 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral [2]rotaxane host systems that recognise halide anions in aqueous solvent mixtures†
James M. Mercurioa, Fergus Tyrrella, James Cooksonb and Paul D. Beer*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
bJohnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading, RG4 9NH, UK
First published on 3rd October 2013
Abstract
Four pyridine N-oxide axle containing [2]rotaxanes have been synthesised via an anion templated threading-followed-by-stoppering strategy and shown to be the first examples of neutral interlocked host systems capable of recognising halide anions in aqueous solvent mixtures.
Inspired by the fundamental roles that negatively charged species play in a vast array of chemical, biological, medical and environmental processes, the field of anion supramolecular chemistry has expanded enormously in recent years.1 Incorporating numerous non-covalent interactions, such as electrostatics, hydrogen bonding, Lewis acid–base,2 anion–π interactions,3 and more recently halogen bonding4 into acyclic and macrocyclic host frameworks has allowed for a panoply of anion receptors to be developed. However, the challenge of raising the degree of recognition to that of biotic systems remains a significant one. In an effort to meet this challenge we have embarked on the anion templated construction of positively charged interlocked host molecules and demonstrated their ability to bind anions in aqueous solvent media.5,6 In this communication, we report the first examples of neutral interlocked [2]rotaxane host systems that are capable of recognising halide anions in aqueous solvent mixtures.
We have used the pyridine N-oxide motif as an axle component in the synthesis of [2]rotaxane structures through alkali metal and lanthanide metal cation-templation,7,8 whereas hydrogen bonding interactions have been exploited recently in [2]pseudorotaxane assemblies with pyridine N-oxide threading derivatives.9 The macrocyclic component of the target rotaxane host system was designed to contain two isophthalamide motifs, which serve to facilitate interpenetration with a 3,5-bis-amide pyridine N-oxide thread, where the stability of the resulting pseudorotaxane assembly would be augmented via anion binding, in particular with chloride (Fig. 1).
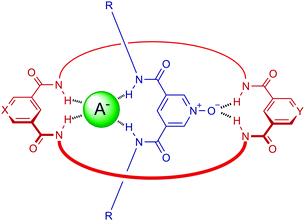 | ||
| Fig. 1 Anion templated pseudorotaxane assembly between a bis-isophthalamide macrocycle and 3,5-bis-amide pyridine N-oxide thread. | ||
The preparation of four novel bis-isophthalamide macrocycles 7–10 was achieved via a common multi-step pathway as described in the ESI† (see S2). Preliminary 1H NMR experiments were performed to investigate pseudorotaxane formation between macrocycles 7–9 and a 3,5-bis(hexylamide) pyridine N-oxide derivative 117 alone, and in the presence of one equivalent of TBACl, in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CDCl3–CD3CN (see ESI,† S4.1). Importantly, both in the absence and presence of chloride, the macrocycle hydroquinone protons δ are shifted upfield significantly when pyridine N-oxide thread 11 is added to macrocycle 8. This is the result of aromatic donor–acceptor interactions between the macrocycle and threading species, indicative of pseudorotaxane formation. It is noteworthy that the magnitude of this perturbation is relatively larger when chloride is present, giving evidence for a templating effect. In addition, the internal isophthalamide protons α and χ can be seen to shift downfield. This arises from hydrogen bonding interactions between these protons and the pyridine N-oxide oxygen donor atom and hydrogen bonding interactions with the same oxygen donor atom and chloride when the halide anion is present. Analogous 1H NMR pseudorotaxane titration experiments with macrocycles 7 and 9 revealed similar hydroquinone and isophthalamide perturbations, suggesting interpenetrative formation with 11.
