Porous aromatic frameworks: Synthesis, structure and functions
Teng
Ben
b and
Shilun
Qiu
*a
aState Key Laboratory of Inorganic Synthesis & Preparative Chemistry, Jilin University, Changchun, China. E-mail: sqiu@jlu.edu.cn; Fax: (+86)431-8516-8589; Tel: (+86)431-8516-8589
bDepartment of Chemistry, Jilin University, Changchun, China
First published on 31st May 2012
Abstract
The creation of ultrahigh surface area materials are of great interest in academia and industry. In recent years, porous aromatic frameworks (PAF) were discovered and their porosity and properties were also explored. They are characterized by a rigid aromatic open-framework structure constructed by covalent bonds. The building block design, network formation method and relationship between functions and secondary building units were compiled in this highlight. In addition, advantages and challenges of predicted PAF derivatives were also discussed.
 Teng Ben | Teng Ben received his PhD in 2002 from Jilin University, China, on polymer science. After graduation, he joined the faculty at Jilin University, working with Prof. Zhongwen Wu as Lecturer. In 2005, he moved to a research group, led by Prof. Eiji Yashima, at Nagoya University in Japan as a postdoctoral researcher. In 2008, he moved back to Jilin University as an Associate Professor and was promoted to Professor in 2010. His research interests include the fundamental understanding of host–guest interactions in nanoporous materials, gas storage and separation using porous organic frameworks. |
 Shilun Qiu | Shilun Qiu received his Ph.D. in chemistry from Jilin University, China, in 1988. He joined the University De Haute-Alsace, France, and Hokkaido University Sapporo, Japan, for postdoctoral research. In 1994, he was promoted to be a full professor in the Department of Chemistry, Jilin University. He won the Second Grade Award for the State Natural Science Award of China in 2008. His recent research interests focus on the studies of molecular engineering, synthesis and catalysis of micro- and mesoporous materials, rational synthesis and hydrogen storage of porous organic framework materials and host–guest chemistry, in particular, nano-composites in porous materials. |
Introduction
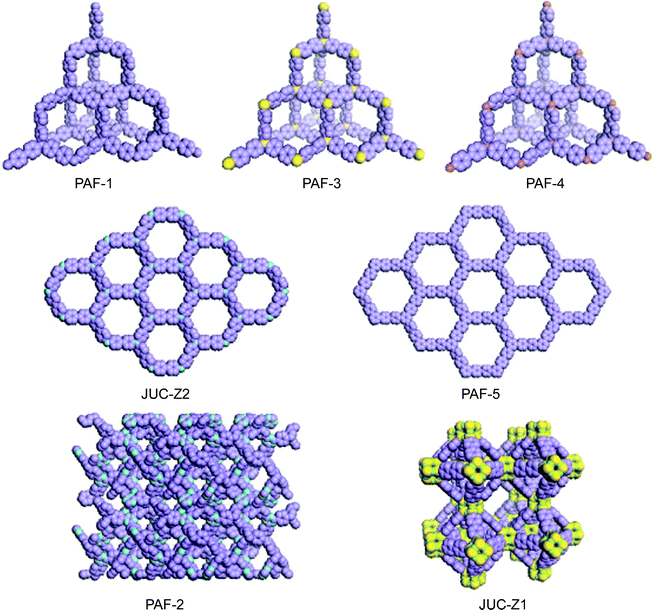
The pore is the central structural motif in host–guest functional materials, such as zeolites, and is also the ubiquitous object in nature from microscopic to macroscopic points of view. Most importantly, the porous structure provides an inherently accessible space, for guests, where chemical reactions or physical sorption can take place; therefore, they are always addressed as open frameworks. Accordingly, if one of the open frameworks could be selectively synthesized, assembled, or constructed for molecules, supramolecules, or polymers, they should form continuously communicating pore passages. Inspired by the success of sophisticated natural and artificial zeolites that are of key importance for their elaborate functions in modern chemical industry involving molecular storage, recognition, separation and catalytic activity,1,2 chemists have been challenged to develop porous organic–inorganic hybrids and organic frameworks, supramolecules, and oligomers with a controlled functional pore, not only to mimic zeolite structures and functions but also for their potential applications in materials science, such as molecular separation,3 clean energy storage,4,5 photoelectric materials,6,7 molecular motors,8 and catalysis.9 Based on such ideas, in the last decades, porous organic frameworks (POFs) such as metal–organic frameworks (MOFs),10–12 covalent organic frameworks (COFs),13–15 polymers of intrinsic microporosity (PIMs),16,17 conjugated microporous polymers (CMPs),18–23 hyper-crosslinked polymers (HCPs)24,25 and porous aromatic frameworks (PAFs)26–33 have been prepared and the functions of nano pores are also explored. The first example of introducing organic frameworks into pore materials was reported by Kitagawa et al. in 1997.34 With that, Yaghi et al. and Williams et al. established the gas sorption method to detect the pore structure of POFs by Langmuir and Brunauer–Emmett–Teller (BET) theory, respectively, in 1998 and 1999, which was proven to be a standard of characterization for the porosity of microporous organic frameworks.35,36 Since then, gas sorption experiments were also used to explore the surface area and pore size of organic porous polymers. In 2002, Budd and McKeown described the first PIM material with a surface area of about 950 m2 g−1.37 3 years later, in 2005, Yaghi and coworkers synthesized the first crystalline covalent linked organic framework.13 The surface area of a COF could achieve 4200 m2 g−1 which surpassed all of the porous