
All solvents and chemicals were purchased from Sigma-Aldrich and used without further purification unless otherwise indicated. Fmoc-Lys(mtt)-OH was purchased from EMD Chemicals, Inc. Methoxy PEG succinimidyl carboxymethyl ester, MW 2000 (PEG2K-NHS ester) was purchased from JenKem Technology Co., LTD. Cy5-NHS ester was synthesized by Jillian Smith according to a published procedure.36 The MCF-10A cell lines were purchased from ATCC and cultured under the guidelines provided by ATCC. All cell culture media were purchased from Invitrogen, Inc.2.1. Size and zeta potential measurements
Samples for transmission electron microscopy (TEM) measurements were dropped onto PELCO formvar/carbon coated copper grids (Ted Pella, Inc.). After 15 min, the excess colloid was removed by filter paper and 2% uranyl acetate was then dropped on the same grid and incubated for another 2 min. Then the excess solution was again removed by filter paper. The resulting grids were imaged by a FEI Spirit Lab6 TEM. Hydrodynamic diameters and zeta potentials were determined by Malvern Zetasizer Nano using a He–Ne laser at 633 nm. Surface plasmon resonance bands of gold nanoparticles were measured by Varian Cary 100.2.2. Synthesis of ODN capped gold nanoparticles ODN-AuNP and ODNnc-AuNP
The ODN capped gold nanoparticles were synthesized according to a reported procedure.15 Briefly, sodium citrate was added to boiling chloroauric acid solution to yield gold nanoparticles about 11 nm in size.37 TGGTGTGCTTTGTGGATG-O-C3H6-SS-C3H6-OH and ACCACACGAAACACCTAC-O-C3H6-SS-C3H6-OH were purchased from IDT and reduced with tris(2-carboxyethyl)phosphine at room temperature for one hour to produce ODN-SH and ODNnc-SH respectively. The ODNs were then isolated by ethanol precipitation and redissolved in water. These freshly prepared thiolated ODN or ODNnc solutions were added to the citrate gold nanoparticles (3 nmol per 1 mL of 7 nM citrate·AuNP). After 20 min, 10% SDS solution was added to bring the mixture to 0.1% SDS. Phosphate buffer (0.1 M, pH = 7.4) was added to bring the mixture to 0.01 M phosphate and sodium chloride (2.0 M) was added in 30 min intervals to bring the final mixture to 0.3 M NaCl. The final mixture was gently shaken for 24 h in the dark under room temperature. The gold NP were centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rcf, 30 min; 3×) and resuspended in PBS.
500 rcf, 30 min; 3×) and resuspended in PBS.2.3. Molar extinction coefficients of the gold NPs
Citrate·AuNP or ODN-AuNP (10 μL) was dissolved in 90 μL of freshly made aqua regia at RT. The mixture was diluted to 5 mL with 1% nitric acid and the gold concentration was analyzed by Perkin Elmer Elan DRC II ICP-MS using Bi as external reference and standard gold solutions. The concentration of gold nanoparticles was calculated as [Au]/(4NAρπr3/3) where [Au] is the concentration of gold from ICP-MS analysis, ρ is the density of gold (19.3 g cm−3), r is the average radius of the AuNP measured by TEM (5.1 nm) and NA is Avogadro's number. The absorbance of the gold nanoparticles was then measured at 520 nm for citrate·AuNP or 524 nm for ODN-AuNP as a function of concentration, and the molar extinction coefficients were obtained from a linear least squares fit of the absorbance vs. the particle concentration (Fig. S1‡).2.4. ODN loading of the ODN gold NP
ODN-AuNP (0.5 mL of 10 nM) was dissolved in 2 mM KCN solution and 1 μL of this solution was mixed with 1 μL of [32P]-ATP, 2 μL of 10x kinase buffer, 1 μL of T4 polynucleotide kinase solution (10 U) and 15 μL of water, incubated at 37 °C for 1 h and then boiled for 10 min. Authentic ODN of known concentration was also labeled following the same procedure. 32P-labeled ODN (50 nM) and 1 μL of the radiolabeled ODN from the dissolved ODN-AuNP solution were then separately incubated with known amounts of antisense PNA in Tris buffer (10 mM, pH 7.5) and NaCl (50 mM), boiled for 5 min and slowly cooled down to room temperature. The mixtures were electrophoresed on a 10% native PAGE and the radiolabeled bands were imaged on a Biorad Personal Molecular Imager and quantified by Biorad Quantity One software. A calibration curve was obtained by a linear least squares fit of the observed ratio of the shifted ODN·PNA duplex band and free ODN band to the actual ratio (Fig. S2‡). The calibration curve was then used to determine the concentration of ODN obtained from the dissolved ODN-AuNP solution. The average ODN loading per ODN-AuNP was then determined from the concentration of the ODN divided by the concentration of the ODN-AuNP determined from the absorbance at 520 nm.2.5. General procedure for PNA synthesis
