Chemically orthogonal trifunctional Janus beads by photochemical “sandwich” microcontact printing†
Tobias
Kaufmann
a,
Christian
Wendeln
a,
M. Talha
Gokmen‡
b,
Stefan
Rinnen
c,
Maria M.
Becker
a,
Heinrich F.
Arlinghaus
c,
Filip
Du Prez
*b and
Bart Jan
Ravoo
*a
aOrganisch-Chemisches Institut and CeNTech, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: b.j.ravoo@uni-muenster.de; Fax: +49 251 8336557; Tel: +49 251 8333287
bDepartment of Organic Chemistry, Ghent University, Krijgslaan 281 S4, 9000 Gent, Belgium. E-mail: Filip.DuPrez@UGent.be; Fax: +32 9 2644972; Tel: +32 9 2644503
cPhysikalisches Institut, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Straße 10, 48149 Münster, Germany. E-mail: arlinghaus@wwu.de; Fax: +49 251 8333682; Tel: +49 251 8339064
First published on 6th November 2012
Abstract
The combination of topographic and chemical orthogonality on polymer particles by site selective immobilization of functional thiols via thiol–ene chemistry provides a trifunctional particle surface with azide and acid functionalities on opposing poles and alkenes in the equatorial area. These Janus beads are accessible for site selective orthogonal chemical reactions as well as biomolecular recognition on the same particle.
Janus beads are particles that display different properties on opposing sides.1,2 These particles are often spherical beads, however the concept is not restricted to centrosymmetric geometries. Possible applications of such anisotropic particles include the catalysis of two distinct chemical reactions in both phases of an oil–water emulsion,3 display applications4 or detection of multiple proteins on barcoded “Janus chips”.5
The preparation of bifunctional particles can be roughly categorized into two main approaches: on the one hand, the particle synthesis can be exploited to realize anisotropic products. Such methods often employ either co-jetting6 or phase separation of two building blocks.7,8 On the other hand, numerous strategies have been developed to site selectively post-functionalize originally isotropic particles. These strategies include vapor deposition of metals9,10 or polymers,11 site specific chemical derivatization of beads trapped in a protecting matrix12–14 and printing.15–17 Especially the technique of microcontact chemistry has the beneficial features of linking “ink molecules” covalently and site specifically onto the surface of the particle by using minimal amounts of the functional material. Furthermore this technique is easy and quick in execution, yet versatile with respect to tolerable functional groups.18,19 We recently introduced the method of “sandwich” microcontact printing as a tool for Janus bead preparation20 and demonstrated its broad applicability.21
In this report we show another versatile application of this technique by adopting the principles of orthogonal chemistry. The concept of orthogonal chemical modifications is based on the introduction of two or more different functional groups in a system, allowing further post-functionalization by specific ligation reactions. The reactions have to be selected in such a way that all functional groups involved tolerate each other and that exclusive addressing of every functional group is assured. During the last few years, orthogonal chemistry has for example been applied to modify polymers,22–24 dendrons and dendrimers25,26 and also surfaces.27–29 In a recent paper we described the preparation of multifunctional orthogonal surfaces by the sequential immobilization of heterobifunctional crosslinkers using microcontact printing.30 Trifunctional surfaces with alkene, acid and azide groups were prepared by printing an azide-thiol linker and an acid thiol linker on alkene-modified glass and silicon substrates using thiol–ene chemistry. In this communication, we use these hetero-bifunctional linkers for the preparation of unprecedented chemically orthogonal trifunctional polymer Janus beads. Opposing sides of these beads with acid and azide groups are suitable for specific ligation with amines by peptide chemistry and with alkynes by copper catalyzed alkyne–azide cycloaddition (CuAAC), respectively. Remaining alkene groups in unmodified areas of the Janus beads can be reacted with thiols using thiol–ene chemistry.