polymer networks at that time.14 Sherrington et al. showed the “Davankov type” hypercrosslinked polymer with a surface area up to 2000 m2 g−1 in 2006.38,39 One year later, a first conjugated microporous polymer (CMP), was discovered by Cooper et al. which expressed multiple functions with a high surface area.18In 2009, our group developed a method to synthesise the first long range ordered porous aromatic framework (PAF) with dia topology (PAF-1),26 which had a record surface area (SBET = 5640 m2 g−1) at that time and exceptional physicochemical stability via a nickel(0)-catalyzed Yamamoto-type Ullmann cross-coupling.40,41 Besides, PAF-1 also showed very high uptake of carbon dioxide (1.3 g g−1 at 40 bar, 298 K) to make it a good candidate for carbon dioxide storage. This new type of three-dimensional homogeneous, rigid and open-network structure gives a “new generation” PAF with several unusual, even peculiar properties. This highlight describes the significant progress in the development of PAF, with a particular focus on the relationship between structure design, synthesis method and properties. The average size of crystalline domains in PAF are very small resulting in broad reflection peaks in the powder X-ray diffraction. Although ab initio structure determination could not be performed in this case, the target of synthesis, theoretical simulation and X-ray diffraction may present perfect matches in chemically reasonable models with such like dia, hcb and LTA topologies (Fig. 1).
 | ||
| Fig. 1 Structure model of synthesized and simulated PAF. (C, purple; N, blue; Si, yellow, O, green, Ge, brown). | ||
Molecular design of PAF
Before the appearance of PAF, there was plenty of research on the synthesis and applications of crystalline zeolite analogues such as MOF and COF. A practical drawback of MOF and COF is that most of them possess limited physicochemical stability. By contrast, there is also a growing range of porous polymer networks comprising stable covalent C–C, C–H and C–N bonds with high physicochemical stability. Unfortunately, the highest surface area of covalent linked porous polymer networks were among 3000 m2 g−1 at that time. The emergence of PAF first provides an ideal microporous material combining ultrahigh surface area and high physicochemical stability.The original idea for the synthesis of PAF-1 came from the structure and properties of diamond, in which each carbon atom is tetrahedrally connected to four neighboring atoms by covalent bonds. Breaking the C–C covalent bond of diamond and inserting rigid phenyl rings should allow sufficient exposure of the faces and edges of phenyl rings with the expectation of increasing the internal surface areas share dia topology. Before starting the synthesis, we systematically calculated the surface area and density of PAF-301, PAF-1 (PAF-302), PAF-303 (see Fig. 5 later in the manuscript) by employing a multiscale simulation method combining first-principles calculations and grand canonical Monte Carlo simulation. Results indicated that PAF-301 shares a simulated Langmuir surface area of 2350 m2 g−1 (simulated BET surface area, 1880 m2 g−1) with a simulated density of 0.8364 g cm−3 and PAF-1 shows a Langmuir surface area of 7000 m2 g−1 (BET surface area, 5640 m2 g−1) with a density of 0.315 g cm−3. The simulation result of PAF-303 indicated that its pore sizes were so large and was broken into the mesoscale range. Based on the simulation results, PAF-1 shows a record surface area which has a broad potential application in hydrogen storage and carbon capture.
Another example is the synthesis of JUC-Z1 (Fig. 1), which may have LTA topology.28 Linde type A (LTA) is a classic and critical porous aluminosilicate zeolite that is widely used in the chemical industry as an adsorber, detergent builder and container for combined materials.42–44 Its unit cell contains 24 tetrahedrally coordinated T atoms to form truncated octahedral (β-cage) and truncated cuboctahedra (α-cage) with cubic symmetry Fm-3c. The framework of LTA forms a CsCl-type structure, that is, the α-cage is surrounded by an eight β-cage (and vice versa). Our previous cooperative work with Sugiyama et al. had proved by AFM that double four ring (D4R) is the crystal growth unit for LTA.45 Linking D4R along the octant direction can generate LTA and ACO net.46 The difference of two such nets could be distinguished by the pore size distribution. The LTA net has at least two kinds of cages and the ACO net has only one type of channel. Our basic idea is to alternate the bridging Si–O–Si between D4R by rigid organic moieties. After synthesising JUC-Z1, we found a new type of hybrid materials that shows an unexpected ability to selectively adsorb benzene which is totally different with LTA.