All PNAs and conjugates were synthesized on an Expedite 8900 PNA synthesizer on 2 μmol of Fmoc-PAL-PEG-PS according to the standard automated Fmoc PNA synthesis procedure38 utilizing commercial PNA and peptide monomers (Panagene Inc., Korea). Following the final step of synthesis, the resin was washed with dry CH2Cl2 (2 × 3 mL), followed by drying under a stream of N2. The resin was then shaken in a vial with trifluoroacetic acid (160 μL) and m-cresol (40 μL) at room temperature for 1.5 h twice to release and deprotect the PNA. The solution from each deprotection was separated from the resin, combined and added to ice-cold Et2O (1.5 mL). The resulting precipitate was collected by centrifugation and purified by reverse-phase HPLC on a Waters XBridge Prep C18 column (130 Å) with buffer A (0.1% TFA in H2O) and buffer B (0.1% TFA in CH3CN) on a Beckman System Gold instrument equipped with a UV-vis array detector. The flow rate was 2.5 mL min−1 and the gradient was 1% buffer B per min starting from 5% buffer B. The fractions were collected and concentrated to dryness in a SpeedVac (Savant) and characterized by UV-vis and MALDI-TOF which was carried out on a PerSpective Voyager mass spectrometer with α-cyano-4-hydroxycinnamic acid as the matrix and insulin as the internal reference.2.6. Synthesis of Cy5-PNA
PNA CATCCACAAAGCACACCA was synthesized with removal of the amino terminal Fmoc group. Cy5-NHS ester (20 μmol) was dissolved in anhydrous DMF (100 μL) and mixed with DIPEA (2 μmol). The mixture was incubated with the solid support under room temperature for 2 h. Then the solid support was separated and washed with DMF. The Cy5-PNA was cleaved and purified by the general procedure described above. MALDI: exact [M + H]+ expected 4413.0, found 4415.2.2.7. Synthesis of PNA-DOTA and PNA-Cy5
CATCCACAAAGCACACCA-Lys(Mtt) was synthesized with deprotection and capping of the N terminal amine with 5% acetic anhydride and 6% lutidine in DMF. The Mtt protecting group was removed by washing the solid support with 1 mL of 1% TFA in DCM several times, 5 min each time, until no yellow color was detected. The solid support was then washed with DMF until the eluant was no longer acidic, as measured by wet pH paper. For PNA-Cy5 synthesis, Cy5-NHS ester (20 μmol) and DIPEA (2 μmol) were dissolved in 100 μL DMF and incubated with the solid support at room temperature for 2 h. For PNA-DOTA synthesis, tri-tert-butyl 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate (20 μmol), EDC (20 μmol) and DIPEA (2 μmol) in 100 μL DMF was incubated with the solid support under room temperature overnight. After both syntheses, the reaction mixtures were removed and the solid supports were washed with 1 mL of DMF four times. PNA-DOTA or PNA-Cy5 was cleaved and purified by HPLC as described above. MALDI of PNA-DOTA: [M + Na]+ expected 5365.3, found 5364.8. PNA-Cy5 did not give rise to an identifiable MALDI peak but was characterized by UV, fluorescence spectroscopy, and ability to hybridize with complementary 32P-labeled ODN by native PAGE. UV: A642 (Cy5)/A260 (PNA), obsvd 1.13, calcd 1.37 using estimated extinction coefficients for Cy5 at 642 nm (250000 L M−1 cm−1) and PNA at 260 nm (182000 L M−1 cm−1).2.8. Synthesis of PNA-PEG3
CATCCACAAAGCACACCA-Lys(Mtt)-bAla-bAla-Lys(Mtt)-bAla-bAla-Lys(Mtt) was synthesized followed by removal of the amino terminal Fmoc group, the amino terminus was acetylated with 5% acetic anhydride and 6% lutidine in DMF. The PNA was cleaved from the solid support and purified by HPLC. PEG2K-NHS ester (10 μmol) was dissolved in 100 μL DMF and mixed with 1 μL of DIPEA. Ac-PNA-(Lys-bAla)2Lys (0.0963 μmol) was dissolved in 10 μL DMF and added to the PEG solution, 1 μL at a time. Following each addition, the pH of the reaction of the mixture was checked with wet pH paper and additional DIPEA was added to maintain the basicity. The reaction mixture was incubated at 55 °C for 20 min before the next aliquot was added. After all the PNA was added, the reaction mixture was left at room temperature overnight and then precipitated with ether. The crude product was purified by HPLC. PNA-PEG3 did not give rise to identifiable MALDI and was analyzed by its ability to hybridize with 32P labeled ODN by native PAGE.2.9. Fluorescence quenching studies
Various amounts of 36.6 μM Cy5-PNA were added to 60 μL of 5.23 nM ODN-AuNP or ODNnc-AuNP or citrate·AuNP in 1× PBS. The mixtures were incubated at room temperature for 5 min and then the fluorescence was measured at 663 nm with excitation at 633 nm. The fluorescence of Cy5-PNA in 1× PBS was measured at each concentration as control.2.10. Fluorescence melting temperature study of Cy5-PNA·ODN-AuNP
Temperature-dependent fluorescence intensity measurements of Cy5-PNA·ODN-AuNP were made with excitation at 633 nm and emission detection at 663 nm. The Cy5-PNA·ODN-AuNP complex (0.27 μM Cy5-PNA, 4.7 nM ODN-AuNP in 1× PBS buffer) was heated from 25° to 80 °C at 1 °C per min and held at 80 °C for 5 min and then cooled down to 25 °C (Fig. S3A‡). After 5 min, the heating-cooling cycle was repeated. The temperature dependent fluorescence of Cy5-PNA alone was also acquired (Fig. S3B‡). The raw data was then converted to fraction single strand by using the average observed fluorescence for Cy5-PNA·ODN-AuNP as a function of temperature (Fobsvd) and using a linear fit of the fluorescence curve prior to melting for the temperature dependence of the duplex form (Fduplex), and using the fluorescence curve of the Cy5-PNA for the temperature dependence of the single strand form (Fsingle). The fraction single strand at a given temperature was then calculated as (Fobsvd − Fduplex)/(Fsingle − Fduplex). Thermodynamic parameters were derived from van't Hoff analysis as shown in Fig. S4.‡2.11. Absorbance melting temperature study of Cy5-PNA·ODN
The absorbance at 260 nm of the complex from Cy5-PNA (26.4 μM) and ODN (40 μM) in 1× PBS buffer was monitored from 25° to 85 °C at 1 °C per min, and after holding at 85 °C for 5 min, from 85° to 25 °C. After 5 min, the heating-cooling cycle was repeated. The data was then converted to fraction single strand by using the average observed absorbance for Cy5-PNA·ODN as a function of temperature (Aobsvd) and linear fits to the initial and final parts of the melting curve for the temperature dependence of the duplex (Aduplex) and single strand (Asingle) forms respectively. The fraction single strand at a given temperature was then calculated as (Aobsvd − Aduplex)/(Asingle − Aduplex). Thermodynamic parameters were derived as shown in Fig. S4.‡2.12. Cellular uptake of ODN-AuNP
MCF10A cells were seeded in 35 mm glass bottom dish and allowed to grow to 70% confluence. PNA-Cy5 (1 μL of 1.18 μM) and 15 μL of 10 nM ODN-AuNP was incubated at 50 °C for 10 min and after cooling to RT, was added to 135 μL of fresh complete media containing 0.5 μg mL−1 of FAM-dextran (MW 25000, Invitrogen). The cell media was then replaced with this mixture and incubated at 37 °C with 5% CO2 for 6 h. After 5 h, 2 μL of 10 μg mL−1 Hoechst 3342 was added to the mixture. 1 h later, the mixture was removed and the cells were washed with 1× PBS buffer 5 times and then incubated with 2 mL of fresh media. The cellular uptake was visualized by Nikon A1 confocal microscope.2.13. Reduction of nonspecific cell binding by PEGylation
MCF10A cells were seeded in a 12 well plate and grown to 90% confluence. PNA-PEG3 was mixed with ODN-AuNP in various ratios. The mixtures were incubated at room temperature for 30 min and added to the cells with complete cell media. The final concentration of AuNP was 0.9 nM. After 6 h of incubation at 37 °C, cell media was removed and the cells were washed with PBS twice. The cells were trypsinized, counted with hemacytometer and centrifuged. Each cell pellet was digested with 100 μL nitric acid and hydrogen peroxide mixture (9:1, v/v) overnight followed by addition of 200 μL aqua regia. The resulting solution was then diluted to 7 mL with 1% nitric acid and analyzed by Perkin Elmer Elan DRC II ICP-MS using Bi as external reference.2.14. Radiolabeling of the PNA
Copper-64 (t1/2 = 12.7 h, β+ = 17%, β− = 40%) was produced on the Washington University Medical School CS-15 cyclotron by the 64Ni(p,n)64Cu nuclear reaction at a specific activity of 1.85–7.40 GBq μg−1 at the end of bombardment.39 PNA-DOTA (320 pmol) was incubated with 64Cu (185 MBq) in 100 μL of 0.1 M ammonium acetate buffer (pH 5.5) at 65 °C for 1 h, with a yield of 52.5 ± 2.4%. The PNA-DOTA·64Cu was purified by solid-phase extraction after ethylenediaminetetraacetic acid (EDTA, 10 mM in 50 mM pH 7.4 phosphate buffer) and the specific activity was 301 ± 14 MBq nmol−1.40 The PNA-DOTA used for annealing with ODN and ODN-AuNP was radiolabeled and purified following the same procedure with the specific activity of 285 ± 11 MBq nmol−1. Then 37 MBq of PNA-DOTA·64Cu was mixed with ODN. Another 37 MBq (9.1 nmol) of PNA-DOTA·64Cu was mixed with ODN-AuNP (0.52 nmol) followed by addition of PNA-(PEG2K)3 (27.3 nmol). The mixture was purified with a Zeba spin desalting column (Pierce).2.15. Bio-distribution studies