:1 CDCl3–CD3CN (see ESI,† S4.1). Importantly, both in the absence and presence of chloride, the macrocycle hydroquinone protons δ are shifted upfield significantly when pyridine N-oxide thread 11 is added to macrocycle 8. This is the result of aromatic donor–acceptor interactions between the macrocycle and threading species, indicative of pseudorotaxane formation. It is noteworthy that the magnitude of this perturbation is relatively larger when chloride is present, giving evidence for a templating effect. In addition, the internal isophthalamide protons α and χ can be seen to shift downfield. This arises from hydrogen bonding interactions between these protons and the pyridine N-oxide oxygen donor atom and hydrogen bonding interactions with the same oxygen donor atom and chloride when the halide anion is present. Analogous 1H NMR pseudorotaxane titration experiments with macrocycles 7 and 9 revealed similar hydroquinone and isophthalamide perturbations, suggesting interpenetrative formation with 11.
Quantitative 1H NMR titration experiments monitoring the hydroquinone protons δ of the respective macrocycle gave titration data (see ESI,† S4.2) from which 1:1 stoichiometric association constants10 were determined by WinEQNMR211 (see ESI,† S4.3). All the macrocycles were found to form stable interpenetrative assemblies with 11 in 1:1 CDCl3–CD3CN, through the combination of favourable pyridine N-oxide – isophthalamide macrocycle hydrogen bonding and aromatic donor–acceptor interactions. Importantly, the apparent association constants obtained for pseudorotaxane formation in the presence of chloride were found to be >104 M−1 in the same solvent mixture demonstrating a significant enhancement of the stability of interpenetrative assembly when the macrocycle binds the halide anion.
The synthesis of the target interlocked [2]rotaxane systems was achieved by a copper(I) catalysed cycloaddition azide–alkyne (CuAAC) click stoppering reaction, as shown in Scheme 1. Equimolar amounts of bis-azide axle precursor 12,8 the respective macrocycle and TBACl, together with two equivalents of alkyne-functionalised terphenyl stopper 1312 and three equivalents of DIPEA were stirred in chloroform solution in the presence of a catalytic amount of Cu(MeCN)4PF6 and TBTA at room temperature for 24 hours. After purification by preparative thin layer chromatography, [2]rotaxanes 14–17 were isolated in 9%, 17%, 16% and 16% yields respectively and characterised by 1H NMR spectroscopy and high resolution mass spectrometry, with their interlocked nature confirmed by two-dimensional 1H–1H ROESY spectroscopy (see ESI,† S5). It is important to note that when the syntheses were repeated in the absence of chloride, no rotaxane product was isolated which serves to highlight the crucial templating role the halide anion is playing in the mechanical bond forming process.
![Neutral [2]rotaxane syntheses via an anion templated threading-followed-by-stoppering strategy.](/image/article/2013/CC/c3cc47076h/c3cc47076h-s1.gif) | ||
| Scheme 1 Neutral [2]rotaxane syntheses via an anion templated threading-followed-by-stoppering strategy. | ||
The rotaxanes possess two isophthalamide binding sites and so can in principle adopt two different conformations via the macrocycle component undergoing a pirouetting motion around the pyridine N-oxide axle (Scheme 2). Evidence for this dynamic process was obtained from a low temperature VT 1H NMR investigation of rotaxane 15 in 1:1 CDCl3–CD3OD. Significant broadening of both the macrocycle and axle isophthalamide protons and, importantly, the macrocycle hydroquinone protons is observed as the temperature of the sample is cooled from 298 K to 198 K (see ESI,† S6.1) indicating the interlocked structure is dynamic on the NMR timescale and at room temperature is switching between the two possible conformations.