Yamamoto type Ullmann cross-coupling reaction
After careful design of the second building unit (SBU), a proper growing method should be chosen to link all SBUs into an open framework. Based on our targeted network, reactions for the formation of the C–C bond are always taken into account. Especially for the synthesis of PAF-1, optimized nickel(0)-catalyzed Yamamoto type Ullmann cross-coupling was used. The Yamamoto coupling in general is an aryl–aryl coupling of aryl-halogenide compounds mediated mostly by stoichiometric amounts of bis(1,5-cyclooctadiene)nickel(0) (Ni(COD)2 for short). The polymerization is advantageous as a self-polycondensation with just a single, halogen-functionalized SBU can be used to form an organic framework. The mechanism of Yamamoto coupling is shown in Scheme 1.40 Firstly, the oxidative addition is triggered between Ni(0)Lm and halogen-functionalized monomers. After the disproportionation of the two complexes (I) and (II) of nickel, reductive elimination of complex (III) lead to regeneration of Ni(0)Lm and an addition product. During the recycling period of the coupling reaction, stoichiometric Ni(0)Lm is consumed when an aryl–aryl bond is formed. That is, the Ni(COD)2 had to be used in excess (1.2–2.8 equiv per aryl–aryl bond formed) to allow a high yield of the desired product. Compared with other C–C coupling reactions such as Sonogashira–Hagihara routes47–56 and Suzuki cross-coupling,57–66 Yamamoto coupling shows the unexpected halogen elimination ability of the ending group. This makes it unique to prepare ultrahigh porosity solids because heavy ending halogen atoms evidently decrease the surface area. By using Yamamoto type Ullmann cross coupling, PAF-1 was synthesized successfully and for the first time, a covalent linked porous organic framework showed an unexpected high surface area of 5640 m2 g−1 by BET theory. Furthermore, we continuously synthesized PAFs by the same method containing quadricovalent Si (PAF-3) and Ge (PAF-4).29 These PAF are thermally stable up to 465 °C for PAF-3 and 443 °C for PAF-4. As PAF-1, PAF-3 also shows a high surface area (up to 2932 m2 g−1) and excellent adsorption abilities for carbon dioxide, hydrogen and methane. The synthesis of PAF-3 via Yamamoto coupling was first reported by Cooper et al. in 2010 (SBET = 1102 m2 g−1).67 Recently, this structure was synthesized by an optimized Yamamoto coupling again, known as PPN-4 (Fig. 2), and a world record BET surface area of 6461 m2 g−1 was revealed.68 Combined with such an impressive surface area, PPN-4 can adsorb 2121 mg g−1 CO2 (212 wt%) at 50 bar/295 K, 158 mg g−1 hydrogen at 77 K/80 bar and 389 mg g−1 methane at 298 K/55 bar. This result proved again that Yamamoto coupling is an efficient method for the synthesis of ultrahigh surface area microporous networks.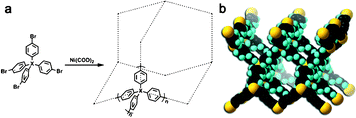 | ||
| Fig. 2 a) Synthetic route for PPN-3 (X: Adamantane), PPN-4 (X: Si), PPN-5 (X: Ge), and PAF-1 (X: C). b) The default noninterpenetrated diamondoid network of PPN-4 (black, C; pale grey, H; grey, Si). Reproduced from ref. 68 with permission from Wiley. | ||
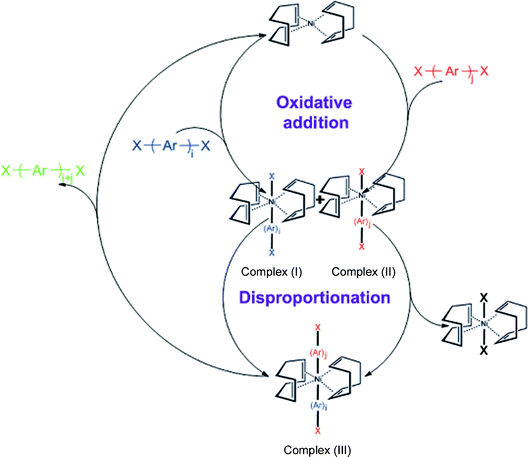 | ||
| Scheme 1 Mechanism of Yamamoto coupling. | ||
Another microporous organic framework named PAF-11 which shares dia topology structure was further explored via Suzuki cross-coupling reaction by Yuan et al. The targeted PAF-1132 was linked by a quaterphenyl group. That is, the pore size of PAF-11 should theoretically be mesoporous. Results show the yield is lower than that of PAF-1 and the CP/MAS NMR and FT-IR revealed that a bromo capping species were found. As an inevitable result, the apparent surface area derived from the BET model is 704 m2 g−1 which is much lower than PAF-1. Compared with the very narrow pore size distribution observed in PAF-1, a very widespread pore size distribution of PAF-11 between 0.5 nm and 5 nm was found. Interpenetrating as well as low efficiency synthesis are main reasons for the low surface area and multiple pore size distribution of PAF-11, the doubtless fact should be noted that Yamamoto type Ullmann cross-coupling is more efficient and has greater specificity for aryl–aryl coupling.
