Redetermination of crystal structure of Ag(II)SO4 and its high-pressure behavior up to 30 GPa†‡
Mariana
Derzsi
*a,
Armand
Budzianowski
a,
Viktor V.
Struzhkin
*b,
Przemysław J.
Malinowski
c,
Piotr J.
Leszczyński
a,
Zoran
Mazej
d and
Wojciech
Grochala
*ac
aCentre for New Technologies, The University of Warsaw, Zwirki i Wigury 93, 02079 Warsaw, Poland. E-mail: mariana@cent.uw.edu.pl; armand@cent.uw.edu.pl; pleszcz@cent.uw.edu.pl; wg22@cornell.edu; Fax: +48 225540801; Tel: +48 225540828
bGeophysical Laboratory, Carnegie Institution, Washington, DC 20015 USA. E-mail: struzhkin@gl.ciw.edu; Fax: +1 2024788901; Tel: +1 2024788900
cFaculty of Chemistry, The University of Warsaw, Pasteur 1, 02093 Warsaw, Poland. E-mail: malin@chem.uw.edu.pl; Fax: +48 228225996; Tel: +48 228220211
dDepartment of Inorganic Chemistry and Technology, Jožef Stefan Institute, Jamova 39, Ljubljana 1000, Slovenia. E-mail: zoran.mazej@ijs.si; Fax: +386 12519385; Tel: +386 14773900
First published on 8th November 2012
Abstract
Here we redetermine the crystal structure of Ag(II)SO4, an unusual d9 system, at 1 atm from powder X-ray data and we report hydrostatic pressure X-ray diffraction experiments on Ag(II)SO4 inside the diamond anvil cell. AgSO4 crystallizes in the monoclinic C2/c cell, with a = 12.8476(2) Å, b = 13.6690(4) Å, c = 9.36678(19) Å, β = 47.5653(13)°, and V = 1214.04(5) Å3 (Z = 16). AgSO4 exhibits bulk modulus, B0, of 36.9 GPa, and undergoes sluggish decomposition at ∼23 GPa yielding a high-pressure phase of Ag2S2O7 (K2S2O7-type), with the substrate and product coexisting at 30 GPa. Theoretical calculations within Density Functional Theory for the C2/c cell nicely reproduce the observed trend for lattice constants as well as the B0 values of AgSO4, and suggest that the rigidity of the infinite [Ag(SO4)] chains as well as the Jahn–Teller effect for the Ag(II) cation persist even at 30 GPa.
Introduction
Although the precise mechanism of high-TC superconductivity1 remains a subject of debate,2 it is generally believed that the synergistic phonon (BCS-like)-spin mechanisms contribute to the enhancement of the critical superconducting temperature, TC,3–5 for oxocuprates. The antiferromagnetic interactions between the spin-½ 3d9 Cu(II) cations within [CuO2] sheets in high-TC cuprates can be considered as unusually strong with J as large as −129 meV6 and ordering temperatures reaching 539 K for CaCuO2.7 The recently discovered iron pnictide superconductors also exhibit 2D antiferromagnetism in their undoped state, with large ordering temperatures up to 559 K for K0.8Fe1.6Se2,8 and J up to −50 meV for La(O,F)FeAs.9 Searches for antiferromagnetism with similarly large interactions in magnetic insulators with other 3d spin-½ transition metal ions have not been successful to date; it is therefore instructive to more carefully examine materials possessing 4d electrons.10Strong antiferromagnetic interactions in 4d systems are exemplified by a high-pressure form of CaRu(IV)O311 and by Ag(II)SO4;12 both compounds exhibit 1D metal–ligand chains reminiscent of those found for CuO, which host very strong antiferromagnetic interactions. The latter one, black in colour, is of particular interest due to (i) an unusually strong antiferromagnetic superexchange constant of −18.7 meV with superexchange taking place via a two-atomic O⋯O bridge,13 (ii) the narrow bandgap at the Fermi level of ca. 1 eV,11,14 (iii) the pronounced Ag(d)–O(p) mixing in its electronic structure,11,15 (iv) selective (resonance) enhancement of the Ag–O stretching and O–S–O bending modes in the Raman spectra,16 and (v) anomalies regarding its thermal decomposition, which takes place via electron transfer between the SO42− anion and Ag2+ cation and leads to the evolution of O2.17 Here, guided by a rule of a thumb that the application of external pressure on low-dimensional networks usually leads to increased structural dimensionality, we report the combined X-ray diffraction study of Ag(II)SO4 in a diamond anvil cell (DAC) combined with theoretical DFT calculations at pressures up to 30 GPa. To our best knowledge, this is also the first experimental high-pressure study for any compound of divalent silver in a pressure range exceeding 6 GPa.18
Results and discussion
1. Redetermination of the crystal structure of Ag(II)SO4
In the original study of Ag(II)SO4 we reported a small triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) cell derived from powder X-ray study, with a = 4.6923(1) Å, b = 4.7535(1) Å, c = 8.0125(2) Å, α = 103.403(1)°, β = 76.478(1)°, γ = 118.078(1)°, Z = 2 and V = 151.761(6) Å3.12 This cell has helped us to understand the main features of chemical bonding and rationalize the 1D antiferromagnetic ordering observed experimentally for AgSO4.
cell derived from powder X-ray study, with a = 4.6923(1) Å, b = 4.7535(1) Å, c = 8.0125(2) Å, α = 103.403(1)°, β = 76.478(1)°, γ = 118.078(1)°, Z = 2 and V = 151.761(6) Å3.12 This cell has helped us to understand the main features of chemical bonding and rationalize the 1D antiferromagnetic ordering observed experimentally for AgSO4.