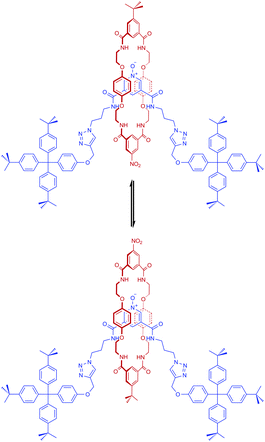 | ||
| Scheme 2 Two possible conformations of rotaxane 15 resulting from molecular pirouetting of the macrocycle around the N-oxide axle. | ||
1H NMR anion titration experiments were undertaken to assess the anion binding affinities of the four [2]rotaxanes with nitro functionalised [2]rotaxane 15 studied initially. Upon addition of increasing amounts of TBACl to a solution of 15 in 1:1 CDCl3–CD3OD protons α, χ, b and f were observed to shift downfield significantly (Fig. 2).
![1H NMR spectra of (A) rotaxane 15 plus 3 equivalents of TBACl; (B) rotaxane 15 plus 1 equivalent of TBACl; and (C) rotaxane 15 (500 MHz, 1 : 1 CDCl3–CD3OD, 298 K, [host] = 2 mM).](/image/article/2013/CC/c3cc47076h/c3cc47076h-f2.gif) | ||
| Fig. 2 1H NMR spectra of (A) rotaxane 15 plus 3 equivalents of TBACl; (B) rotaxane 15 plus 1 equivalent of TBACl; and (C) rotaxane 15 (500 MHz, 1:1 CDCl3–CD3OD, 298 K, [host] = 2 mM). | ||
These downfield shifts are indicative of polarising hydrogen bonding interactions between these protons and the chloride anion. The shifting of the axle internal isophthalamide proton b and triazole proton f suggests the axle wraps around and encapsulates the anion, whereas perturbation of macrocycle isophthalamide protons α and χ indicates that anion recognition is occurring at both isophthalamide binding sites of 15 in a dynamic manner. This was confirmed by low temperature VT 1H NMR of a 1:1 mixture of [2]rotaxane 15 and TBACl (see ESI,† S6.2) where similar broadening in the aromatic region of the 1H NMR spectra was observed to that in the free rotaxane, indicating that the conformation of 15 is not locked upon chloride binding and both isophthalamide binding sites are accessible to the halide anion.
Similar downfield shifts of protons α, χ, b and f were observed for the other rotaxane systems with chloride and various other anions. The titration data monitoring the internal isophthalamide proton α (see ESI,† S7) was analysed using the WinEQNMR211 curve fitting software to give the 1:1 stoichiometric association constants reported in Table 1.13
| 14 | 15 | 16 | 17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1:1 CDCl3–CD3OD, 298 K, [host] = 2 mM, errors in parentheses. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cl− | 2720(280) | 2790(240) | >104 | 3010(340) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Br− | 1330(90) | 2050(280) | >104 | 2090(30) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I− | 295(22) | 667(28) | 1700(190) | 635(84) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H2PO4− | 129(33) | 152(7) | 112(7) | 671(54) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
All the rotaxanes exhibit strong binding of halides, especially with chloride and bromide, which are preferentially bound over iodide and dihydrogen phosphate.14 This suggests the interlocked binding cleft within these host systems is of complementary size for the smaller chloride and bromide anions, whereas larger iodide and dihydrogen phosphate anions are presumably too large to penetrate the interlocked binding pocket. It is noteworthy that the strength of halide binding for these neutral interlocked host systems is comparable to that of previously reported charged [2]rotaxanes containing a pyridinium axle component and isophthalamide macrocycle.15 Taking into account the substantial chloride and bromide association constant values observed, in particular with rotaxane 16, 1H NMR halide anion titrations were repeated in the more competitive 45:45:10 CDCl3–CD3OD–D2O aqueous solvent mixture (see ESI,† S8) and the determined association constants are shown in Table 2. Even in an aqueous solvent mixture, the binding of chloride and bromide is still remarkably strong for the neutral [2]rotaxanes, with the strength of halide binding again being comparable to that of previously reported charged pyridinium axle containing rotaxane systems.5 Impressively, chloride and bromide anions are still bound the strongest, in spite of the Hofmeister series bias16 favouring iodide on the basis of relative ease of desolvation, which provides further compelling evidence of the rotaxane host binding domains being of a complementary size-match for the smaller halides.
| 14 | 15 | 16 | 17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 45:45:10 CDCl3–CD3OD–D2O, 298 K, [host] = 2 mM, errors in parentheses. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cl− | 501(27) | 475(15) | 552(8) | 487(39) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Br− | 311(14) | 442(6) | 466(9) | 324(17) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I− | 59(3) | 92(4) | 139(10) | 175(32) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In summary, four neutral pyridine N-oxide functionalised [2]rotaxanes have been prepared via a chloride anion templated threading-followed-by-stoppering methodology. 1H NMR anion titration experiments revealed these [2]rotaxanes are the first examples of neutral interlocked host systems to be capable of recognising halide anions in aqueous solvent mixtures, with a selectivity preference for chloride and bromide anions over iodide and dihydrogen phosphate resulting from their complementary sized interlocked binding domains for the smaller halide anions.
J.M.M. thanks the EPSRC for a DTA studentship and Johnson Matthey for a CASE Award.
Notes and references
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC; Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC; J. Perez and L. Riera, Chem. Soc. Rev., 2008, 37, 2658–2667 RSC; P. A. Gale, S. E. Garcia-Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC; G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571–586 CrossRef CAS PubMed.
- T. W. Hudnall and F. P. Gabbai, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef CAS PubMed.
- B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417–2428 RSC; O. B. Berryman, F. Hof, M. J. Hynes and D. W. Johnson, Chem. Commun., 2006, 506–508 RSC.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed; P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef PubMed; T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- L. M. Hancock, L. C. Gilday, S. Carvalho, P. J. Costa, V. Félix, C. J. Serpell, N. L. Kilah and P. D. Beer, Chem.–Eur. J., 2010, 16, 13082–13094 CrossRef CAS PubMed.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed; N. H. Evans, E. S. H. Allinson, M. D. Lankshear, K.-Y. Ng, A. R. Cowley, C. J. Serpell, S. M. Santos, P. J. Costa, V. Felix and P. D. Beer, RSC Adv., 2011, 1, 995–1003 RSC; Y. Li, K. M. Mullen, J. Sardinha, V. Felix and P. D. Beer, Dalton Trans., 2011, 40, 12180–12190 RSC.
- L. M. Hancock and P. D. Beer, Chem. Commun., 2011, 47, 6012–6014 RSC.
- F. Zapata, O. A. Blackburn, M. J. Langton, S. Faulkner and P. D. Beer, Chem. Commun., 2013, 49, 8157–8159 RSC.
- M. Chen, S. Han, L. Jiang, S. Zhou, F. Jiang, Z. Xu, J. Liang and S. Zhang, Chem. Commun., 2010, 46, 3932–3934 RSC ; while this work was in progress, azide, cyanate and thiocyanate anions were reported to assist the assembly of [2]pseudorotaxanes, see: V. Valderrey, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2012, 134, 10733–10736 CrossRef CAS PubMed.
- The association constants obtained from titrations with 1:1 TBACl/thread 11 are apparent association constants, given that a three component system is being studied, for detailed explanation see: M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292–2302 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- V. Aucagne, K. D. Hanni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed.
- It is noteworthy that the same association constant values, within experimental error, are obtained by monitoring macrocycle internal isophthalamide proton χ and axle internal isophthalamide proton b.
- 1H NMR anion titrations were also carried out with TBAOAc but WinEQNMR2 could not fit the data.
- M. R. Sambrook, P. D. Beer, M. D. Lankshear, R. F. Ludlow and J. A. Wisner, Org. Biomol. Chem., 2006, 4, 1529–1538 Search PubMed; J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469–12476 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2011, 133, 7344–7347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthetic procedures, additional characterisation and titration data. See DOI: 10.1039/c3cc47076h |
| This journal is © The Royal Society of Chemistry 2013 |