Properties and applications of PAF
Compared with other ultrahigh surface area solids such as porous carbons, porous silicas, zeolites, MOF and microporous polymers, PAF show very high surface areas and excellent physicochemical stability. PAF-1 is the first example of porous aromatic frameworks with a recorded surface area of 5640 m2 g−1 at that time. Based on its powder X-ray diffraction (PXRD) patterns, PAF-1 shows a long range order and, to a certain extent, an amorphous nature as well as the TEM results revealing that all the pores are wormlike but with uniform pore sizes. Currently, PPN-4 shows the highest surface area and is predominantly amorphous. These results broke a misleading notion that exceptionally high surface areas are the preserve of highly ordered molecular networks. We believe that rigid biphenyl frameworks, three dimensional dia topology and the elimination of the ending bromo group (heavy atoms) are key to preparing high surface area materials. So many porous polymer networks have been reported with lower surface areas and some of those also share dia topology, so they are worthy of exploration again by a more efficient synthesis route.Along with a high surface area, PAF also appear to have high physicochemical stability which makes them unique ultrahigh surface area materials. Most PAFs we have reported do not dissolve in organic solvents. Moreover, they can sustain in boiling water and cold acid or base solution. This is due to its covalent bonding nature and crosslinked rigid phenyl framework. High pressure hydrogen storage of PAF-1, PAF-3 and PAF-4 was determined at 77 K and the excess hydrogen uptake capacity is 7.0 wt% at 48 bar, 5.5 wt% at 60 bar, and 4.2 wt% at 60 bar for PAF-1, PAF-3 and PAF-4, respectively.26,29 The heat of adsorption related to hydrogen-adsorbent (QstH2 for short) interactions were calculated from the hydrogen adsorption isotherms recorded at 77 K and 87 K. Calculated by the Clausius–Clapeyron equation, it appears to be 5.4 kJ mol−1, 6.6 kJ mol−1 and 6.3 kJ mol−1 for PAF-1, PAF-3 and PAF-4 respectively. Accordingly, the low pressure uptakes of hydrogen are 186 cm3 g−1, 232 cm3 g−1 and 169 cm3 g−1 for PAF-1, PAF-3 and PAF-4, respectively. The results coming from the high pressure and low pressure hydrogen storage and QstH2 show that for high pressure gas storage, Qst is not so important to determine how many gas molecules could be stored. The energy for high pressure could remedy the weak interaction between gases and porous frameworks. In the case of high pressure gas storage, surface area is more important because it reveals how much porosity a has. A greater surface area means a greater volume of gas can be stored. But in the case of low pressure gas storage, especially when ambient condition gas storage (1 bar, room temperature) was applied, both surface area and Qst should be considered. Usually, the Qst is more important and results often show higher gas uptake material express higher Qst at ambient conditions.
PAF not only have high surface areas and excellent physicochemical stabilities, but also exceptional selectivity for green house gases. At 273 K and 1 bar, PAF-1, PAF-3 and PAF-4 exhibit very high selectivity to adsorb carbon dioxide and methane over hydrogen, nitrogen, oxygen and argon. In particular, PAF-3 shows extraordinary selectivity for the adsorption of CO2 over nitrogen with a value of 87/1 as well as of methane, which is about 30 times higher than that of nitrogen. That is, PAF could efficiently recognize and enrich greenhouse gases from dry air which have potential applications in carbon capture and storage. To prove this, a CO2 reversible adsorption (25 °C) and regeneration (80 °C) experiment was designed and performed. As shown in Fig. 3, PAF-3 is the best one which shows a very high average weight change of 4.40 wt% during the cycles. When considering the high stability of PAF-3, this carbon dioxide adsorption procedure exhibits better energy savings and cost-effectiveness.
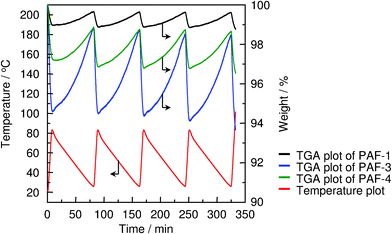 | ||
| Fig. 3 CO2 cyclic adsorption (25 °C) and regeneration (85 °C) of PAF-1 (black), PAF-3 (blue) and PAF-4 (green) (red is temperature). Reproduced from ref. 29 with permission from the Royal Society of Chemistry. | ||
The former results reveal the fantastic gas storage and carbon capture properties of PAF. Among the PAF reported, PAF-1 has the best physicochemical stability and ultrahigh surface area. Unfortunately, the relatively low Qst between PAF-1 and gases is one of the practical drawbacks for PAF-1 when applying at ambient conditions. Very recently, Zhou et al. presented a post-synthetic way to improve the heat of adsorption of PAF-1 by introducing sulfone groups onto the biphenyl frameworks.69 By reacting it with chlorosulfonic acid, PAF-1 was modified to give sulfonate grafted acid (PPN-6-SO3H) and lithium salt (PPN-6-SO3Li) species. After sulfonation, the surface area of PPN-6-SO3H and PPN-6-SO3Li reduced to as low as 1254 m2 g−1 and 1186 m2 g−1 but the Qst increased to as high as 30.4 and 35.7 kJ mol−1. Strong interactions between the sulfonate-graft samples and carbon dioxide lead to a very high uptake capacity, with values of 13.1 and 13.5 wt% (equivalent to 3.6 and 3.7 mmol g−1) for PPN-6-SO3H and PPN-6-SO3Li, respectively. Calculated by the ideal adsorption solution theory (IAST) of Myers and Prausnitz,70–77 at postcombustion carbon capture procedure conditions (typically 15% CO2 and 85% N2), sulfonate-graft samples showed unexpectedly high adsorption selectivities for carbon dioxide and nitrogen which surpass the selectivity of the NaX zeolite.78 Thus, these materials hold considerable promise for postcombustion carbon capture applications.