Trifunctional bead surfaces were prepared by printing functional thiol inks onto isotropic alkene terminated polymer particles as depicted in Scheme 1. As substrates, monodisperse epoxy polymer beads of 5 μm or 170 μm diameter were used. Their preparation by, respectively, precipitation polymerization and microfluidics can be found elsewhere.21 The epoxy beads were perfunctionalized with alkene groups by reaction with allyl mercaptane (see ESI†). The steps for successful surface modification with functional thiols include the following: a first flat stamp made of poly(dimethyl siloxane) (PDMS) is covered with azido-thiol ink 1 solution (80 mM of 1 in ethanol including 40 mM dimethoxyphenyl acetophenone (DMPA), see ESI†), subsequently dried in a stream of argon and fixed into a UV-transparent object carrier connected to a mechanical press. A second flat stamp is loaded with the acid-thiol 2 (see ESI†) as outlined above and a monolayer of alkene functionalized polymer beads is applied. The first stamp is brought into conformal contact with the polymer beads from the top and the stamp–bead–stamp sandwich is irradiated with light of 365 nm using a UV-LED for 5 min. The trifunctional polymer beads were cleaned by washing with solvents of different polarity (water, dimethyl formamide, dichloromethane, diethyl ether) and subsequent extraction in hot ethanol for 9 h.
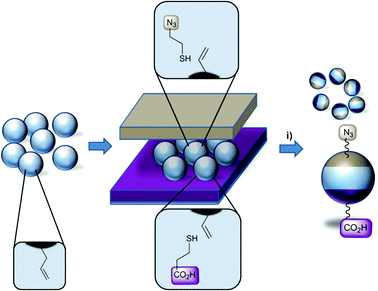 | ||
| Scheme 1 Schematic illustration of the preparation of chemically orthogonal Janus beads by “sandwich” microcontact printing of functional thiols on alkene bead surfaces; (i) 80 mM thiol 1 and 2, 40 mM DMPA in ethanol, UV-LED (365 nm), 5 min. | ||
As a proof of concept experiment, acid and azide terminated poles of the beads were specifically brought to reaction with dansyl cadaverine via a peptide coupling reaction or rhodamine alkyne using CuAAC chemistry, respectively. After detection of each immobilized fluorophore individually (see Fig. S1, ESI†), both poles were derivatized in sequential reactions (peptide coupling followed by CuAAC) (Fig. 1). As can be observed from these images, both fluorophores were exclusively immobilized on opposing poles of the beads due to selective reaction of dansyl cadaverine with surface bound carboxylic acid moieties and rhodamine alkyne with surface azide groups. Large polymer beads of 170 μm diameter (a, c) are selectively modified as well as small polymer beads of 5 μm diameter (b, d). The constructed 3D-volume plot of stacked confocal laser scanning microscopy (CLSM) images (d) highlights the exclusiveness of modification on opposing poles and a 360° rotation (see video, ESI†) reveals the size of the printed patches on the small beads. Furthermore, it should be noted that the “yield” of the two sequential reactions of surface bound alkenes with azide thiol during μCP and subsequent CuAAC with rhodamine alkyne from solution is obviously very high, as the rhodamine modified patches can even be detected by light microscopy (c).
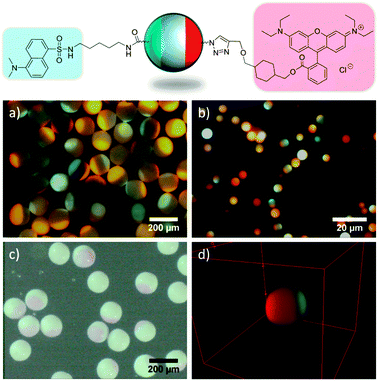 | ||
| Fig. 1 Fluorescent Janus particles obtained by coupling dansyl cadaverine and rhodamine alkyne onto chemically orthogonal beads. Fluorescence microscopy images (a, b), light microscopy image (c) and CLSM 3D volume plot (d). The diameter of the particle in (d) is 5.6 μm. | ||
In order to apply an additional analytical method to verify the site selective orthogonal chemical attachment of ink molecules to the polymer beads, ethynyl trifluorotoluene (see ESI†) was coupled to the azide-decorated side of the beads via CuAAC and the Janus beads were imaged by Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS). We emphasize that ToF-SIMS imaging of the beads is highly challenging due to the curvature and poor conductance of the polymer microparticles. ToF-SIMS indicated the formation of F− and of CN− fragments selectively on one side of the beads (see Fig. S2, ESI†). The fluoride signal indicates the successful regiospecific immobilization of the fluorine containing alkyne, whereas the CN− fragment can be attributed to the immobilization of the nitrogen containing azide-thiol linker in the same area. These measurements confirm the results obtained by fluorescence microscopy.
