High signal contrast gating with biomodified Gd doped mesoporous nanoparticles†
Wen-Yen
Huang
,
Gemma-Louise
Davies
and
Jason J.
Davis
*
Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: jason.davis@chem.ox.ac.uk; Fax: +44 (0)1865 272 690; Tel: +44 (0)1865 275 914
First published on 8th November 2012
Abstract
Internally Gd doped mesoporous nanoparticles have been prepared and exhibit unprecedented relaxivities that are retained on external biomodification. In tuning diffusive water access, image contrast can be reversibly switched in the presence of a specific protein target.
Among the spatially resolved clinical imaging modalities now available, magnetic resonance imaging (MRI) remains the most convenient and widely used of those which are non-invasive.1 During the past decade a great deal of progress has been made in enhancing the natively low image contrast available within protic MRI relaxivity with molecular, biological or nanoparticle based formulations based on chelated gadolinium, through aggregation/rotation based changes or alterations in metal hydration number.1,2 Such attempts include those which are biovectorable and thus capable of identifying and reporting on specific physiology and pathology.3–11 A range of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and diethylenetriaminepentaacetic acid (DTPA) derivatives have, for example, been bound to peptides, monoclonal antibodies or proteins capable of targeting specific cell receptors.3,4,12
Mesoporous silica nanoparticles (MSNs) offer much potential to both high contrast imaging and the delivery of therapeutics and can be chemically modified with relative ease.13–17 Indeed, the chemical capping of MSNs has been investigated as a means of triggering drug delivery.18 Particles <200 nm in size are generally believed to be able to avoid reticuloendothdlial system (RES),19,20 an ability greatly aided through the use of masking hydrophilic coatings (e.g. polyethylene glycol; PEG).21 To date, there exist several examples of doped MSNs capable of generating marked T1 or T2 contrast.2,22 Notably, although central to any targetted imaging application, the effects of particle biomodification on this have not previously been considered. Since the diffusive access of water through particle nanopores to internalised magnetic elements is both theoretically expected23,24 and experimentally25,26 verified to underpin T1 relaxivity (and thus image contrast), the initial motivation of this work was to generate <100 nm pegylated and biomodified nanoparticles with retained internal water access and high relaxivity. The ability of an external protein to selectively respond to specific partner proteins in solution was subsequently used to gate image contrast.
Silica nanoparticles are generally believed to be non-toxic in vitro and in vivo.27 To avoid the known toxic risk associated with free Gd3+,28 kinetically inert Gd–DOTA complexes were utilised herein.29 We have previously demonstrated that the covalent tethering of these macrocycles to MSNs can be achieved with negligible Gd loss over a 4 week period30 (also see below). The appendage of paramagnetic centres to MSNs instills not only advantages associated with increased rotational correlation time,11 but also those engendered by the high surface area and water access features of a mesoporous scaffold,23,25,26 yielding constructs with markedly high relaxivity.24,25,30 In order to maximise the sensitivity of this to processes occurring on the outer surface of the particles we have biased the location of covalently bound Gd–DOTA internally (a space likely to be associated with restricted water mobility, increased diffusional (τD) and rotational (τR) correlation times and altered water proton residence lifetime (τm)31–34) through the use of a modified co-condensation procedure.30,35 This methodology generates imaging agents with extraordinarily high relaxivities30 (Fig. S1, ESI†).