Besides, PAF also show molecular recognition properties between organic molecules and water. PAF-5,31 PAF-1132 and JUC-Z128 are hydrophobic materials with high organic small molecule uptake such as methanol and benzene against water which could be used to eliminate harmful small organic molecules produced by industry. Based on the aromatic nature of PAF-2,27 it shows high selective adsorption of aromatic benzene against aliphatic hexane. Moreover, PAF-633 could uptake 0.35 g g−1 of ibuprofen, and drug release results show a large initial burst release within 10 h. The release behavior of PAF-6 is comparable to that of MCM-41,79 which has potential applications in drug release systems.
Recently, we have designed and synthesized an electroactive microporous organic framework (JUC-Z2).30 The SBU of JUC-Z2 is triphenylamine, which is a very popular building block for conducting polymers because they are able to form stable aminium radical cations with low ionization potentials and relatively high mobility. JUC-Z2 was comprised of stacked organic network sheets with an hcb topology forming a well-defined uniform micropore (pore size distribution is 1.2 nm), high surface area (SBET = 2081 m2 g−1) and high physicochemical stability (>440 °C). Most important of all, after doping with I2, JUC-Z2 exhibits typical p-type semiconductive properties and \ resistivity of 9 × 10−1 Ω cm with a Hall mobility of 0.811 cm2 V−1 S−1, and a carrier concentration of 8.26 × 1018 cm−3. Interestingly, JUC-Z2 was found to be capable of express reversible and stable electrochemical redox behaviors for supporting electrolytes composed of hydrophilic small anions such as tetrabutylammonium hexafluorophosphate (Bu4NPF6), tetrabutylammonium perchlorate (Bu4NClO4) or tetrabutylammonium tetrafluoroborate (Bu4NBF4). As far as we know, JUC-Z2 is the first example of an electroactive microporous organic framework. Furthermore, electrochemical recognition properties of electrolytes with different sizes was also found in oligomeric JUC-Z2 (Oligo-JUC-Z2 for short). As shown in Fig. 4, Oligo-JUC-Z2 shows different electrochemical responses in degassed acetonitrile solutions containing tetrabutylammonium hexafluorophosphate (Bu4NPF6), tetraethylammonium toluene-4-sulfonate (ET4NTOS), and 10-camphorsulfonic acid (CSA). The reason for such unique electrochemical recognition is that JUC-Z2 is a p-doping type semiconductive POF, whose electrochemical redox process is dependent on either adsorption or desorption of electrolyte anions to maintain the framework charge neutrality (Scheme 2). The micropores of Oligo-JUC-Z2 (1.2 nm) permit free mobility of adsorption and desorption for small anions such as PF6− (5.12 Å), ClO4− (5.0 Å) and BF4− (4.54 Å) to balance the charge of the framework, which lead to stable and reversible electrochemical behavior. The uniform micropore of Oligo-JUC-Z2 could be very easily blocked by the ET4NTOS molecules and show irreversible electrochemical behavior. Similarly, the charge of the micropores cannot be balanced by a large CSA (9 Å) anion, as a result, Oligo-JUC-Z2 lost its electroactivity. The above mentioned experiments prove that JUC-Z2 possess novel electrochemical features, totally different from previously electrosynthesized conducting polymers that can exhibit the large and stable voltammetric response for either the CSA− or TOS− anion.80,81 In summary, the electrochemical redox behavior of the designed electrochemically accessible pores testify the conversion between electric and the chemical energy for the micropore of JUC-Z2 depend on matching attributes between the micropores and ion guest. Combined with its p-type semiconductive nature, JUC-Z2 shows potential application in sensors, electroluminescent devices and solar cells, etc.
