Hydrothermal synthesis and structural characterisation of [H2DABCO]3[Cu16Cl22]: a new copper(I) chloride framework†
Edward R.
Williams
and
Mark T.
Weller
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: m.t.weller@bath.ac.uk; Tel: +44 (0) 1225 386444
First published on 19th October 2012
Abstract
A H2DABCO templated copper(I) chloride framework of the stoichiometry [Cu16Cl22]6− formed from linked CuCl4, CuCl3 and CuCl2 units crystallizes in a non-centrosymmetric space group.
Porous materials are of increasing interest due to their range of possible applications, including ion exchange, gas storage–adsorption and catalysis.1–3 Typically such materials consist of vertex linked oxo-tetrahedra, such as zeolites and zeotypes,4,5 oxo-tetrahedra combined with other metal-centred polyhedra6,7 and metal-centred polyhedra linked via carboxylic acid components (MOFs).8 Examples of frameworks consisting solely of metal–halide polyhedra are notably rarer, particularly when formed in combination with a large cation or an organic templating agent with the aim of inducing porosity; the few examples known include members of the scandium–fluoride–amine system9 and CsCe2F8[F·H2O].10 Copper(I) chloride is an important catalytic component in a range of organic reactions:11–13 a microporous copper chloride material could therefore be of particular interest in the catalysis of certain organic transformations, particularly if chirality can be introduced through the adoption of a non-centrosymmetric space group. Very few copper halide frameworks have been synthesised previously but examples include the two-dimensional [{Cu2(μ-X)2(μ-phenazine)}∞] (X = I, Br, Cl)14 and the three-dimensional [Cu(en)2]2Cu7Cl11 where copper species form both the cationic template, [Cu(en)2]2+, and the negatively charged framework, [Cu7Cl11]4− among others.15–18 We report here the synthesis and structure of a new three dimensional copper(I) chloride framework of stoichiometry [Cu16Cl22]6− templated on di-protonated 1,4-diazobicyclo[2.2.2]octane (DABCO) cations and adopting the space group P212121.
Commercially available materials of reagent grade, that did not require further purification, were used in the synthesis of this material. HPF6 (65 wt%, 0.32 mL, 2.52 mmol) was added to a mixture of CuCl2·2H2O (0.0773 g, 0.45 mmol) and 1,4-diazabicyclo[2.2.2]octane (0.1234 g, 1.10 mmol) in a 23 mL Teflon™ liner. The reaction mixture was left to incubate for 30 minutes before heating in a steel autoclave at 175 °C for 48 hours. The presence of HPF6 and little water produces highly acidic reaction conditions under which the DABOC is di-protonated and also partially decomposes via oxidation by Cu(II). This generates Cu(I) in the reaction medium; a similar process involving U(VI) and U(IV) has been reported previously.19 The autoclave was allowed to cool slowly to room temperature and the solid product removed from solution by filtration, washed with distilled water and dried in air at room temperature.
Single-crystal X-ray diffraction (SXD) data were collected at 100 K on a Rigaku FR-E+ Very High Flux diffractometer, using Mo Kα (λ = 0.71073 Å) radiation. The structure was solved and refined using the WinGX package20 by direct methods21 using SHELXS-97.22 The structure was refined in the space group P212121, initially yielding a Flack parameter of ∼0.5 before completion of the structure refinement as a racemic twin, BASF = 0.52(2).23 The H2DABCO template position was fully determined and idealised hydrogen positions on this template molecule were calculated.
Scanning electron microscopy (SEM) was carried out using a JEOL JSM 5910 SEM fitted with an Oxford Instruments Inca Energy-Dispersive Spectrometry (EDS) analysis system. Semi-quantitative EDS data confirmed the presence of Cu and Cl, in levels approximately consistent with the formula above (idealised ratio 0.73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cf. experimental 0.53:1). Powder X-ray diffraction data collected on a bulk sample showed a very good match to the calculated pattern from the SXD derived structural model indicating sample purity in excess of 80% (see ESI†); this was consistent with assessment of sample purity using optical microscopy.
:1 cf. experimental 0.53:1). Powder X-ray diffraction data collected on a bulk sample showed a very good match to the calculated pattern from the SXD derived structural model indicating sample purity in excess of 80% (see ESI†); this was consistent with assessment of sample purity using optical microscopy.
The product crystallises in the orthorhombic P212121 space group as very pale yellow-green block-shaped crystals of size 0.15 × 0.10 × 0.05 mm3. The crystallographic information is summarised in Table 1. The structure is formed from CuCl4 tetrahedra and CuCl3 trigonal planes, forming di-protonated DABCO filled channels and pores within the structure, with the channels running along the a- and b-axes having diameters of approximately 7 Å, Fig. 1.
![Structure of [H2DABCO]3Cu16Cl22 viewed down the a- and b-axes. DABCO molecules removed for clarity, Cu in blue, Cl in green.](/image/article/2013/CE/c2ce26068a/c2ce26068a-f1.gif) | ||
| Fig. 1 Structure of [H2DABCO]3Cu16Cl22 viewed down the a- and b-axes. DABCO molecules removed for clarity, Cu in blue, Cl in green. | ||
| Formula | Cu16Cl22C18N6H36 |
|---|---|
| Formula weight | 2139.28 |
| T/K | 100 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Racemic twin, BASF | 0.52(2) |
| a/Å | 12.8722(5) |
| b/Å | 12.9237(4) |
| c/Å | 29.6414(21) |
| V/Å3 | 4931.04(4) |
| Z | 4 |
| Density/g cm−3 | 2.88 |
| F(000) | 4118.9 |
| Reflections collected | 23562 |
| Unique reflections | 11287 |
| R-indices all data | R 1 = 0.066 wR2 = 0.141 |
| R-indices obs. Data | R 1 = 0.059 wR2 = 0.138 |
| GOOF | 1.130 |
Copper(I) is known to adopt a number of coordination environments in its compounds, ranging from two-fold linear species, to three-fold trigonal planar and tetrahedral geometries.15,24,25 Each of these coordination environments is observed in the complex [Cu16Cl22]6− framework. The three-dimensional framework is built from triplets of vertex sharing CuCl4 tetrahedra, linked via a μ3-Cl to form infinite sinusoidal chains parallel to the a-axis. The CuCl4 tetrahedra at each apex of the wave-like chains are further linked to two edge-sharing CuCl4 tetrahedra and two CuCl3 trigonal planes. The resulting infinite chains are in turn linked to form the three-dimensional structure by a CuCl4 tetrahedron, a linear CuCl2 unit and a CuCl3 trigonal plane pair, in addition to a disordered copper trimer, with two possible structural conformations. For the preferred (85% occupied) motif, the trimer forms as a CuCl4 tetrahedron with two CuCl3 trigonal planes bridging one tetrahedral edge, and average Cu–Cu distance of 2.827 Å (Fig. 2). Disorder arises from the tetrahedral copper site 15% of the time shifting by 1.398 Å to form a different tetrahedral species, presumably slightly less energetically favourable due to the elongation of one Cu–Cl bond of length to 2.686(1) Å. To prevent short Cu–Cu distances, the two trigonal planar copper sites in this unit also shift, one to form another adjacent trigonal plane, which is again less favourable due to an elongated Cu–Cl bond (2.682(1) Å), the other to form a linear Cl–Cu–Cl species along the edge of the original trigonal plane (Fig. 2). This linear species is asymmetric with bond distances of 1.959(1) and 2.134(1) Å. Bond valence calculations were used to confirm the oxidation states of these Cu sites, as well as confirming the fully bonded nature of the chloride anions within the disordered trimer.
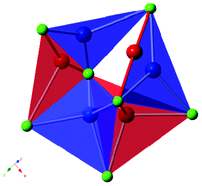 | ||
| Fig. 2 The two possible copper arrangements within the disordered component of the structure (Cu1–3), the preferred conformation is shown as blue polyhedra and the less favoured shown in red. | ||
The copper environments observed within the structure are summarised in Table 2. A number of copper sites showed distinct distortion, in particular the tetrahedral environments. The majority of these displayed a significant elongation, approximately 0.2 Å, of two of the Cu–Cl bonds, destroying the tetrahedral symmetry and tending towards linear CuCl2.
| Copper environments (A/B denote disordered sites) | Bond distances (Å) short bonds; elongated bonds |
|---|---|
| Cu(1A)Cl3 | 2.263, 2.287, 2.301 |
| Cu(1B)Cl2 | 1.960, 2.133 |
| Cu(2A)Cl3 | 2.243, 2.322; 2.549 |
| Cu(2B)Cl3 | 2.083, 2.189; 2.686 |
| Cu(3A)Cl4 | 2.324, 2.361, 2.376, 2.386 |
| Cu(3A)Cl4 | 2.297, 2.333, 2.369; 2.688 |
| Cu(4)Cl3 | 2.231, 2.235; 2.454 |
| Cu(5)Cl4 | 2.282, 2.308; 2.469, 2.480 |
| Cu(6)Cl3 | 2.224, 2.344, 2.349 |
| Cu(7)Cl4 | 2.271, 2.280; 2.433, 2.516 |
| Cu(8)Cl3 | 2.231, 2.294, 2.339 |
| Cu(9)Cl2 | 2.145, 2.150 |
| Cu(10)Cl4 | 2.258, 2.262; 2.449, 2.574 |
| Cu(11)Cl4 | 2.272, 2.278, 2.384; 2.681 |
| Cu(12)Cl4 | 2.295, 2.30; 2.378, 2.450 |
| Cu(13)Cl3 | 2.253, 2.280, 2.282 |
| Cu(14)Cl4 | 2.229, 2.290, 2.327, 2.396 |
| Cu(15)Cl4 | 2.281, 2.285; 2.343, 2.469 |
| Cu(16)Cl4 | 2.268; 2.373, 2.386, 2.472 |
Di-protonated DABCO molecular cations reside within the channels; presumably the strongly acidic synthesis conditions favour the formation of this template rather than the more normally found [HDABCO]+. The template molecular cations are arranged within the pores which form a fully interconnected three dimensional channel system, Fig. 3. The arrangement of the template species corresponds to the 21 screw axes along the a and b directions producing straight almost circular orthogonal channels of diameter ∼7.2 Å.
![Structure of [H2DABCO]3Cu16Cl22 viewed down the a (top)- and b (bottom)-axes showing the DABCO molecule positions and delineating the pore connectivity. On the left hand side of each diagram the CuCln polyhedra are shown, while these are omitted on the left in the otherwise identical view; shortest intermolecular DABCO interactions (C–C distances between 3 and 6 Å) are shown as dashed lines.](/image/article/2013/CE/c2ce26068a/c2ce26068a-f3.gif) | ||
| Fig. 3 Structure of [H2DABCO]3Cu16Cl22 viewed down the a (top)- and b (bottom)-axes showing the DABCO molecule positions and delineating the pore connectivity. On the left hand side of each diagram the CuCln polyhedra are shown, while these are omitted on the left in the otherwise identical view; shortest intermolecular DABCO interactions (C–C distances between 3 and 6 Å) are shown as dashed lines. | ||
Conclusions
A new copper(I) chloride framework with the stoichiometry [Cu16Cl22]6− has been synthesised and characterised structurally from single crystal X-ray diffraction data. The framework is templated on di-protonated DABCO molecular cations whose arrangement within the structures pores generates orthogonal channels of 7 Å in diameter with 21 rotational symmetry. The framework is constructed by linking various CuCln, n = 2, 3, 4, units and displays a small degree of disorder associated with two possible linkage configurations of these building motifs. The material crystallises as 1:1 racemic twin and further investigations are focusing on producing a single enantiomeric form through, for example the use of a chiral, substituted DABCO template.
References
- S. Natarajan and S. Mandal, Angew. Chem., Int. Ed., 2008, 47, 4798–4828 CrossRef CAS.
- Z. H. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church, Z. T. Kang, H. W. Abernathy, C. J. Summers, M. L. Liu and K. H. Sandhage, Nature, 2007, 446, 172–175 CrossRef CAS.
- X. B. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS.
- R. M. Barrer, Hydrothermal Chemistry of Zeolites, Academic Press, London, 1982 Search PubMed.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS.
- M. Takahashi, S. Tobishima, K. Takei and Y. Sakurai, J. Power Sources, 2001, 97–98, 508–511 CrossRef CAS.
- Z. Lin, A. Ferreira and J. Rocha, J. Solid State Chem., 2003, 175, 258–263 CrossRef CAS.
- D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS.
- N. F. Stephens and P. Lightfoot, Solid State Sci., 2006, 8, 197–202 CrossRef CAS.
- J. Rouse and M. T. Weller, Dalton Trans., 2009, 10330–10337 RSC.
- M. Kozuka, A. Inoue, T. Tsuchida and M. Mitani, Adv. Synth. Catal., 2006, 348, 953–966 CrossRef CAS.
- Z. Lu and S. M. Ma, Adv. Synth. Catal., 2007, 349, 1225–1230 CrossRef CAS.
- J. Ren, S. S. Liu, Z. Li, X. L. Lu and K. C. Xie, Appl. Catal., A, 2009, 366, 93–101 CrossRef CAS.
- M. Munakata, T. Kurodasowa, M. Maekawa, A. Honda and S. Kitagawa, J. Chem. Soc., Dalton Trans., 1994, 2771–2775 RSC.
- J. R. D. DeBord, Y. J. Lu, C. J. Warren, R. C. Haushalter and J. Zubieta, Chem. Commun., 1997, 1365–1366 RSC.
- J. D. Martin, A. M. Dattelbaum, T. A. Thornton, R. M. Sullivan, J. C. Yang and M. T. Peachey, Chem. Mater., 1998, 10, 2699–2713 CrossRef CAS.
- A. A. Thorn, R. D. Willett and B. Twamley, Inorg. Chem., 2008, 47, 5775–5779 CrossRef CAS.
- W. G. Haije, J. A. L. Dobbelaar and W. J. A. Maaskant, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1485–1487 CrossRef.
- C. L. Cahill and P. C. Burns, Inorg. Chem., 2001, 40, 1347–1351 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Release 97-2, University of Göttingen, Germany, 1997 Search PubMed.
- H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55, 908–915 CrossRef.
- J. L. Fulton, M. M. Hoffmann and J. G. Darab, Chem. Phys. Lett., 2000, 330, 300–308 CrossRef CAS.
- J. K. Burdett and O. Eisenstein, Inorg. Chem., 1992, 31, 1758–1762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CIF for [H2DABCO]3[Cu16Cl22]; experimental and calculated powder X-ray diffraction patterns. CCDC reference number 890546. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ce26068a |
| This journal is © The Royal Society of Chemistry 2013 |