In a more sophisticated experiment, protein-modified Janus beads were prepared. Three different biomolecules were used as protein binding sites on polymer beads: a biotin alkyne derivative, an amino-α-mannoside and an amino-β-lactoside (see ESI† for chemical structures). These ligands are known to bind specifically to different proteins, namely streptavidin, concanavalin A (ConA) and peanut agglutinin (PNA) respectively. Binding one of these ligands to one pole of the chemically orthogonal Janus beads resulted in highly preferential adsorption of the respective proteins on one side of the beads (see Fig. S3, ESI†). In a following step, two of the reactive bioligands were combined on one bead by sequential reaction of biotin alkyne (by CuAAC) followed by either the amino-α-mannoside or the amino-β-lactoside (by peptide coupling). Subsequent incubation of the biofunctionalized Janus beads in a mixed solution of fluorescently labeled proteins (DyLight 405-labeled streptavidin and rhodamine-labeled PNA or DyLight 405-labeled streptavidin and fluorescein-labeled ConA) resulted in simultaneous yet site specific protein immobilization on the beads (Fig. 2). Fig. 2 clearly demonstrates the site specific immobilization of the ligands as well as their selectivity towards proteins. We note that amino-functionalized carbohydrates were immobilized on acid patches by two different strategies. Whereas the amino-α-mannoside was bound to the acid patch of the beads via peptide coupling reaction using O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), the amino-β-lactoside was coupled to the beads in a two step procedure: acid functionalized beads were first activated by reaction with dicyclohexyl carbodiimide and N-hydroxysuccinimide and subsequently exposed to a solution of the β-lactoside in dimethyl formamide. As can be seen from the protein binding experiments, both chemical reactions worked equally well.
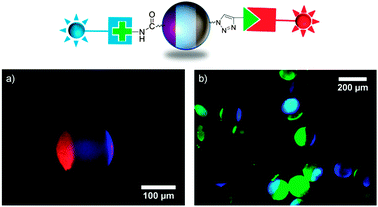 | ||
| Fig. 2 Schematic illustration of protein Janus beads. Overlay of fluorescence images of rhodamine-labeled PNA bound to amino-β-lactoside and DyLight 405-labeled streptavidin bound to biotin alkyne (a) and fluorescein-labeled ConA bound to amino-α-mannoside and DyLight 405-labeled streptavidin bound to biotin alkyne (b). | ||
A third functionality is present in the form of non-contacted alkenes on the bead surface as well as in the bead interior. These were addressed by thiol–ene reaction of a mannoside thiol on large beads (170 nm diameter) that were previously coupled with dansyl cadaverine by peptide coupling and rhodamine alkyne by CuAAC. The surface bound mannoside was incubated with fluorescein-labeled ConA to visualize the carbohydrate functionalization. However, fluorescence microscopy (see Fig. S4, ESI†) does not show selective protein attachment in the form of a “belt” structure in non-contacted regions, but rather a homogeneous distribution of ConA in the beads. This is ascribed to the porous nature of the employed polymer particles. Although no site selective attachment can be observed, this result may be valuable when considering applications in heterogeneous catalysis of chemical reactions, where a high surface area equipped with a catalyst would be desirable. Successful protein attachment clearly demonstrates the accessibility of remaining alkenes on the surface and in the interior of the beads.
In summary, we could demonstrate that three different chemical functionalities can be introduced selectively in different surface regions of small and large polymer microbeads. The combination of topographic and chemical orthogonality on polymer particles is unprecedented and provides access to a wide range of Janus particles.
B.J.R. is grateful to the Deutsche Forschungsgemeinschaft for financial support. F.D.P. acknowledges the Belgian Program on Interuniversity Attraction Poles initiated by the Belgian State, Prime Minister's Office (Program P7/05) for financial support. Prof. H. J. Galla and P. Seelheim (Westfälische Wilhelms-Universität Münster) are acknowledged for access to and help with CLSM.
Notes and references
- J. Du and R. K. O'Reilly, Chem. Soc. Rev., 2011, 40, 2402–2416 RSC.
- J. Yoon, K. J. Lee and J. Lahann, J. Mater. Chem., 2011, 21, 8502–8510 RSC.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef CAS.
- T. Nisisako, T. Torii, T. Takahashi and Y. Takizawa, Adv. Mater., 2006, 18, 1152–1156 CrossRef CAS.
- D. Dendukuri, D. C. Pregibon, J. Collins, T. A. Hatton and P. S. Doyle, Nat. Mater., 2006, 5, 365–369 CrossRef CAS.
- K.-H. Roh, D. C. Martin and J. Lahann, Nat. Mater., 2005, 4, 759–763 CrossRef CAS.
- R. Erhardt, A. Böker, H. Zettl, H. Kaya, W. Pyckhout-Hintzen, G. Krausch, V. Abetz and A. H. E. Müller, Macromolecules, 2001, 34, 1069–1075 CrossRef CAS.
- I. K. Voets, A. de Keizer, P. de Waard, P. M. Frederik, P. H. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. M. Leermakers and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2006, 45, 6673–6676 CrossRef CAS.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381–384 CrossRef CAS.
- Q. Chen, J. K. Whitmer, S. Jiang, S. C. Bae, E. Luijten and S. Granick, Science, 2011, 331, 199–202 CrossRef CAS.
- R. T. Chen, B. W. Muir, G. K. Such, A. Postma, K. M. McLean and F. Caruso, Chem. Commun., 2010, 46, 5121–5123 RSC.
- C. Casagrande and M. Veyssié, C. R. Acad. Sci., Ser. II, 1988, 306, 1423–1425 Search PubMed.
- X. Y. Ling, I. Y. Phang, C. Acikgoz, M. D. Yilmaz, M. A. Hempenius, G. J. Vancso and J. Huskens, Angew. Chem., Int. Ed., 2009, 48, 7677–7682 CrossRef CAS.
- K. Kamalasanan, S. Jhunjhunwala, J. Wu, A. Swanson, D. Gao and S. R. Little, Angew. Chem., Int. Ed., 2011, 50, 8706–8708 CrossRef CAS.
- O. Cayre, V. N. Paunov and O. D. Velev, Chem. Commun., 2003, 2296–2297 RSC.
- Z. Li, D. Lee, M. F. Rubner and R. E. Cohen, Macromolecules, 2005, 38, 7876–7879 CrossRef CAS.
- S. Jiang and S. Granick, Langmuir, 2009, 25, 8915–8918 CrossRef CAS.
- B. J. Ravoo, J. Mater. Chem., 2009, 19, 8902–8906 RSC.
- C. Wendeln and B. J. Ravoo, Langmuir, 2012, 28, 5527–5538 CrossRef CAS.
- T. Kaufmann, M. T. Gokmen, C. Wendeln, M. Schneiders, S. Rinnen, H. F. Arlinghaus, S. A. F. Bon, F. E. Du Prez and B. J. Ravoo, Adv. Mater., 2011, 23, 79–83 CrossRef CAS.
- T. Kaufmann, M. T. Gokmen, S. Rinnen, H. F. Arlinghaus, F. E. Du Prez and B. J. Ravoo, J. Mater. Chem., 2012, 20, 6190–6199 RSC.
- M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS.
- S. K. Yang and M. Weck, Macromolecules, 2007, 41, 346–351 CrossRef.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- M. B. Steffensen and E. E. Simanek, Angew. Chem., Int. Ed., 2004, 43, 5178–5180 CrossRef CAS.
- L. Röglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102–112 CrossRef.
- S. G. Im, K. W. Bong, B.-S. Kim, S. H. Baxamusa, P. T. Hammond, P. S. Doyle and K. K. Gleason, J. Am. Chem. Soc., 2008, 130, 14424–14425 CrossRef CAS.
- N. Gupta, B. F. Lin, L. M. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 CrossRef CAS.
- A. Pulsipher, N. P. Westcott, W. Luo and M. N. Yousaf, J. Am. Chem. Soc., 2009, 131, 7626–7632 CrossRef CAS.
- C. Wendeln, S. Rinnen, C. Schulz, T. Kaufmann, H. F. Arlinghaus and B. J. Ravoo, Chem.–Eur. J., 2012, 18, 5880–5888 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, general procedures and surface analytical data. See DOI: 10.1039/c2cc36483b |
| ‡ Currently staff scientist at Life Technologies, Svelleveien 29, 2004 Lillestrom, Norway. |
| This journal is © The Royal Society of Chemistry 2013 |