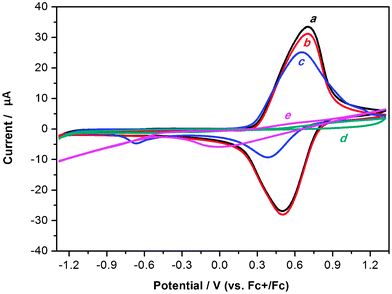 | ||
| Fig. 4 Cyclic voltammograms of Oligo-JUC-Z2 powder film on Pt microelectrode in degassed acetonitrile solution containing 0.1 M Bu4NPF6 at (a) the first cycle and (b) fifth cycle; 0.1 M ET4TOS at (c) the first cycle and (d) fifth cycle; (e) 0.1 M CSA at the first cycle. Scan rate: 20 mV s−1. Reproduced from ref. 30 with permission from the Royal Society of Chemistry. | ||
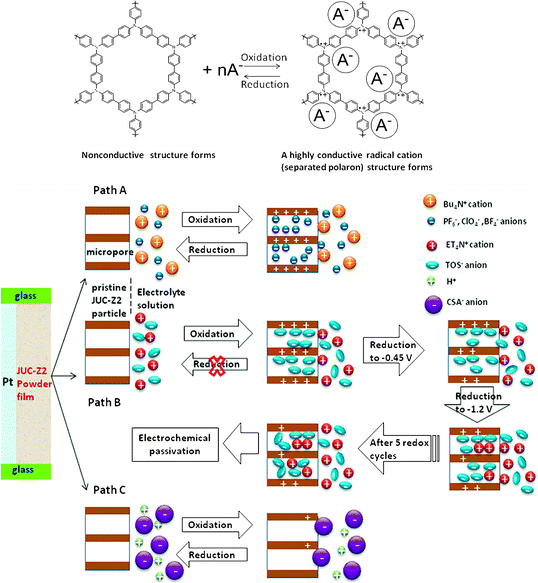 | ||
| Scheme 2 Illustration of electrochemical redox behavior of pristine JUC-Z2 and its different redox processes occurring in either Bu4NPF6 (Bu4NClO4, Bu4NBF4), ET4TOS or HCSA supporting electrolyte solutions. Reproduced from ref. 30 with permission from the Royal Society of Chemistry. | ||
Outlook and conclusion
Concluding the paper, we state once again that there are very good reasons to consider porous aromatic frameworks as the new generation of a whole new class of organic networks with an intrinsic nanoporosity. They are characterized by a rigid aromatic open-framework structure constructed by covalent bonds that remain accessible to small molecules driven both by weak interactions and, for the first time we proved, by the electrochemical induced charge interaction. Because of the extremely large apparent specific surface areas measured by conventional gas adsorption techniques, and the high physicochemical stability, PAFs have extended usage lifetime under relatively harsh conditions.In the last ten years, there has been great progress in the molecular engineering of ultrahigh surface area organic frameworks. The records of surface area, heat of adsorption, and selectivity of molecular recognition are continually broken. With the development of molecular simulation methods and computer techniques, more and more structures which have high possibilities for applications in carbon capture, gas storage, molecular recognition and separation were revealed. It seems PAF have great potential to be a more fantastic host material when functional groups are introduced onto the aromatic frameworks. Cao et al. and our own group have evaluated the hydrogen storage performance of designed porous materials PAF-30X (X = 1–4) (Fig. 5) with dia topological structures by multiscale simulation methods.82 Results show that hydrogen uptake of PAF mainly depends on their densities and free volumes. Simulated results show PAF-304 and PAF-303 possess higher gravimetric hydrogen uptake than PAF-1. In particular, the simulated gravimetric hydrogen uptake of PAF-304 reaches 6.53 wt% at room temperature and 100 bar, which is the highest among all of the present porous materials. The structure of PAF-304 is also known as PAF-11, which was synthesized by Suzuki cross coupling with a relatively low surface area of 704 m2 g−1. The contradiction between PAF-11 and theoretical calculations come from the inefficient synthesis method and the possibilities of interpenetration of aromatic frameworks. It is worth to exploring again by more carefully designs SBU and synthesis methods. Sun et al. and our own group also predict a lithium tetrazolide grafted PAF (Fig. 6) with dia topology and simulated results show very promising hydrogen uptake reaches 4.9 wt% at 233 K and 10 MPa, which exceeds the 2010 DOE target of 4.5 wt%.83 Jiang et al. designed a new PAF by introducing polar organic groups into the biphenyl unit (Fig. 7) and investigated their separating power toward carbon dioxide by using grand-canonical Monte Carlo (GCMC) simulations.84 Simulated results indicated that PAF-1 functionalized with tetrahydrofuran-like ether groups shows a high adsorption capacity for carbon dioxide (10 mmol g−1, 1 bar, r.t.) and high selectivity for CO2/H2, CO2/CH4 and CO2/N2 mixtures at ambient conditions. Whether the above mentioned novel PAF could be effectively synthesized, the theoretical simulation and calculation provide an effective method to guide the developing direction of more attractive PAF.
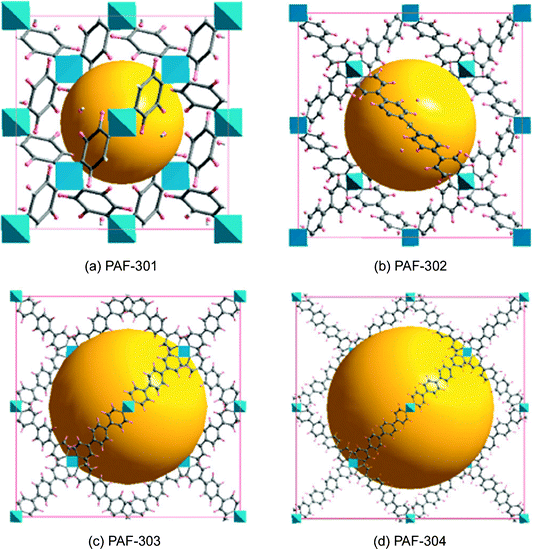 | ||
| Fig. 5 Unit cells of PAF, (a) PAF-301, (b) PAF-302, (c) PAF-303, and (d) PAF-304, derived from topology design and geometry optimization with the force field method. Here, gray and pink spheres represent carbon and hydrogen atoms, respectively, while the blue polyhedron represents the tetrahedrally bonded carbon atoms. In addition, the yellow sphere denotes the pores in 3D PAF. Reproduced from ref. 82 with permission from the American Chemical Society. | ||