Simultaneously with the experimental study12 the theoretical prediction of the crystal structure of AgSO4 was published.19 That was an unprejudiced ab initio screening of various ABX4 polytypes with the aim to propose the lowest energy structure of AgSO4. In the outcome of the calculations we have suggested a tetragonal I41/a cell with Z = 16, a = 10.56 Å and c = 13.115 Å as the lowest energy polymorph of AgSO4. Our further study has now shown that a small monoclinic distortion of the tetragonal cell leads to an even lower energy C2/c polymorph and provides a better model for structural refinement in comparison with the previous P cell.12 Structure refinement of the monoclinic C2/c cell with Z = 16 using the powder X-ray data (Table 1) yields a more stable and more accurate Rietveld fit than the former triclinic cell, as reflected by the values of Rp = 2.93% and Rwp = 3.84%. The previous refinement for the P cell yielded Rp = 6.12% and Rwp = 6.26%, and required considerable constraints on S–O bond lengths and O–S–O angles.12 The DFT-calculated lattice parameters (a = 12.955 Å; b = 13.787 Å; c = 9.480 Å; β = 48.07°; V = 1259.79 Å3) and bond lengths for the C2/c cell are in excellent agreement with the experimental values; the errors for lattice vectors do not exceed +1.2%, that on volume is +3.8%, while the β angle is overestimated only slightly, by +1.1%.20,21
| Empirical formula | AgO4S |
|---|---|
| M | 203.932 |
| T/K | 293 |
| λ/Å | 1.5418 |
| Crystal system, space group | Monoclinic, C2/c (no. 15) |
| Unit cell dimensions | a = 12.8476(2) Å; b = 13.6690(4) Å; c = 9.36678(19) Å; β = 47.5653(13)° |
| V/Å3 | 1214.04(5) |
| Z/FU | 16 |
The main difference between the P and C2/c cells is in the mutual orientation of the sulfate anions. The C2/c cell contains two crystallographically independent SO4 ions and four different Ag(II) cations as contrasted to one SO4 and one Ag(II) ion present in the smaller P cell (Z = 2). This feature of the C2/c cell permits sulfate anions to be arranged in a more diverse fashion than for the former P cell (Fig. 1).33 However, the topology of the interatomic connections is similar for both cells: (i) each Ag(II) is coordinated by four oxygen atoms from four distinct sulfate anions (adopting a slightly distorted square planar 2 + 2 coordination), (ii) there are no terminal oxygen atoms in the structure, and (iii) in both cases the infinite 1D [Ag(SO4)]∞ chains run along one crystallographic direction (Fig. 1). The presence of these chains allowed us previously to rationalize the 1D antiferromagnetic ordering observed experimentally for AgSO4.
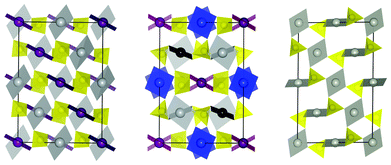 | ||
| Fig. 1 Comparison of AgSO4C2/c (middle) with AgSO4P (left) and high-temperature form of PdSO434C2/c (right). Both, AgSO4 (P) and HT-PdSO4 were transformed to the AgSO4 (C2/c) type cell using matrices: (1,-1,1; 3,1,-1; 1,1,1) and (-1,-2,-1; −1,0,1; -1,1,0), respectively, and projected along c for comparison. The square planar coordination of AgII (grey, purple, blue, black) and PdII (grey) together with SO4 tetrahedra (yellow) is shown. | ||
Interestingly, all these structural features are characteristic also for high temperature (HT) form of PdSO4 (Fig. 1).22 AgSO4 and HT-PdSO4 have an identical heavy atoms sublattice; the main differences between the two are that: (i) the metal–oxygen distances are by ca. 0.1 Å shorter for Pd(II) than for Ag(II), in accordance with the low-spin d8vs. d9 electronic configuration of these cations, and (ii) the arrangement of the sulfate anions is less diverse for the latter compound, at a smaller C2/c cell with Z = 4 for PdSO4 and only one crystallographically independent Pd(II) cation.22 Interestingly, in our theoretical quest for AgSO419 the HT-PdSO4 polytype was the second lowest energy polymorph of AgSO4, placed only 3.7 meV per FU above the I41/a structure. The previously published P cell is in fact the PdSO4-type cell showing a tiny triclinic distortion.
The volume of AgSO4 (C2/c) equals 75.878(3) Å3 per one formula unit (FU) and it is slightly smaller than the respective ‘molecular’ volume of the HT-PdSO4 (79.95 Å3).34 However, it is very close to the volume of the low-temperature form of PdSO4 (75.52 Å3, here, atomic positions were not determined).22 The structural polymorphism of AgSO4 and its relation to that observed for PdSO4 will be discussed in a separate contribution.
The β angle of the C2/c cell of AgSO4 is close to 45°, which suggests that this cell is related to some higher-symmetry cell. Indeed, when the C2/c cell is transformed using [(0 1 0) (1 0 -2) (-1 0 0)] matrix, a quasi-tetragonal cell results with a′ = 13.670 Å, b′ = 13.828 Å, c′ = 12.847 Å, α = 90.852°, β = 90.058(1)°, γ = 89.969°, and Z = 32 (Fig. 2). The cell angles are very close to 90° while a ≈ b < c thus revealing the similarity of the transformed cell to that of the NaCl prototype (Fig. 2). Hence AgSO4 crystallizes in the tetragonally distorted variant of the NaCl-type structure with the infinite [Ag(SO4)]∞ chains propagating along the shortest c′ vector.
![Comparison of the crystal structures of AgSO4 transformed using [(0 1 0) (1 0 -2) (-1 0 0)] matrix in three different projections along a′, b′ and c′ (only Ag and S atoms are showed for clarity) and of the 2 × 2 × 2 supercell of NaCl (in one projection). Ag, Na – gray, S, Cl – yellow balls.](/image/article/2013/CE/c2ce26282g/c2ce26282g-f2.gif) | ||
| Fig. 2 Comparison of the crystal structures of AgSO4 transformed using [(0 1 0) (1 0 -2) (-1 0 0)] matrix in three different projections along a′, b′ and c′ (only Ag and S atoms are showed for clarity) and of the 2 × 2 × 2 supercell of NaCl (in one projection). Ag, Na – gray, S, Cl – yellow balls. | ||
2. High-pressure behaviour and the equation of state of Ag(II)SO4
Application of external pressure usually leads to increased structural (and often electronic) dimensionality. Therefore, we have examined the evolution of the crystal structure of quasi-1D AgSO4 in the DAC at pressures up to 30 GPa using the synchrotron X-ray radiation (Fig. 3). The extreme reactivity of the fine powder of AgSO4 to moisture represents a formidable challenge for high-pressure measurements. Here, only one out of three DAC loadings was successful. In the first two loadings only Ag(I)HSO4 (i.e. the product of reaction of AgSO4 with water vapour) was loaded.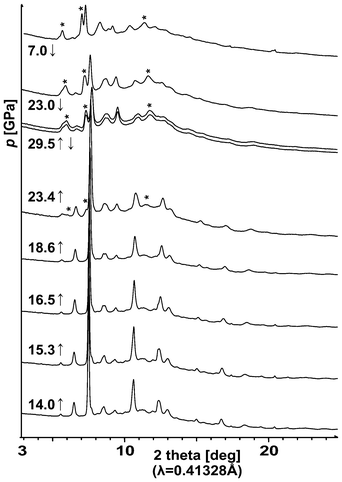 | ||
| Fig. 3 Evolution of the X-ray powder diffraction patterns for AgSO4 on compression (↑) up to 29.5 GPa and then on decompression (↓) to 7 GPa. Starlets indicate the most intense reflections from the high-pressure phase of Ag2S2O7, which is quenchable to 7 GPa (cf. Section 4). | ||
We have been able to index all the measured powder diffraction patterns based on the C2/c cell of AgSO4 and subsequently refine the values of the lattice vectors, while taking into account a preferred orientation of crystallites enclosed within DAC; the obtained values of the unit cell constants for the related NaCl-type unit cell of AgSO4 are shown in Fig. 4.
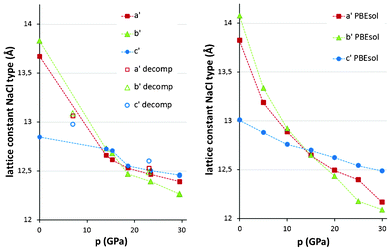 | ||
| Fig. 4 Evolution of the unit cell constants for the NaCl-type unit cell of AgSO4 as a function of external pressures: experiment (left) and theory (right). Full symbols – compression, open symbols – decompression. | ||
The first point on the compression path is at 14 GPa. The quasi-tetragonal AgSO4 turns at this pressure to a quasi-cubic NaCl-type with the lattice constants becoming very similar to each other, while the cell angles do not change markedly from 90°. Further compression to 29.5 GPa leads to a rather uniform decrease of all lattice constants with unit cell angles departing from 90° by no more than ±2°. The lattice constants drop to less than 12.5 Å at 29.5 GPa, corresponding to a small reduction of c′ (direction of propagation of the [AgSO4] infinite chains) from ∼12.85 Å at 1 atm, and a much more pronounced reduction of a′ and b′ from ∼13.67 Å and ∼13.83 Å. This testifies to the rigidity of the 1D [Ag(SO4)] chains and relative compressibility in directions perpendicular to the chains. DFT calculations very nicely reproduce the pressure trend for all lattice constants (Fig. 4).
Three new reflexes may be detected in the X-ray diffraction pattern measured at 23.4 GPa on compression (Fig. 3) which indicates that AgSO4 either decomposes or it undergoes a crystallographic phase transition. The nature of this transformation will be discussed in Section 4.23
It may seem unusual that a soft AgSO4, which contains a large Ag(II) cation, does not exhibit any crystallographic phase transition prior to decomposition, while its lighter congener, the harder CuSO4, which contains a smaller Cu(II) cation, shows a structural phase transition at a pressure as low as 5 GPa.24 The reason for that may be that AgSO4 and CuSO4 are not isostructural at 1 atm. In this aspect, AgSO4 also differs from many other metal sulfates, which exhibit a rich spectrum of structural phase transitions (cf. ESI†) at rather low pressures (p < 10 GPa). The high-pressure behaviour thus adds to other peculiarities of AgSO4 (see Introduction).
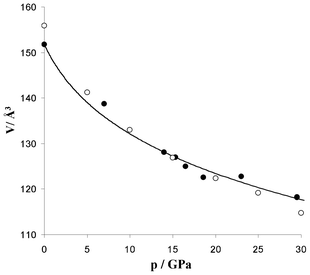 | ||
| Fig. 5 The pressure (p)–volume (V) data for AgSO4 (experiment – filled, theory – empty circles) and the fit of the experimental data to the Birch–Murnaghan EOS (solid line); data both on compression and decompression. V is given per 2 FUs, to compare with the old P cell. | ||
The equation of state (EOS) for AgSO4 (both experimental and theoretical data) and the fit of the Birch–Murnaghan EOS25 to experimental points for the C2/c form is presented in Fig. 4. The volume compressibility of AgSO4 is similar to that of BaSO4 as both compounds show comparable, ca. 20%, volume reductions, at ca. 22 GPa.26 The bulk modulus of AgSO4, B0, estimated from the 3rd order Birch–Murnaghan EOS equals 36.9 GPa; DFT calculations yield a slightly larger value of B0 = 39.5 GPa (B0′ equals 10.6 GPa and 6.2 GPa for experimental and DFT data, respectively, see ESI†).
3. The crystal structure of AgSO4 (C2/c) and Jahn–Teller effect for Ag(2+) at high-pressure: DFT view
Since the quality of the X-ray diffraction data was insufficient for determination of the atomic positions for light atoms (S, O) of the C2/c phase, the discussion of this section will be based on theoretical results. For a proper description of the Jahn–Teller effect it was necessary to include a spin polarization to the DFT calculations. Importantly, the inclusion of spin polarization had a negligible effect on unit cell vectors (<1%). Several magnetic models within the C2/c unit cell were built for which a full structure optimization was performed; they all resulted in the same type and magnitude of the Jahn–Teller effect. Here, results for the lowest energy magnetic models are presented: P and P21/c for 0 GPa and 30 GPa, respectively. The magnetic interactions in AgSO4 are very complex, however, and will be analyzed in detail elsewhere.
At 0 GPa all four crystallographically independent Ag(II) cations exhibit a close to square planar coordination with short Ag–O distances ranging from 2.120 Å to 2.147 Å. When the long apical Ag–O distances are considered, two distinct coordination spheres can be distinguished with half of the Ag(II) cations taking each coordination type (Fig. 6). In one case the Ag(II) cation is axially coordinated with two oxygen atoms (forming a distorted elongated 4 + 2 octahedron with average Ag–Oeq distance equal to 3.135 Å) and in the other case the Ag(II) cation is coordinated with three pairs of oxygen atoms with average Ag–Oeq distances equal to 2.967 Å, 3.164 Å and 3.238 Å, respectively (forming a ten-fold 4 + 6 coordination). The nearly square planar coordination of Ag(2+) is preserved up to 30 GPa except for a small 2 + 2 distortion of the [AgO4] square (here, Ag is in the extended 10-fold coordination). The four shortest Ag–O distances are not very compressible, with the reduction of 2.5%, from 2.136 Å to 2.083 Å (averaged calculated values for all types of Ag atoms, cf.Fig. 6). Simultaneously, the long apical Ag–O distances experience huge reduction by over 0.7 Å (∼32%), from 3.135 Å to 2.368 Å for the first type of Ag center, and by nearly 0.6 Å (∼23%), from 3.126 Å to 2.536 Å (averaged values) for the second type of the Ag center.
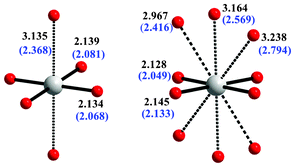 | ||
| Fig. 6 The two distinct Ag(II) coordination spheres present in AgSO4. The average Ag–O distances calculated for AgSO4 at 1 atm and at 30 GPa (blue font, in parentheses) are shown. | ||
The ratios of the apical to horizontal Ag–O bond lengths at 30 GPa are close to 1.14 and 1.24 for the octahedral and 10-fold coordination, respectively, which corresponds to a still substantial elongation of the [AgO6] octahedron.27 This signifies the persistence of the Jahn–Teller effect for the Ag2+ cation at this pressure with important consequences for magnetism: the unpaired electron will reside on the local d(x2 − y2) orbital of Ag rather than on the d(z2) orbital, just like it is observed for AgSO4 at 1 atm. The robustness of the Jahn–Teller effect for Ag(II) in compressed AgSO4 is similar to that of Cu(II) in various compounds at high-pressure.28
4. Pressure-induced decomposition of AgSO4 yielding Ag2S2O7
As mentioned in Section 2, three new large reflexes and a number of smaller ones appear in the X-ray diffraction pattern measured at 23.4 GPa, which indicates that AgSO4 either decomposes or undergoes a crystallographic phase transition. The new phase coexists with the C2/c form up to the highest measured pressure of 29.5 GPa, suggesting that the transformation is sluggish. The new phase is seen also on decompression down to 7 GPa with intensities similar to those at 29.5 GPa, hence it is quenchable.Actually, AgSO4 is a thermodynamically unstable (metastable) compound at ambient conditions, which thermally decomposes via an exothermic reaction at T > 120 °C yielding Ag2S2O7 (Na2S2O7-type structure).17 The equilibrium overpressure of O2 over AgSO4 at 123.5 °C exceeds 6 × 1012 atm29 and it is impressive that this compound is metastable for months at room temperature. Thus, we have examined a possibility that AgSO4 undergoes a pressure-induced decomposition to Ag2S2O7, with ½ O2 either being liberated in this reaction or oxidizing the surface of the platinum gasket at elevated pressure. We have considered both the ambient-pressure Na2S2O7-type structure as well as a more compact K2S2O7-type.17
Guided by these considerations, we have performed a two-phase structural refinement with AgSO4 (C2/c) and the K2S2O7-type Ag2S2O7 (also C2/c, starting at DFT-calculated parameters, cf. ESI†). A nice match between the theoretical and experimental volumes of Ag2S2O7 in the K2S2O7-type was obtained, as illustrated in Fig. 7.
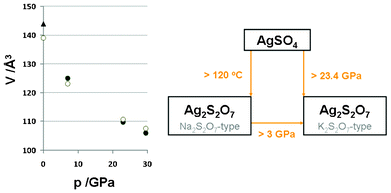 | ||
| Fig. 7 Left: the pressure (p)–volume (V) data points for the high-pressure K2S2O7-type form of Ag2S2O7 (experiment-filled, theory-empty circles); data taken on decompression. V is given per 1 FUs, to compare with the 2 FUs of parent AgSO4 (Fig. 5). The triangle stands for the 1 atm Na2S2O7-type. Right: the diagram of temperature- and pressure-induced phase transformations of AgSO4 and of Ag2S2O7. The value of 3 GPa for a supposed pressure of phase transformation of Ag2S2O7 comes from DFT calculations (an extrapolation using common tangent method), and the quenchable character of the HP-form of Ag2S2O7 down to at least 7 GPa has been confirmed by experiment. | ||
The HP phase has a calculated volume per formula unit, V0, of 138.93 Å3 at 0 GPa; this is ca. 3.5% smaller than the corresponding volume of the ambient-pressure Na2S2O7-type form, 143.948(5) Å3.17 This is consistent with a better packing of the K2S2O7 polytype as compared to the Na2S2O7 one. Fig. 7 summarizes the observed thermal and pressure-induced instability of AgSO4, which decomposes to Ag2S2O7 in either the Na2S2O7-type form (>120 °C), or in the K2S2O7-type form (>∼23 GPa).
Concluding this section we should say that experimental evidence suggests that the application of high pressure exceeding ∼23 GPa triggers the decomposition of metastable AgSO4 and yields the high-pressure form of Ag2S2O7. No phase transitions of AgSO4 precede decomposition.
Conclusions
We have reinvestigated the crystal structure and got insight into high-pressure behaviour of silver(II) sulfate up to 295![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atm. Based on theoretical calculations and the X-ray diffraction data we have been able to propose the new C2/c unit cell for AgSO4 (Z = 16), which more correctly describes this compound at 1 atm than the previously proposed P cell (Z = 2).12 The C2/c unit cell may be transformed to the quasi-tetragonal NaCl-type representation with the infinite [Ag(SO4)] chains running parallel to c′.
000 atm. Based on theoretical calculations and the X-ray diffraction data we have been able to propose the new C2/c unit cell for AgSO4 (Z = 16), which more correctly describes this compound at 1 atm than the previously proposed P cell (Z = 2).12 The C2/c unit cell may be transformed to the quasi-tetragonal NaCl-type representation with the infinite [Ag(SO4)] chains running parallel to c′.
AgSO4 exhibits a rather small bulk modulus, B0, of 36.9 GPa. This compound undergoes a pressure-induced decomposition at pressure above ∼23 GPa yielding the high-pressure phase of Ag2S2O7, which is quenchable to 7 GPa. The theoretical DFT calculations confirm the decomposition scenario. AgSO4 differs from other transition metal sulfates by the fact that they typically require less than 5 GPa for a phase transformation, while AgSO4 does not show any phase transition preceding its decomposition.
Theoretical calculations within Density Functional Theory nicely reproduce the observed lattice parameters, their pressure evolution, unit cell volume, and the value of bulk modulus for AgSO4. DFT results suggest that rigidity of the infinite [Ag(SO4)] chains as well as the Jahn–Teller effect (manifested as elongation of the [AgO6] octahedra) are preserved at 30 GPa; the unpaired electrons reside mostly within the d(x2 − y2) orbitals of Ag.
Experimental procedures and theoretical calculations
All operations with AgSO4 were performed in an Ar-filled glovebox (MBraun). Synthesis of AgSO4 was performed according to the published procedure,12 and the high purity of the specimen was confirmed with X-ray diffraction and Raman spectroscopy.X-ray diffractometry at 1 atm
Our experimental setup has been described elsewhere.12,30 Further details of the crystal structure of AgSO4 at 1 atm may be obtained from Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (Fax: +49 7247-808-666; Email: crysdata@fizkarlsruhe.de) on quoting the ICSD number 424926.High-pressure experiments
A sample of AgSO4 (ca. 50 × 20 × 10 μm) was loaded into the DAC with the culet of 300 μm diameter. Loading of DAC with the AgSO4 powder has been performed using a glovebox. Due to the extreme sensitivity of the material, we decided not to grind the powder manually. Instead, a conglomerate of small grains was picked up with a FEP spatula and loaded as a whole to the high-pressure chamber, the colour of the sample being indicative of its chemical composition (Ag(II)SO4 is black, while Ag(I) salts are colourless).One of the seats was WC and the second cBN to get the maximum range in 2θ, and an opening angle of about 40°. Rhenium foil was used as a gasket material, where inside a hole about 100–120 μm diameter the platinum foil was pre-indented; the inner diameter size of the hole was about 60–80 μm. Neon was used as a pressure medium. X-Ray data were collected at Sector 16BM-D beamline of HPCAT, at the Advanced Photon Source, Argonne National Laboratory, using 0.41328 Å radiation and using the MAR345 image plate. After each pressure increase or decrease the pressure was allowed to relax for at least 15 min. Pressure was measured with platinum as an internal standard, usually before the sample has been measured. Reflections from Pt are also visible in some of sample patterns (see ESI†). The sample to detector distance was ∼300 mm; the measurement time was 120 s per image while at very high pressure and at decompression (twice at 29.5 GPa, at 23 GPa at 7 GPa) the recording time was increased to 300 s per image. The sample to detector distance and detector tilt angle was calibrated with the NIST standard CeO2 powder pattern. The data were integrated azimuthally using FIT2D31 and analysed by CheckCell, Eva and TOPAS.32
The measured XRD patterns do not correspond to a powder sample with a perfectly random orientation of crystallites, but rather to a conglomerate of very small single-crystalline grains. Initial indexing of the diffraction patterns was performed for simplicity using the small P unit cell (this triclinic cell has identical Ag sublattice as the C2/c cell) with TOPAS, followed by Pawley and eventually structure refinement of the lattice vectors (see ESI† for details). The lattice vectors were then matrix-transformed to those corresponding to the C2/c unit cell and to the NaCl-type representation (ESI†). The preferred orientation was applied and it was found to be crucial for a correct description of the powder patterns.
DFT calculations
The periodic DFT calculations were done using the Vienna ab initio simulation package (VASP)33 within generalized gradient approximation (GGA),34 PBEsol exchange-correlation functional revised for solids35 and projector-augmented wave method (PAW).36 Parameters for full geometry optimization were: SCF convergence criterion 10−7 eV, ionic convergence 10−5 eV, kpoint spacing of 0.3 Å−1 (using the Monkhorst–Pack scheme), valence electrons were described by plane waves with a kinetic energy cutoff of 600 eV. For the spin-polarized calculations GGA(PBEsol) + U method was used with U(Ag4d) = U(O2p) = 4 eV, U(S3p) = 2 eV, and J(all atoms) = 1 eV. The calculations for 12 compact polymorphs19 of AgSO4 were performed at 0 GPa, 10 GPa, and their results extrapolated to 25 GPa using a linear tangent method, indicating that any pressure-induced phase transitions should not occur below 30 GPa. We found that taking magnetic interactions into account was important for ranking of structures in enthalpy.Acknowledgements
The project ‘Quest for superconductivity in crystal-engineered higher fluorides of silver’ is operated within the Foundation for Polish Science ‘TEAM’ Programme co-financed by the EU European Regional Development Fund. WG also acknowledges support from the NCN project AgCENT (2011/01/B/ST5/06673). Portions of this work were performed at HPCAT (Sector 16), Advanced Photon Source (APS), Argonne National Laboratory. HPCAT is supported by CIW, CDAC, UNLV and LLNL through funding from DOE-NNSA, DOE-BES and NSF. APS is supported by DOE-BES, under Contract No. DE-AC02-06CH11357. V. V. S. acknowledges financial support from the Department of Energy under grant # DEFG02-02ER45955. This work was partly supported by the Slovenian Research Agency (ARRS) within the research program P1-0045 Inorganic Chemistry and Technology. DFT calculations were performed at ICM supercomputers within grant G34-10. Kind assistance of beam scientist, Dr. Daijo Ikuta, is gratefully acknowledged.References
- G. M. Bednorz and A. K. Müller, Z. Phys. B: Condens. Matter, 1986, 64, 189 CAS.
- (a) P. W. Anderson, P. A. Lee, M. Randeria, T. M. Rice, N. Trivedi and F. C. Zhang, J. Phys.: Condens. Matter, 2004, 16, R755 Search PubMed; (b) P. A. Lee, N. Nagaosa and X. G. Wen, Rev. Mod. Phys., 2006, 78, 17 CrossRef CAS; (c) Q. J. Chen, J. Stajic, S. Tan and K. Levin, Phys. Rev., 2005, 412, 1 Search PubMed.
- (a) A. Lanzara, P. V. Bogdanov, X. J. Zhou, S. A. Kellar, D. L. Feng, E. D. Lu, T. Yoshida, H. Eisaki, A. Fujimori, K. Kishio, J. I. Shimoyama, T. Noda, S. Uchida, Z. Hussain and Z. X. Shen, Nature, 2001, 412, 510 CrossRef; (b) J. Lee, K. Fujita, K. McElroy, J. A. Slezak, M. Wang, Y. Aiura, H. Bando, M. Ishikado, T. Masui, J. X. Zhu, A. V. Balatsky, H. Eisaki, S. Uchida and J. C. Davis, Nature, 2006, 442, 546 Search PubMed.
- (a) H. Krakauer, W. E. Pickett and R. E. Cohen, Phys. Rev. B: Condens. Matter, 1993, 47, 1002 Search PubMed; (b) B. Batlogg, R. J. Cava, A. Jayaraman, R. B. Vandover, G. A. Kourouklis, S. Sunshine, D. W. Murphy, L. W. Rupp, H. S. Chen, A. White, K. T. Short, A. M. Mujsce and E. A. Rietman, Phys. Rev. Lett., 1987, 58, 2333 CrossRef CAS; (c) X. J. Chen, V. V. Struzhkin, Z. G. Wu, H. Q. Lin, R. J. Hemley and H. K. Mao, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3732 CrossRef CAS.
- (a) P. Monthoux, D. Pines and G. G. Lonzarich, Nature, 2007, 450, 1177 CrossRef CAS; (b) N. D. Mathur, F. M. Grosche, S. R. Julian, I. R. Walker, D. M. Freye, R. K. W. Haselwimmer and G. G. Lonzarich, Nature, 1998, 394, 39 CrossRef CAS; (c) E. Manousakis, Rev. Mod. Phys., 1991, 63, 1 CrossRef CAS; (d) J. P. Carbotte, E. Schachinger and D. N. Basov, Nature, 1999, 401, 354 Search PubMed.
- (a) D. Vaknin, E. Caignol, P. K. Davies, J. E. Fischer, D. C. Johnston and D. P. Goshorn, Phys. Rev. B, 1989, 39, 9122 CrossRef CAS; (b) Y. Tokura, S. Koshihara, T. Arima, H. Takagi, S. Ishibashi, T. Ido and S. Uchida, Phys. Rev. B: Condens. Matter, 1990, 41, 11657 CrossRef CAS.
- A. Lombardi, M. Mali, J. Roos, D. Brinkmann and I. Mangelschots, Phys. Rev. B: Condens. Matter, 1996, 54, 93 Search PubMed.
- B. Wei, H. Qing-Zhen, C. Gen-Fu, M. A. Green, W. Du-Ming, H. Jun-Bao and Q. Yi-Ming, Chin. Phys. Lett., 2011, 28, 086104 Search PubMed.
- Z. -Y. Lu, F. Ma and T. Xiang, J. Phys. Chem. Solids, 2011, 72, 319 Search PubMed.
- (a) W. Grochala and R. Hoffmann, Angew. Chem., Int. Ed., 2001, 40, 2742 CrossRef CAS; (b) W. Grochala, J. Mater. Chem., 2009, 19, 6949 RSC; (c) W. Grochala, A. Porch and P. P. Edwards, Solid State Commun., 2004, 130, 137 CrossRef CAS.
- Y. Shirako, H. Satsukawa, X. X. Wang, J. J. Li, Y. F. Guo, M. Arai, K. Yamaura, M. Yoshida, H. Kojitani, T. Katsumata, Y. Inaguma, K. Hiraki, T. Takahashi and M. Akaogi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 174411 Search PubMed.
- P. J. Malinowski, M. Derzsi, Z. Mazej, Z. Jagličić, B. Gaweł, W. Łasocha and W. Grochala, Angew. Chem., Int. Ed., 2010, 49, 1683 CAS.
- Note that the value of J divided by 2 was erroneously given in the previous paper (ref. 12). Cf. also: X. Zhang, T. Jia, T. Liu, Z. Zeng and H. Q. Lin, J. Appl. Phys., 2012, 111, 07E136 Search PubMed.
- The value of activation energy for electronic conductivity of Ag(II)SO4 equals ∼1.0 eV at −50 °C and ∼1.25 eV at +30 °C: R. Jurczakowski, P. J. Malinowski, Z. Mazej and W. Grochala, unpublished impedance spectroscopy data.
- M. Derzsi, J. Stasiewicz and W. Grochala, J. Mol. Model., 2011, 17, 2259 CrossRef.
- M. Derzsi, P. J. Malinowski, Z. Mazej and W. Grochala, Vib. Spectrosc., 2011, 57, 334 CrossRef CAS.
- P. J. Malinowski, M. Derzsi, A. Budzianowski, P. J. Leszczyński, B. Gaweł, Z. Mazej and W. Grochala, Chem.–Eur. J., 2011, 17, 10523 Search PubMed.
- (a) See high-pressure study of AgF2: B. G. Müller, Nature, 1979, 66, 519 Search PubMed; (b) Conclusions from this study severely contradict theoretical calculations unless substantial disorder of fluorine sublattice is considered: J. Romiszewski, W. Grochala and L. Z. Stolarczyk, J. Phys.: Condens. Matter, 2007, 19, 116206 Search PubMed; (c) T. Jaroń and W. Grochala, Phys. Status Solidi RRL, 2008, 2, 71 Search PubMed.
- M. Derzsi, K. Dymkowski and W. Grochala, Inorg. Chem., 2010, 49, 2735 CrossRef CAS.
- Results obtained with the improved PBE functional for solids (PBEsol): J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 Search PubMed.
- The former P cell may be transformed to the new monoclinic cell using the transformation matrix [(1 -1 1) (3 1 -1) (1 1 1)], while preserving the positions of Ag atoms, however, positions of sulfate anions in the transformed cell and in the experimental C2/c cell differ to some extent.
- High-temperature C2/c form, possibly metastable at ambient conditions, is called M-form in the original paper, while the low-temperature form is denoted as N-form: T. Dahmen, P. Rittner, S. Böger-Seidl and R. Gruehn, J. Alloys Compd., 1994, 216, 11 Search PubMed.
- The analysis based on intensity of reflexes is difficult because of the possibility of (varying) preferred orientation of the small sample enclosed within DAC.
- C. W. F. T. Pistorius, Z. Kristallogr., 1961, 116, 220 Search PubMed.
- (a) F. D. Murnaghan, Proc. Natl. Acad. Sci. U. S. A., 1944, 30, 244; (b) F. Birch, Phys. Rev., 1947, 71, 809 CrossRef CAS.
- W. Crichtons, M. Merlini, M. Hanfland and H. Müller, Am. Mineral., 2011, 96, 364 Search PubMed.
- Compare with the typical values for the [AgF6] octahedra at 1 atm: W. Grochala, Phys. Status Solidi B, 2006, 243, R81 Search PubMed.
- F. Aguado and F. Rodríguez, High Pressure Res., 2006, 26, 319 Search PubMed.
- As calculated from an equilibrium constant of 2.5 × 109, which was derived from the enthalpy of decomposition (−30.7 kJ mol−1 at 1 K min−1) and entropy term of gaseous ½ O2(g) at 123.5 K (−40.7 kJ mol−1).
- Z. Mazej, E. Goreshnik, Z. Jagličić, B. Gaweł, W. Łasocha, D. Grzybowska, T. Jaroń, D. Kurzydłowski, P. Malinowski, W. Koźminski, J. Szydłowska, P. Leszczyński and W. Grochala, CrystEngComm, 2009, 11, 1702 RSC.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14, 235 CrossRef.
- DIFFRAC.EVA v.1.2, Software for phase identification and quantitative phase analysis, Bruker AXS, 2011 Karlsruhe, Germany, CheckCell – http://www.inpg.fr/LMGP, TOPAS v 4.2, Total Pattern Analysis Solution Software, Bruker AXS, 2009, Karlsruhe, Germany.
- (a) G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS; (b) G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS; (c) G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef.
- P. E. Blöch, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
Footnotes |
| † This work is dedicated to Prof. Ho–Kwang (Dave) Mao at his 70th birthday and in recognition of his outstanding contribution to high pressure sciences. |
| ‡ Electronic supplementary information (ESI) available: Pressure evolution of X-ray diffraction patterns, refined lattice vectors, database of structural phase transitions for sulfates, and results of DFT calculations. See DOI: 10.1039/c2ce26282g |
| This journal is © The Royal Society of Chemistry 2013 |