All animal studies were performed in compliance with guidelines set forth by the NIH Office of Laboratory Animal Welfare and approved by the Washington University Animal Studies Committee. Biodistribution studies were performed in male C57BL/6 mice weighing 20–25 g (n = 4 per group) and about 370 kBq of PNA-DOTA·64Cu, PNA-DOTA·64Cu·ODN and PNA-DOTA·64Cu/PEG3·ODN-AuNP in 100 μL saline (APP Pharmaceuticals) was injected via the tail vein. The mice were anesthetized with inhaled isoflurane and were re-anesthetized before being euthanized by cervical dislocation at each time point (1 h, 4 h, and 24 h) post injection (p.i.). Organs of interest were collected, weighed and counted in a gamma counter (Beckman 8000). Standards were prepared and measured along with the samples to calculate the percentage of the injected dose per gram of tissue (% ID g−1).413. Results and discussion
3.1. ODN-gold nanoparticle synthesis
Citrate gold nanoparticles (citrate·AuNP) were synthesized by standard methods37 and were 10.2 ± 0.8 nm in diameter as measured by TEM (Fig. 3a). Dynamic light scattering gave a very similar hydrodynamic radius of 10.3 ± 2.6 nm (Fig. 4). The particles had an expected highly negative zeta potential of −22.5 ± 2.8 mV. Conjugation of the gold nanoparticles to a 3′-thiolated ODN, ODN-SH (Fig. 2), by standard methods,15 gave ODN-AuNP. TEM of a negatively stained sample showed particles with a diameter of about 16.1 ± 0.9 nm consisting of a dark core and a bright outer circle corresponding to the ODN layer (Fig. 3b). Dynamic light scattering gave a similar hydrodynamic diameter of 16.2 ± 4.1 nm (Fig. 4). The thickness of the ODN layer was about 2.5–2.8 nm as deduced from the difference in size of the ODN-AuNP and the citrate-AuNP from TEM and DLS experiments. The thickness of the ODN layer is less than half the length of a helical 18-mer ODN (6.3 nm) indicating that the ODN is not fully extended on the gold surface and may be partially contacting the surface. The zeta potential of the ODN-AuNP decreased to −31.7 ± 0.1 mV due to the large number of negatively charged phosphate groups contributed by the ODN. |
| | Fig. 3 TEM pictures of gold NPs at various stages of construction and assembly. TEM of (a) citrate·AuNP, (b) ODN-AuNP, and (c) PNA-PEG3/PNA-DOTA·ODN-AuNP with a PNA-PEG/PNA-DOTA ratio of 3:1. All three samples were stained with uranyl acetate. Panel (c) was assembled from four representative pictures. The scale bars represent 20 nm. | |
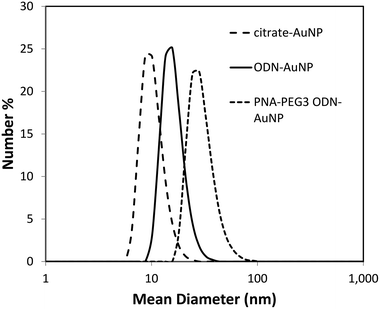 |
| | Fig. 4 Dynamic light scattering analysis of gold NP at various stages of construction. | |
3.2. Sequence specific self-assembly with PNA
Gold has been known as an effective fluorescent quencher and the fluorescent quenching efficiency decreases drastically as the distance between the fluorophore and the gold surface increases.42 We utilized this unique property to demonstrate the self-assembly of the ODN-AuNP with PNA. We synthesized a 15-mer PNA that was labeled with Cy5 at its amino end, Cy5-PNA (Fig. 2), which is complementary (antisense) to the ODN on the AuNP. Upon PNA-ODN hybridization, the fluorophore is positioned three nucleotides away from the gold surface, or about one nm if the PNA-ODN duplex were perpendicular to the surface.43 This distance is close enough to cause significant fluorescent quenching.42 As shown in Fig. 5, the fluorescence of the Cy5 was almost completely quenched when the Cy5-PNA was mixed with ODN-AuNP and remained quenched until the concentration of the Cy5-PNA-mer was about 90 times the concentration of the gold nanoparticle. Beyond this point, the fluorescence started to increase linearly indicating that all the complementary binding sites on the gold NP had been saturated. The gold NP without the ODN shell, however, also quenched the fluorescence of the PNA. To demonstrate that the quenching was due to sequence specific binding of the PNA to the ODN shell, and not simply to the gold surface, a gold NP bearing the non-complementary ODN, ODNnc (Fig. 2), was also synthesized. When titrated with Cy5-PNA very little quenching was observed (Fig. 5).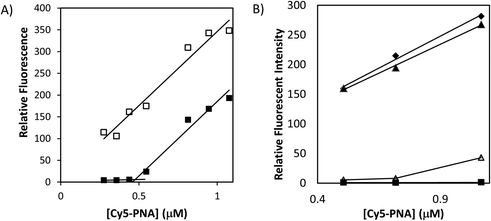 |
| | Fig. 5 Fluorescence titration of ODN-AuNPs with Cy5-PNAs. (A) Fluorescence of increasing concentration of Cy5-PNA in the presence (■) or absence (□) of 5 nM ODN-AuNP. (B) Fluorescence of increasing concentration of Cy5-PNA in (◆) buffer alone, or the presence of: (△) 10 nM ODN-AuNP; (▲) 10 nM ODNnc-AuNP; (■) 10 nM citrate-AuNP. | |
To determine the stability of the PNA·ODN hybrid on the gold NP, temperature dependent fluorescence measurements were made between room temperature and 85 °C. Following an initial heating to 85 °C to degas the sample, the fluorescent heating and cooling curves for ODN-AuNP·Cy5-PNA were quite reproducible and indicated that the complexes had a melting temperature of 70 °C (Fig. 6). This can be compared to a melting temperature of 71.5 °C for ODN·Cy5-PNA determined by temperature dependent absorbance measurements. Thermodynamic parameters for the stability of the two duplexes was obtained from van't Hoff analysis of the melting temperature curves for a two state all or none melting model for the temperature range of 52–72 °C (the most linear increase in ΔG) (Fig. S4‡). For the free ODN and AuNP bound ODN, ΔH° was −53.2 ± 1.0 and −38.6 ± 1.2 kcal mol−1, and ΔS was 133 ± 3 and 82.3 ± 6 cal deg−1 mol−1, respectively. From these values, the Kd's for PNA binding to the free and AuNP bound ODN at 37 °C were extrapolated as 4.2 and 0.7 nM respectively. The Kd for the AuNP bound ODN was about 6 times lower, likely due to the polydentate effect.
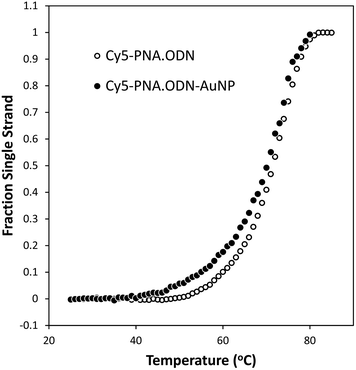 |
| | Fig. 6 Melting curves for Cy5-PNA·ODN-AuNP and Cy5-PNA·ODN. The fraction single strand form for Cy5-PNA·ODN-AuNP (●) from analysis of temperature dependent Cy5 fluorescence measurements at 0.270 μM PNA and 0.423 μM ODN bound to AuNP, and for Cy5-PNA·ODN (○) from analysis of temperature dependent absorbance measurements at 26.4 μM PNA and 40 μM ODN. Data from two heating/cooling cycles were used to generate each melting temperature curve. Determination of the thermodynamic parameters for duplex formation can be found in Fig. S4.‡ | |
3.3. PEGylation of the ODN-gold NP
For imaging specific targets in vivo it is important that the NPs are not recognized by the MPS (mononuclear phagocyte) system, often referred to in the literature as the RES (reticulo-endothelial) system, or taken up by non-target cells.44 It has been found that PEGylation can greatly reduce binding of NP to proteins and confer stealth character.45 To study the interaction of the PNA·ODN-AuNP with cells and the ability of PEGylation to reduce non-specific binding and uptake, we synthesized a fluorescently labeled PNA for tracking the NP and a PEGylated PNA for conferring stealth character. To minimize quenching of the fluorescence, Cy5 was conjugated to the carboxy terminus of PNA to give PNA-Cy5 (Fig. 2) via a carboxy-terminal lysine group so that the fluorophore would be directed away from the surface of the gold NP. To create a PEGylated PNA with sufficient PEG to cover the surface, the PNA was derivatized with three PEG2K molecules via acylation of three lysines at the carboxy terminus of PNA and spaced apart by two beta-alanines to give PNA-PEG3 (Fig. 2). The ODN-AuNP was hybridized with a stoichiometric amount of PNA with differing ratios of PNA-PEG3:PNA-Cy5. TEM analysis under negative staining of 75/25 ratio of PNA-DOTA/PNA-PEG3·ODN-AuNP showed a dark center of about 12 nm, representing the gold core, surrounded by a narrower bright band than was seen with the ODN-AuNP, and a large and diffuse dark area of average diameter 41.7 ± 1.1 nm that must correspond to the ODN-PNA duplex linked to the PEG2K layer (Fig. 3C).Dynamic light scattering gave a hydrodynamic radius of 30.2 ± 9.1 nm which when subtracted from the original diameter of 10.1 nm for the citrate-AuNP gives an increased diameter of 17.1, or an increased radius of 8.5 nm (Fig. 4). The increased radius corresponds very well to 6 nm for the PNA-ODN helix (18 bp × 0.33 nm bp−1)43 plus 3.8 nm corresponding to the Flory radius of PEG2K.46 Simple calculations suggest that there is ample surface area to accommodate the PNA·ODN duplex and the three PEG2K chains. A PNA·ODN duplex has a diameter of 2.2 nm corresponding to a cross-sectional area of 0.95 nm2 which when multiplied by 100 ODNs/AuNP gives and area of 95 nm2 which is 3 times less than the calculated surface area of 314 nm2 for a 10 nm diameter gold particle. If fully extended, the PNA·ODN duplex would extend the radius of the gold NP by 6 nm, resulting in an outer surface area of 1520 nm2, or 152 nm2 per PNA·ODN duplex. Given that the Flory radius of PEG2K is 3.8 nm and that the center of radius for each PEG2K would be an additional 1 nm further out, then the available surface area is calculated to be 3157 nm2. Since three PEG2K accommodate a cross-sectional surface area of 114 nm2, the available surface would become saturated upon addition of about 28 PNA-PEG3 units. Above this amount, one would expect the PEG chains to adopt a more extended, brush-like structure. In accord with this analysis, the zeta potential of the particle increased to −4.1 ± 0.4 mV as the PEG layer significantly shields the charges.47
3.4. Cellular uptake of the PEGylated ODN-gold NP
The interaction of the AuNPs with a normal human breast cell line (MCF10A) was examined by confocal microscopy and quantified by inductively coupled plasma mass spectrometry (ICP-MS). As expected from previous studies with ODN-AuNP,48 Cy5-PNA·ODN-AuNP was internalized by MCF-10A cells efficiently in complete growth media without any cell permeation enhancing ligand (Fig. 7A). The particle (red) colocalized with FAM-dextran (green) which is considered to enter the cells through an endosomal pathway. The uptake presumably occurs through specific uptake of serum proteins bound to the gold nanoparticle. PEG is known to prevent protein binding to nanoparticles, and upon complexation with PNA-PEG3, the uptake decreased with increasing density of the PNA-PEG3 on the gold NP (Fig. 7B). MCF-10A uptake was reduced by 60% at 60% PEGylation and further reduced by 80% with almost complete PEGylation. The required percentage of PNA-PEG3 could possibly be further driven down by utilizing PEG of higher molecular weight or higher density per PNA.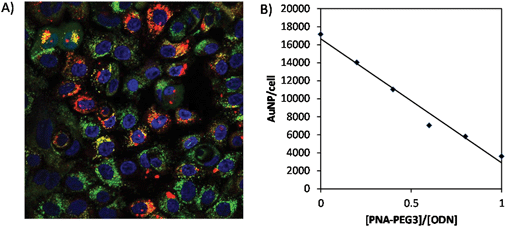 |
| | Fig. 7 Effect of the degree of PEGylation on uptake of PNA-Cy5/PNA-PEG3·ODN-AuNP by MCF-10A cells. (A) Fluorescence imaging of MCF10A cells incubated with PNA-Cy5·ODN-AuNP (red) and FAM-dextran (MW 25000) (green). Cell nuclei were stained with Hoechst 3342 (blue). (B) Quantification of PNA-Cy5/PNA-PEG3·ODN-AuNP uptake by cells by ICP-MS. | |
3.5. Biodistribution studies of the PEGylated gold NP
To determine whether or not the non-covalently assembled PNA-PEG3·ODN-AuNP would remain intact in vivo, we carried out biodistribution studies with a nanoparticle that was radiolabeled with the PET radionuclide 64Cu.49,50 To radiolabel the particle, we synthesized a PNA functionalized with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) which has a high affinity for the radionuclide 64Cu. The DOTA was coupled to carboxy-terminal lysine on the PNA via tri-t-butyl-DOTA ester and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) to give PNA-DOTA (Fig. 2) following deprotection. PNA-DOTA was first radiolabeled with 64Cu, purified, and then hybridized with a complementary ODN and the ODN-AuNP.The biodistribution of the 64Cu·PNA-DOTA associated with the ODN-AuNP (Fig. 8C) was strikingly different than for 64Cu·PNA-DOTA alone (Fig. 8A), or when complexed to the ODN (Fig. 8B) at 1 h, 4 h and 24 h post injection (p.i.). Both 64Cu·PNA-DOTA and 64Cu·PNA-DOTA·ODN were cleared very quickly through the renal system, with some minor accumulation in the liver, lung and gastrointestinal tract (stomach and intestine), and negligible uptake in other organs (Fig. 8A and B). At 1 h p.i., the blood retention of 64Cu·PNA-DOTA and 64Cu·PNA-DOTA·ODN were both less than 0.5% ID g−1 while the kidney retention was about 5%. The liver uptake was constant over 24 h with less than 2% ID g−1. While 64Cu·PNA-DOTA/PNA-PEG3·ODN-AuNP also showed rapid clearance from systemic circulation (0.31 ± 0.09% ID g−1 at 1 h p.i.), it showed much higher retention in liver than 64Cu·PNA-DOTA or its ODN hybrid (Fig. 8C), typical of covalently linked gold nanoparticles of similar size and composition. For example, the biodistribution at 24 h (21% liver, 11% spleen, 4% kidney) was very similar to that reported in mice at 48 h for a 20 nm gold nanoparticle coated with thioctic acid-anchored PEG5K with a hydrodynamic radius of 45 nm (30% liver, 15% spleen and 5% kidney).51 While liver uptake was dominant, it gradually decreased over 24 h from 32 ± 3% ID g−1 at 1 h, to 28 ± 7% ID g−1 at 4 h, and 21 ± 2% ID g−1 at 24 h p.i. The spleen accumulation, however, was stable with about 10% ID g−1 through the study.
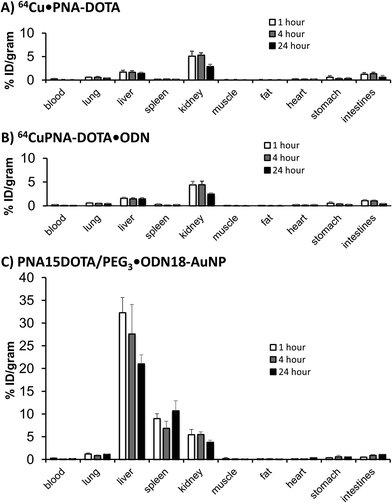 |
| | Fig. 8 Biodistribution studies of AuNP and components. (A) PNA-DOTA·64Cu, (B) PNA-DOTA·64Cu·ODN and (C) PNA-DOTA/PEG3·64Cu·ODN-AuNP for 1 h (a), 4 h (b) and 24 h (c) p.i. | |
The sharp contrast between the biodistribution profile of 64Cu·DOTA/PNA-PEG3·ODN-AuNP and 64Cu·PNA-DOTA by itself or when hybridized to the ODN at different time points demonstrates that the nanoparticle remained largely intact during the initial biodistribution. Since it has been established that gold nanoparticles themselves do not clear from the liver for many weeks,52 the loss of about 50% of the initial dose that accumulated in the liver at 4 h after 24 h may be due to cleavage of the ODN-Au linkage or dissociation of the PNA·ODN duplex. These two mechanisms could be distinguished in the future by using longer, more stable PNA·ODN duplexes. The pharmacokinetics of 64Cu·DOTA/PNA-PEG3·ODN-AuNP would be less than optimal for potential in vivo therapeutic applications due to the rapid depletion of the nanoparticle from the blood and high accumulation in the liver. The rapid clearance may be useful, however, for some diagnostic applications in which a low background would be desirable. Further surface modification of the nanoparticle to resist protein binding and non-specific uptake by the liver will likely be needed before it can be used for efficient cell specific targeting, and is the focus of our further work.
4. Conclusion
We have shown that DNA-directed nucleic acid hybridization with PNA can be used for the rapid combinatorial self-assembly of multifunctional gold nanoparticles that largely maintain their integrity in vivo. The strategy is not limited to gold nanoparticles, but to any nanoparticle bound to oligodeoxynucleotides or their analogs. Nucleic acid hybridization also provides a simple and straightforward method for radiolabeling of the nanoparticle with a PET imaging agent using a purified 64Cu-labeled PNA. Because the stability of the PNA·DNA hybrids used to assemble the nanoparticle is tunable by sequence and length, the stability of the nanoparticles is also expected to be tunable. These initial results demonstrate that nucleic acid-directed self-assembly could be used for the combinatorial synthesis and optimization of nanoparticle-based imaging and therapeutic agents through what amounts to non-covalent click chemistry.Acknowledgements
This work was supported by Kay Yow V Foundation Grant to JST and MJW, and in part by a PEN grant (HHSN268201000046C) and by the Washington University NIH Mass Spectrometry Resource (Grant No. P41 RR000954). We thank J. Smith for synthesizing Cy5-NHS ester. We also thank Nano Research Facility of Washington University for providing the DLS, TEM and ICP-MS facility.References
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS.
- R. Subbiah, M. Veerapandian and K. S. Yun, Curr. Med. Chem., 2010, 17, 4559–4577 CrossRef CAS.
- Z. Liu, F. Kiessling and J. Gatjens, J. Nanomater., 2010 DOI:10.1155/2010/894303.
- F. M. Kievit and M. Zhang, Adv. Mater., 2011, 23, H217–H247 CrossRef CAS.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- E. Lallana, A. Sousa-Herves, F. Fernandez-Trillo, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 1–34 CrossRef CAS.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS.
- N. T. K. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Chem. Soc. Rev., 2012, 41, 2539–2544 RSC.
- A. Patwa, A. Gissot, I. Bestel and P. Barthelemy, Chem. Soc. Rev., 2011, 40, 5844–5854 RSC.
- A. Puri, K. Loomis, B. Smith, J. H. Lee, A. Yavlovich, E. Heldman and R. Blumenthal, Crit. Rev. Ther. Drug. Carrier Syst., 2009, 26, 523–580 CrossRef CAS.
- S. De Koker, R. Hoogenboom and B. G. De Geest, Chem. Soc. Rev., 2012, 41, 2867–2884 RSC.
- M. E. Davis, Mol. Pharm., 2009, 6, 659–668 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- K. V. Gothelf and T. H. LaBean, Org. Biomol. Chem., 2005, 3, 4023–4037 CAS.
- Y. H. Roh, R. C. H. Ruiz, S. Peng, J. B. Lee and D. Luo, Chem. Soc. Rev., 2011, 40, 5730–5744 RSC.
- A. Stadler, C. Chi, D. van der Lelie and O. Gang, Nanomedicine, 2010, 5, 319–334 CrossRef CAS.
- B. Sacca and C. M. Niemeyer, Chem. Soc. Rev., 2011, 40, 5910–5921 RSC.
- J. W. Keum, J. H. Ahn and H. Bermudez, Small, 2011, 7, 3529–3535 CrossRef CAS.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS.
- D. Bhatia, S. Surana, S. Chakraborty, S. P. Koushika and Y. Krishnan, Nat. Commun., 2011, 2, 339 CrossRef.
- H. Lee, A. K. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS.
- A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane and C. A. Mirkin, ACS Nano, 2009, 3, 2147–2152 CrossRef CAS.
- A. E. Prigodich, P. S. Randeria, W. E. Briley, N. J. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2012, 84, 2062–2066 CrossRef CAS.
- M. Rusckowski, T. Qu, F. Chang and D. J. Hnatowich, Cancer, 1997, 80, 2699–2705 CrossRef CAS.
- Y. Wang, F. Chang, Y. Zhang, N. Liu, G. Liu, S. Gupta, M. Rusckowski and D. J. Hnatowich, Bioconjugate Chem., 2001, 12, 807–816 CrossRef CAS.
- J. He, G. Liu, S. Gupta, Y. Zhang, M. Rusckowski and D. J. Hnatowich, J. Nucl. Med., 2004, 45, 1087–1095 CAS.
- J. A. Diaz, D. M. Grewer and J. M. Gibbs-Davis, Small, 2012, 8, 873–883 CrossRef CAS.
- D. T. Nguyen, D.-J. Kim and K.-S. Kim, Micron, 2011, 42, 207–227 CrossRef CAS.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849–2866 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- P. E. Nielsen, Methods Mol. Biol., 2002, 208, 3–26 CAS.
- K. E. Lundin, L. Good, R. Stromberg, A. Graslund and C. I. Smith, Adv. Genet., 2006, 56, 1–51 CrossRef CAS.
- M. V. Kvach, A. V. Ustinov, I. A. Stepanova, A. D. Malakhov, M. V. Skorobogatyi, V. V. Shmanai and V. A. Korshun, Eur. J. Org. Chem., 2008, 2117 Search PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CAS.
- S. A. Thomson, J. A. Josey, R. Cadilla, M. D. Gaul, C. F. Hassman, M. J. Luzzio, A. J. Pipe, K. L. Reed, D. J. Ricca, R. W. Wiethe and S. A. Noble, Tetrahedron, 1995, 51, 6179–6194 CrossRef CAS.
- D. W. McCarthy, R. E. Shefer, R. E. Klinkowstein, L. A. Bass, W. H. Margeneau, C. S. Cutler, C. J. Anderson and M. J. Welch, Nucl. Med. Biol., 1997, 24, 43 CrossRef.
- Y. Liu, D. Abendschein, G. E. Woodard, R. Rossin, K. McCommis, J. Zheng, M. J. Welch and P. K. Woodard, J. Nucl. Med., 2010, 51, 91 Search PubMed.
- Y. Liu, A. Ibricevic, J. A. Cohen, J. L. Cohen, S. P. Gunsten, J. M. Frechet, M. J. Walter, M. J. Welch and S. L. Brody, Mol. Pharm., 2009, 6, 902 Search PubMed.
- G. Schneider, G. Decher, N. Nerambourg, R. Praho, M. H. V. Werts and M. Blanchard-Desce, Nano Lett., 2006, 6, 530–536 CrossRef CAS.
- V. Menchise, G. De Simone, T. Tedeschi, R. Corradini, S. Sforza, R. Marchelli, D. Capasso, M. Saviano and C. Pedone, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12021–12026 CrossRef CAS.
- C. D. Walkey and W. C. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- D. E. Owens 3rd and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef.
- A. K. Kenworthy, K. Hristova, D. Needham and T. J. McIntosh, Biophys. J., 1995, 68, 1921–1936 CrossRef CAS.
- T. L. Doane, C. H. Chuang, R. J. Hill and C. Burda, Acc. Chem. Res., 2012, 45, 317–326 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3821 CrossRef.
- E. C. Cho, C. Glaus, J. Chen, M. J. Welch and Y. Xia, Trends Mol. Med., 2010, 16, 561–573 CrossRef CAS.
- Y. Liu and M. J. Welch, Bioconjugate Chem., 2012, 23, 671–682 CrossRef CAS.
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
Footnotes |
| † This work is dedicated to Dr Michael J. Welch, who passed away on May 6th, 2012. |
| ‡ Electronic supplementary information (ESI) available: Determination of the molar extinction coefficient for the gold NPs and the average ODN loading of the ODN·AuNP, and the fluorescence melting curves for Cy5-PNA·ODN-AuNP. See DOI: 10.1039/c3bm60070j |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.