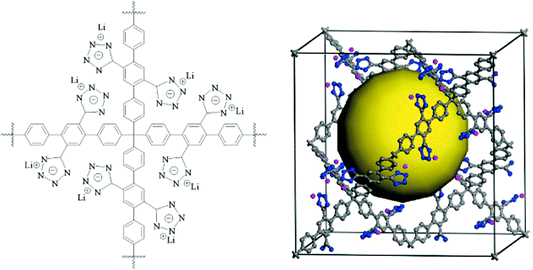 | ||
| Fig. 6 The building block (left) and unit cell (right) of the proposed porous aromatic framework containing Li(+)-CHN4(−) moieties. The yellow ball denotes the free volume. Reproduced from ref. 83 with permission from the American Chemical Society. | ||
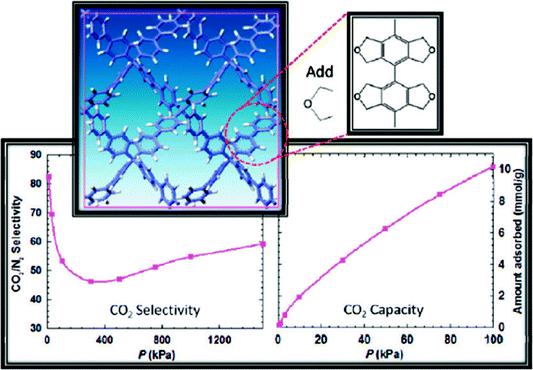 | ||
| Fig. 7 The unit cell of the proposed porous aromatic framework containing tetrahydrofuran-like moieties and the simulation pattern of CO2/N2 selectivity and CO2 capacity. Reproduced from ref. 84 with permission from the American Chemical Society. | ||
At last, it should be noted that PAF, especially synthesized by Yamamoto type Ullmann cross-coupling, are costly to deploy on a large scale as carbon capture adsorbents. There is still a great challenge to find a more convenient and low cost synthesis method to prepare PAF. Besides, the incorporation of useful and novel functions within robust PAF with ultrahigh surface areas remains a formidable challenge and an exciting opportunity for material chemists.
Acknowledgements
This work was supported by the National Basic Research Program of China (2011CB808703, 2012CB821700), National Natural Science Foundation of China (Grant nos. 91022030), “111” project (B07016), International Cooperation Project of Ministry of Science and Technology (2006DFA41190). Jilin Science and Technology Department Project (20106021). The authors gratefully acknowledge Prof. Guangshan Zhu for fruitful discussions.References
- F. Schuth, K. S. W. Sing and J. Weitkamp, Handbook of Porous Solids; Wiley-VCH: New York, 2002 Search PubMed.
- V. Valtchev, S. Mintova and M. Tsapatsis, Ordered Porous Solids: Recent Advances And Prospects; Elsevier B. V.: Oxford, 2009 Search PubMed.
- J. Li, J. Sculley and H. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS.
- M. P. Suh, H. J. Park, T. K. Prasad and D. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS.
- P. Makowski, A. Thomas, P. Kuhn and F. Goettmann, Energy Environ. Sci., 2009, 2, 480 CAS.
- X. Ding, J. Guo, X. Feng, Y. Honsho, J. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 1289 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439 CrossRef CAS.
- A. Gomotti, S. Bracco, P. Valsesia, M. Beretta and P. Sozzani, Angew. Chem., Int. Ed., 2010, 49, 1760 CrossRef.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- K. L.Mulfort, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, Chem.–Eur. J., 2010, 16, 276 CrossRef.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. Chang, Y. K. Hwang, V. Marsaud, P. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef.
- H. M. El-Kaderi, J. R. Hunt, J. L. Medoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- J. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826 CrossRef CAS.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710 CrossRef CAS.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809 CrossRef CAS.
- C. D. Wood, B. Tan. A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193 CrossRef CAS.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457 CrossRef CAS.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291 RSC.
- Y. Peng, T. Ben, J. Xu, M. Xue, X. Jing, F. Deng, S. Qiu and G. Zhu, Dalton Trans., 2011, 40, 2720 RSC.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991 CAS.
- T. Ben, K. Shi, Y. Cui, C. Pei, Y. Zuo, H. Guo, D. Zhang, J. Xu, F. Deng, Z. Tian and S. Qiu, J. Mater. Chem., 2011, 21, 18208 RSC.
- H. Ren, T. Ben, F. Sun, M. Guo, X. Jing, H. Ma, K. Cai, S. Qiu and G. Zhu, J. Mater. Chem., 2011, 21, 10348 RSC.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498 RSC.
- H. Zhao, Z. Jin, H. Su, X. Jing, F. Sun and G. Zhu, Chem. Commun., 2011, 47, 6389 RSC.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725 CrossRef CAS.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571 CrossRef CAS.
- S. S. Chui, S. M. Lo, J. P. Charmant, H. A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780 RSC.
- J. H. Ahn, J. E. Jang, C. G. Oh, S. K. Ihm, J. Cortez and D. C. Sherrington, Macromolecules, 2006, 39, 627 CrossRef CAS.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27 CrossRef CAS.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 1999, 72, 621 CrossRef CAS.
- G. Zhou, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2007, 129, 12211 CrossRef CAS.
- Introduction to Zeolite Science and Practice, ed. H. Van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Elsevier, Amsterdam, 2001 Search PubMed.