Transmission Electron Microscope (TEM; Fig. 1 and Fig. S3, ESI†), dynamic light scattering (DLS; Fig. S2a, ESI†) and BET (Brunauer–Emmett–Teller; Fig. S2c, ESI†) surface area analysis data confirmed the native Gd doped MSNs (Gd–DOTA MSNs) to be well dispersed with a particle diameter of 75.9 ± 6.4 nm, hydrodynamic size of 138.7 nm (PDI: 0.09) and pore sizes of 3.2 ± 1.3 nm. Subsequent external surface modifications were conducted via the stepwise post-grafting of PEG layers (PEG5000 and PEG2000), a process monitored through associated changes in zeta potential (Fig. S2d, ESI†). The assessments confirmed these procedures to be highly reproducible and associated with negligible perturbation of particle stability/dispersion or size (Fig. S2a and S3, ESI†). Importantly, these surface modifications could also be achieved with negligible loss of MR relaxivity (Fig. S4a, ESI†), observations consistent with the high entropy, low packing density and water penetration characteristics of such layers. The effectiveness of subsequent protein modification in mediating clean particle vectoring to target proteins was analyzed in a microarray format on modified glass chips (Fig. S5, ESI†).
 | ||
| Fig. 1 TEM image of representative protein capped Gd–DOTA MSNs (75.9 ± 6.4 nm in diameter; scale bar 50 nm). The schematics depict the native pegylated (left) and sterically capped (right) water access to internalised Gd–DOTA units. | ||
In seeking to analyse the effects of biomodification on particle MR contrast characteristics one may, in the first instance, consider the effects of the biomolecular layer on particle rotational correlation time τc (s) according to τc= (4πηr3)/3kT where r is radius, η the solvent viscosity, (8.94 × 10−4 kg m−1 s−1 for H2O at 298 K), and kT its usual meaning.23 Herein, a ∼6 nm biomolecular coat (BSA)36 is expected to generate a negligible percentage change in τc (from ∼3 × 104 ns to 9 × 104 ns).
Significantly, particle relaxivity can be tuned through the use of variable length spacer sitting between the natively hydroxylated surface and the anchored protein (Fig. 2). Specifically, the direct anchoring of BSA through a short linker (3-aminopropyl)triethoxysilane (APTES) leads to a 78.5 ± 1.5% drop in relaxivity, a reproducible observation we assign to the steric capping of particle pores and thus heavily restricted diffusional water access to internalised Gd centres. In inserting a PEG5000 spacer between the particle and protein, vectoring is enabled with retention of high relaxivity (78.0 ± 6.0% of the non protein capped particle; Fig. 2).37 Interestingly, the use of a shorter PEG2000 spacer leads to a reduced recovery of relaxivity (14.90 ± 0.56 mM−1 s−1, 67.5 ± 5.5% of native value), an observation reflective of its inability to robustly keep the protein coat (tethered or otherwise) away from the particle pores (Fig. 2).
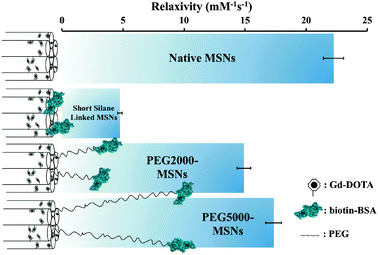 | ||
| Fig. 2 Schematic summary of particle relaxivity tuning through surface chemistry. Non protein capped Gd–DOTA MSNs have open pores, good water accessibility and relaxivities of 22.25 ± 0.83 mM−1 s−1 in water; the direct anchoring of BSA via a short silane linker leads to a dramatic reduction in relaxivity (4.74 ± 0.18 mM−1 s−1); in buffering this biomodification away from the surface with a linker, high relaxivity is recovered (14.90 ± 0.56 mM−1 s−1 for PEG2000, 17.34 ± 0.65 mM−1 s−1 for PEG5000). Error bars here represent cumulative errors arising from triple repeats of relaxivity assessment (across at least three different sample concentrations at 7 T and 20 °C) and ICP quantification. | ||
We have subsequently sought to use this steric gating in fabricating nanoparticles for which image contrast is reversibly switched by the presence or absence of a protein partner (Fig. 3). In establishing this proof of principal we have utilised the steric bulk of streptavidin (STV, 5 nm38 > particle pore size, 3.2 ± 1.3 nm). In the first instance native Gd–DOTA doped MSNs were biotin modified with retention of facile water access (see detailed experimental procedures in ESI†). In the presence of low molar equivalents of streptavidin such particles could be robustly capped without aggregation or significant increase in resolved particle hydrodynamic radius39–41 (see Fig. S1b, ESI†) but a dramatic (>60%) decrease in relaxivity (Fig. 3). The colloidal and relaxivity characteristics of these capped particles are unchanged through three months of storage in water (see Fig. S4c, ESI†), the latter observation, made after particle washing and centrifugation, being consistent with negligible leakage of Gd (the gating characteristics referred to below are also retained at 3 months).
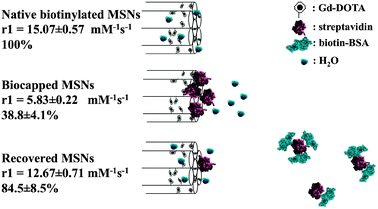 | ||
| Fig. 3 Schematic summary of MSN relaxivity gating. Externally biotinylated Gd-doped MSNs enjoy good water accessibility and a high relaxivity (15.07 ± 0.57 mM−1 s−1) that can be reversibly capped by the steric bulk of a bound STV (5.83 ± 0.22 mM−1 s−1; <40% of the original value). In the presence of low μM biotinylated BSA, the gating protein is competed off the particle surface and relaxivity recovers to 12.67 ± 0.71 mM−1 s−1 (84.5 ± 8.5% of original value). Error here represents cumulative error arising from triple repeats of relaxivity assessment (across at least three different sample concentrations at 7 T and 20 °C) and ICP quantification. | ||
In confirming the specificity of this image contrast capping, the relaxivity characteristics of biotinylated Gd-doped MSNs were unchanged in the presence of molecular biotin, biotin saturated STV or natively unmodified BSA (Fig. S4b, ESI†). Though the solution phase affinity of biotin for STV is very high,38 a substantial size dependent decrease in this at nanoparticle surfaces has been previously noted as has the competitive displacement of STV by solution phase biotin.38,41,42 In the presence of 1000 fold excess (7 μM) of biotinylated BSA, the gating STV is displaced and particle relaxivity recovers to 84.5 ± 8.5% of its original value) (Fig. 3). [Note that, because of the very low biotin coverages (<1 biotin unit per 1000 nm2 of particle surface) employed in this work, we do not expect potentially displacement hindering multivalent/local concentration effects.]42–44
In conclusion, the synthesis of colloidally stable, protein capped and vectoring Gd doped MSNs is reported herein. Through appropriate surface chemistry, these can be externally modified without detrimental loss of very high relaxivity (39.26 ± 1.29 mM−1 s−1 at 3 T, unprecedented for nanoparticles based on a silica scaffold).30,45 Significantly, the Gd loading can be internally biased at the point of synthesis such that diffusive water access, and hence MR contrast, can be sterically gated at the particle periphery. While the integration of suitably long PEG spacers enables protein conjugation to be achieved with retention of an “open gate”, conjugation with short spacers dramatically reduces relaxivity. In the presence of solution phase protein partners these sterically bulky protein gates are competed off and high image contrast returns. Though there exist examples of paramagnetic contrast agents for which contrast has been ionically or biologically tuned through a change in hydration number or rotational correlation time (see Table S1, ESI†), the work herein not only identifies and utilises the importance of water access, but also constitutes the first example of reversible T1 contrast gating exploiting protein recognition. This proof of principal, and an array of potential derivatives, is of significant potential value in furthering the application of nanoparticle chemistry to functional MR imaging.
The authors would like to thank Dr Lowri Cochlin, Department of Physiology, University of Oxford for MRI advice and the Research Complex at Harwell, Oxfordshire, for providing TEM facilities. The authors acknowledge financial support from the Wellcome Trust (WT094114MA).
Notes and references
- W. Y. Huang and J. J. Davis, Dalton Trans., 2011, 40, 6087–6103 RSC
.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC
- L. M. De Leon-Rodriguez, A. Ortiz, A. L. Weiner, S. R. Zhang, Z. Kovacs, T. Kodadek and A. D. Sherry, J. Am. Chem. Soc., 2002, 124, 3514–3515 CrossRef CAS
- R. B. Lauffer and T. J. Brady, Magn. Reson. Imaging, 1985, 3, 11–16 CrossRef CAS
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed., Engl., 1997, 36, 726–728 CrossRef CAS
- W. H. Li, S. E. Fraser and T. J. Meade, J. Am. Chem. Soc., 1999, 121, 1413–1414 CrossRef CAS
- M. P. Lowe, D. Parker, O. Reany, S. Aime, M. Botta, G. Castellano, E. Gianolio and R. Pagliarin, J. Am. Chem. Soc., 2001, 123, 7601–7609 CrossRef CAS
- A. L. Nivorozhkin, A. F. Kolodziej, P. Caravan, M. T. Greenfield, R. B. Lauffer and T. J. McMurry, Angew. Chem., Int. Ed., 2001, 40, 2903–2906 CrossRef CAS
- P. Caravan, N. J. Cloutier, M. T. Greenfield, S. A. McDermid, S. U. Dunham, J. W. M. Bulte, J. C. Amedio, R. J. Looby, R. M. Supkowski, W. D. Horrocks, T. J. McMurry and R. B. Lauffer, J. Am. Chem. Soc., 2002, 124, 3152–3162 CrossRef CAS
- M. O. Breckwoldt, J. W. Chen, L. Stangenberg, E. Aikawa, E. Rodriguez, S. M. Qiu, M. A. Moskowitz and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18584–18589 CrossRef CAS
- J. L. Major, R. M. Boiteau and T. J. Meade, Inorg. Chem., 2008, 47, 10788–10795 CrossRef CAS
- H. X. Guo, H. Q. Yang, F. Gallazzi, E. R. Prossnitz, L. A. Sklar and Y. B. Miao, Bioconjugate Chem., 2009, 20, 2162–2168 CrossRef CAS
- Z. Luo, K. Y. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. H. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS
- E. Climent, A. Bernardos, R. Martinez-Manez, A. Maquieira, M. D. Marcos, N. Pastor-Navarro, R. Puchades, F. Sancenon, J. Soto and P. Amoros, J. Am. Chem. Soc., 2009, 131, 14075–14080 CrossRef CAS
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS
- C. L. Zhu, C. H. Lu, X. Y. Song, H. H. Yang and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 1278–1281 CrossRef CAS
- C. L. Zhu, X. Y. Song, W. H. Zhou, H. H. Yang, Y. H. Wen and X. R. Wang, J. Mater. Chem., 2009, 19, 7765–7770 RSC
- S. Saha, K. C. F. Leung, T. D. Nguyen, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 685–693 CrossRef CAS
- Y. S. Lin, N. Abadeer and C. L. Haynes, Chem. Commun., 2011, 47, 532–534 RSC
- J. Fan, G. Fang, F. Zeng, X. Wang and S. Wu, Small, 2012 DOI:10.1002/smll.201201456
- I. M. Rio-Echevarria, F. Selvestrel, D. Segat, G. Guarino, R. Tavano, V. Causin, E. Reddi, E. Papini and F. Mancin, J. Mater. Chem., 2010, 20, 2780–2787 RSC
- S. Tong, S. J. Hou, Z. L. Zheng, J. Zhou and G. Bao, Nano Lett., 2010, 10, 4607–4613 CrossRef CAS
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS
- S. Takahara, N. Sumiyama, S. Kittaka, T. Yamaguchi and M. C. Bellissent-Funel, J. Phys. Chem. B, 2005, 109, 11231–11239 CrossRef CAS
- F. Carniato, L. Tei, W. Dastru, L. Marchese and M. Botta, Chem. Commun., 2009, 1246–1248 RSC
- K. M. L. Taylor, J. S. Kim, W. J. Rieter, H. An, W. L. Lin and W. B. Lin, J. Am. Chem. Soc., 2008, 130, 2154–2155 CrossRef CAS
- C. A. Barnes, A. Elsaesser, J. Arkusz, A. Smok, J. Palus, A. Lesniak, A. Salvati, J. P. Hanrahan, W. H. de Jong, E. Dziubaltowska, M. Stepnik, K. Rydzynski, G. McKerr, I. Lynch, K. A. Dawson and C. V. Howard, Nano Lett., 2008, 8, 3069–3074 CrossRef CAS
- A. N. Oksendal and P. A. Hals, J. Mag. Res. Imag., 1993, 3, 157–165 CrossRef CAS
- X. Y. Wang, T. Z. Jin, V. Comblin, A. Lopezmut, E. Merciny and J. F. Desreux, Inorg. Chem., 1992, 31, 1095–1099 CrossRef CAS
- J. J. Davis, W.-Y. Huang and G.-L. Davies, J. Mater. Chem., 2012, 22, 22848–22850 RSC
- J. S. Ananta, B. Godin, R. Sethi, L. Moriggi, X. Liu, R. E. Serda, R. Krishnamurthy, R. Muthupillai, R. D. Bolskar, L. Helm, M. Ferrari, L. J. Wilson and P. Decuzzi, Nat. Nanotechnol., 2010, 5, 815–821 CrossRef CAS
- P. H. Fries and E. Belorizky, J. Chem. Phys., 2010, 133, 024504 CrossRef
- S. Aime, L. Frullano and S. G. Crich, Angew. Chem., Int. Ed., 2002, 41, 1017–1019 CrossRef CAS
- R. Sethi, J. S. Ananta, C. Karmonik, M. Zhong, S. H. Fung, X. Liu, K. Li, M. Ferrari, L. J. Wilson and P. Decuzzi, Contrast Media Mol. Imaging, 2012, 7, 501–508 CrossRef CAS
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zurner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS
- J. W. Anderegg, W. W. Beeman, S. Shulman and P. Kaesberg, J. Am. Chem. Soc., 1955, 77, 2927–2937 CrossRef CAS
- The ∼20% relaxivity decrease is assigned to the physisorption of non covalently coupled BSA to non sterically protected areas of the particle surface during the biomodification steps.
- E. V. Piletska and S. A. Piletsky, Langmuir, 2010, 26, 3783–3785 CrossRef CAS
- W. Muller, H. Ringsdorf, E. Rump, X. Zhang, L. Angermaier, W. Knoll and J. Spinke, J. Biomater. Sci., Polym. Ed., 1994, 6, 481–495 CrossRef CAS
- M. Li, K. K. W. Wong and S. Mann, Chem. Mater., 1999, 11, 23–26 CrossRef CAS
- K. Aslan, C. C. Luhrs and V. H. Perez-Luna, J. Phys. Chem. B, 2004, 108, 15631–15639 CrossRef CAS
- X. X. Duan, Y. Li, N. K. Rajan, D. A. Routenberg, Y. Modis and M. A. Reed, Nat. Nanotechnol., 2012, 7, 401–407 CrossRef CAS
- J. H. Myung, K. A. Gajjar, J. Saric, D. T. Eddington and S. Hong, Angew. Chem., Int. Ed., 2011, 50, 11769–11772 CrossRef CAS
- S. Hong, P. R. Leroueil, I. J. Majoros, B. G. Orr, J. R. Baker and M. M. B. Holl, Chem. Biol., 2007, 14, 107–115 CrossRef CAS
- K. M. L. Taylor-Pashow, J. D. Rocca and W. Lin, Nanomaterials, 2011, 2, 1–14 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and Fig. S1–S5 and Table S1. See DOI: 10.1039/c2cc37545a |
| This journal is © The Royal Society of Chemistry 2013 |