- D. W. Breck,Zeolite Molecular Sieves, Wiley, New York, 1974 Search PubMed.
- Y. Nozue, T. Kodaira, S. Ohwashi, T. Goto and O. Terasaki, Phys. Rev. B: Condens. Matter, 1993, 48, 12245 CrossRef.
- S. Sugiyama, S. Yamamoto, O. Matsuoka, H. Nozoye, J. Yu, G. Zhu, S. Qiu and O. Terasaki, Microporous Mesoporous Mater., 1999, 28, 1 CrossRef CAS.
- M. O'Keeffe and O. M. Yaghi, Chem.–Eur. J., 1999, 5, 2796 CrossRef CAS.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- D. L. Trumbo and C. S. Marvel, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 2311 CrossRef CAS.
- E. Hittinger, A. Kokil and C. Weder, Angew. Chem., 2004, 116, 1844 CrossRef Angew. Chem. Int. Ed., 2004, 43, 1808..
- J. D. Mendez, M. Schroeter and C. Weder, Macromol. Chem. Phys., 2007, 208, 1625 CrossRef CAS.
- Z. J. Donhauser, B. A. Mantooth, K. F. Kelly, L. A. Bumm, J. D. Monnell, J. J. Stapleton, D. W. Price, A. M. Rawlett, D. L. Allara, J. M. Tour and P. S. Weiss, Science, 2001, 292, 2303 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 12593 CrossRef CAS.
- D. Venkataraman, S. Lee, J. S. Zhang and J. S. Moore, Nature, 1994, 371, 591 CrossRef CAS.
- W. Zhang and J.S. Moore, Angew. Chem., 2006, 118, 4524 CrossRef Angew. Chem. Int. Ed., 2006, 45, 4416..
- Y. H. Kiang, G. B. Gardner, S. Lee, Z. T. Xu and E. B. Lobkovsky, J. Am. Chem. Soc., 1999, 121, 8204 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 36, 343 Search PubMed.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS.
- H. Zhang, F. Xue, T.C. W. Mak and K. S. Chan, J. Org. Chem., 1996, 61, 8002 CrossRef CAS.
- H. Zhang and K. S. Chan, Tetrahedron Lett., 1996, 37, 1043 CrossRef CAS.
- D. Badone, M. Baroni, R. Cardamone, A. Ielmini and U. Guzzi, J. Org. Chem., 1997, 62, 7170 CrossRef CAS.
- A. L. Casalnuovo and J. C. Calabrese, J. Am. Chem. Soc., 1990, 112, 4324 CrossRef CAS.
- Y. M. A. Yamada, K. Takeda, H. Takahashi and S. Ikegami, Org. Lett., 2002, 4, 3371 CrossRef CAS.
- R. Frenette and R. W. Friesen, Tetrahedron Lett., 1994, 35, 9177 CrossRef CAS.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS.
- T. Ishiyama, Y. Itoh, T. Kitano and N. Miyaura, Tetrahedron Lett., 1997, 38, 3447 CrossRef CAS.
- J. R. Holst, E. Stöckel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531 CrossRef CAS.
- D. Yuan, W. Lu, D. Zhao and H. Zhou, Adv. Mater., 2011, 23, 3723 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H. Zhou, J. Am. Chem. Soc., 2011, 133, 18126 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121 CrossRef CAS.
- Y. S. Bae, O. K. Farha, J. T. Hupp and R. Q. Snurr, J. Mater. Chem., 2009, 19, 2131 RSC.
- A. O. Yazaydin, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425 CrossRef CAS.
- B. S. Zheng, J. F. Bai, J. G. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748 CrossRef CAS.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177 CAS.
- R. Krishna and J. M. van Baten, Phys. Chem. Chem. Phys., 2011, 13, 10593 RSC.
- Y. Belmabkhout and A. Sayari, Adsorption, 2009, 15, 318 CrossRef CAS.
- A. Goj, D. S. Sholl, E. D. Akten and D. I. Kohen, J. Phys. Chem. B, 2002, 106, 8367 CrossRef CAS.
- Y. Belmabkhout, G. Pirngruber, E. Jolimaitre and A. Methivier, Adsorption, 2007, 13, 341 CrossRef CAS.
- R. Mortera, S. Fiorilli, E. Garrone, E. Verne and B. Onida, Chem. Eng. J., 2010, 156, 184 CrossRef CAS.
- I. D. Norris, L. A. P. Kane-Maguire and G. G. Wallace, Macromolecules, 2000, 33, 3237 CrossRef CAS.
- G. Xu, W. Wang, X. Qu, Y. Yin, L. Chu, B. He, H. Wu, J. Fang, Y. Bao and L. Liang, Eur. Polym. J., 2009, 45, 2701 CrossRef CAS.
- J. Lan, D. Cao, W. Wang, T. Ben and G. Zhu, J. Phys. Chem. Lett., 2010, 1, 978–981 CrossRef CAS.
- Y. Sun, T. Ben, L. Wang, S. Qiu and H. Sun, J. Phys. Chem. Lett., 2010, 1, 2753–2756 CrossRef CAS.
- R. Babarao, S. Dai and D. Jiang, Langmuir, 2011, 27, 3451–3460 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |