Triflate anion and ligand influences in silver(I) coordination polymers of four isomeric dipyridyl ketone oximes†
Victoria J.
Argyle
,
Laura M.
Woods
,
Marina
Roxburgh
and
Lyall R.
Hanton
*
Department of Chemistry, Te Tari Hua-Ruanuku, University of Otago, P. O. Box 56, Dunedin, 9054, New Zealand. E-mail: lhanton@chemistry.otago.ac.nz; Fax: +64-3-479-7906; Tel: +64-3-479-7918
First published on 15th October 2012
Abstract
The synthesis and X-ray characterisation of four isomeric dipyridyl ketone oxime ligands, 4,4′-dipyridyl ketone oxime, L44, 3,3′-dipyridyl ketone oxime, L33, 2,4-dipyridyl ketone oxime, L24 and 2,3-dipyridyl ketone oxime, L23 is described. X-ray structural characterisation of the precursor ketones 3,3′-dipyridyl ketone, L1, and 2,4-dipyridyl ketone, L2, is also reported for comparison. Four AgCF3SO3 complexes of the dipyridyl ketone oxime ligands were prepared and structurally characterised, [Ag(L44)2](CF3SO3), 1, {[AgL33](CF3SO3)}∞, 2, [AgL24(CF3SO3)]∞, 3, and [Ag2L23(CF3SO3)2]∞, 4. The structural role of the CF3SO3− anion in the formation of Ag(I) coordination polymer networks, together with the effect of the varying pyridyl substitution patterns of L44, L33, L24 and L23 was investigated. In 1 the CF3SO3− anion was unbound and disordered. It was encapsulated in the channels of a honeycomb 3D H-bonded structure. In 2 the CF3SO3− anion remained unbound, being encapsulated within the centre of a helical polymer chain. In 3 the CF3SO3− anion was weakly bound to the Ag(I) ion and as such decorated the side of a bowed 1-D chain. The two different CF3SO3− anions in 4 adopted a range of binding modes from monodentate, bis-monodentate to tris-monodentate with the Ag(I) ions and gave rise to the formation of an unusual CF3SO3− bridged 2-D network.
Introduction
The chemistry of extended network structures and coordination polymers is an extremely topical and active area of research.1–4 For example, the area of 1D coordination polymers has been thoroughly reviewed recently.5 Judicious choice of organic-linker ligands and transition metal ion nodes has allowed the engineering of a wide variety of coordination networks in recent years.6–10 Thus the main focus has largely been on the binding arrangements of the bridging ligand entities and their interactions with the metal ions. However, the balance between the formations of different coordination networks is often subtle, such that metal-to-ligand ratio,11–13 anion-based interactions11,12,14–24 and solvent effects11,12,14,16,17,19,20,22 can all have an influence on the final structural outcomes. While not unimportant the structural role of a specific counter anion is often of secondary consideration as many studies have probed the comparative effects of a range of different counterions.20,25–32 In part this is due to the fact that many anions are deliberately selected for their “non-coordinating” ability and it is hoped that their necessary incorporation into a coordination complex will have limited consequences for the final array.33 Thus understanding the role played by a specific counterion, especially one which has varying binding abilities and differing binding modes, in concert with subtle changes in the binding arrangements of the ligand entities is of importance in gaining an insight into the formation of network arrays.34,35There have been numerous studies of the coordinating ability of polyatomic monoanions.26,27,36–38 While the results differ slightly depending on the systems in question the following generalisation can be made, that NO3− and CF3CO2− are coordinating anions while ClO4−, BF4− and PF6− are non-coordinating anions. The coordinating ability of the CF3SO3− anion is ambiguous as it appears to sit somewhere in the middle of the series. This gives rise to its varying coordination abilities39–42 and hence its interest in this study. Ag(I) is an ideal metal ion in this context and Ag-coordination polymers are prominent in early investigations of coordination polymers.43–46 A key property is that the Ag(I) ion's d10 configuration imposes few structural requirements on the surrounding ligand and anion entities. A search of the Cambridge Structural Database (CSD) (version 5.33),47,48 provided details of typical Ag(I) cation and CF3SO3− anion interactions. Table 1 displays the mean values and range of bond distances for different types of Ag(I)–OSO2CF3 interactions. From these results it can be concluded that while monodentate bonding interactions between Ag(I) cations and CF3SO3− anions were relatively common, bridging through a single O atom was relatively rare.49 Based on the results in Table 1 a convenient and slightly generous cut-off limit for Ag–O bonding interactions would seem to be 2.7 Å (vide infra).
| Bond/interaction types | Complexes | Observations | Mean/Å | Range/Å |
|---|---|---|---|---|
| a Bonding arrangement involves two different monodentate O donors from the same CF3SO3−. b Bonding arrangement involves three different monodentate O donors from the same CF3SO3−. c Bonding arrangement involves two different O donors from the same CF3SO3−. | ||||
| Ag(I)–O | 230 | 303 | 2.482 | 2.140–2.960 |
| 2 × Ag(I)–Oa | 77 | 186 | 2.457 | 2.204–2.917 |
| 3 × Ag(I)–Ob | 8 | 30 | 2.438 | 2.207–2.630 |
| Ag(I)–O–Ag(I) | 13 | 40 | 2.444 | 2.251–2.799 |
| Ag(I)–O–Ag(I) and Ag(I)–Oc | 6 | 21 | 2.482 | 2.317–2.629 |
In our study of the role of the CF3SO3− anion in association with the subtle changes that different binding arrangements of ligand entities bring about, we chose to investigate the reaction of AgCF3SO3 with four isomeric dipyridyl ketone oxime ligands, namely 4,4′-, 3,3′-, 2,4- and 2,3-dipyridyl ketone oxime (L44, L33, L24, and L23, respectively). These ligands consisted of two pyridyl rings of varying substitution, bound to a central oxime {C=N–OH} moiety and are structural isomers of the commercially available 2,2′-dipyridyl ketone oxime. While this latter ligand has a rich coordination chemistry with a variety of metal ions, including Ag(I),50 the metal chemistry of its isomers is non-existent.
The oxime functional group is a very versatile group capable of binding to metal ions as well as forming a variety of H-bonding interactions, which may be useful in extending coordination polymer arrays.50 Similarly, the pyridyl rings are capable of binding to metal ions and also participating in a variety of H-bonding interactions, for example (pyridine)N⋯H–O(oxime). Within the structure of each ligand there is a degree of conformational freedom about the Cpy–Cox (py = pyridine; ox = oxime) bond. This feature allows the formation of two-bladed chiral propellers in the solid state, whereby each ring is twisted relative to each other by as much as 90°.51 As such, chirality can, in principle, be introduced into the resulting complexes. In the case of both the 2,3- and 2,4-diypridyl oxime, this degree of conformational freedom can be somewhat constrained by the formation of a five-membered chelate ring between the 2-substituted pyridyl ring and the N atom of the oxime moiety. However, the formation of a chelate ring provides a strong anchor from which to build extended arrays and provides an elegant example of the effect of varying substitution patterns within a ligand series.
Herein we report the synthesis and characterisation of four isomeric dipyridyl ketone oxime ligands L44, L33, L24 and L23 and their AgCF3SO3 coordination polymers. X-ray structural analysis showed the differing role played by the CF3SO3− anion in these coordination polymers. In [Ag(L44)2](CF3SO3), 1 the CF3SO3− anion was unbound and disordered. It was encapsulated in the channels of a honeycomb 3D H-bonded network. In {[AgL33](CF3SO3)}∞, 2 the CF3SO3− anion was unbound and encapsulated within a helical polymer chain while in [AgL24(CF3SO3)]∞, 3 the CF3SO3− anion was weakly bound to the Ag(I) ion and decorated the side of an L-shaped 1-D chain. The complex [Ag2L23(CF3SO3)2]∞, 4 formed an unusual CF3SO3− bridged 2-D network.
Experimental section
General
Commercially available 4,4′-dipyridyl ketone was acquired from Chem Bridge and all other precursors were obtained from Sigma Aldrich. Methyl nicotinate was dried by azeotropic distillation using toluene. Methyl isonicotinate was dried by vacuum distillation at 92 °C and 30 mm Hg. Diethyl ether and THF were obtained from a Pure Solv MD-6 solvent purification system. All other reagents were used without purification. Column chromatography was performed using silica gel. Elemental microanalyses were carried out at the Campbell Microanalytical Laboratory, University of Otago. All 1H, 13C and two-dimensional NMR spectra were measured on a 500 MHz Varian UNITYINOVA spectrometer at 298 K. NMR spectra were collected in DMSO and referenced to the solvent signal. High resolution mass spectrometry was recorded on a Bruker micrOTOF-Q spectrometer. Data were presented as m/z values for the parent molecular ion. Infrared (IR) spectra were measured with either a Perkin Elmer Spectrum BX FT-IR System using KBr disks or a Bruker Optics Alpha FT-IR spectrometer (with a diamond Attenuated Total Reflectance (ATR) top-plate).Syntheses of ligands
The procedure for all four ligands L44, L33, L24 and L23 was modified from that described by Blicke and Maxwell.524,4′-Dipyridyl ketone oxime, L44
Hydroxylamine hydrochloride (475 mg, 6.83 mmol) dissolved in water (10 mL) was added to 4,4′-dipyridyl ketone (311 mg, 1.69 mmol) dissolved in ethanol (5 mL). NaOH (276 mg, 6.91 mmol) was slowly added to the solution with vigorous stirring at 0 °C. After 10 min the solution was warmed to room temperature and heated at reflux overnight. The solution was cooled to room temperature, filtered and washed with water to give the product as a white solid (306 mg, 91%). Anal. found: C, 66.39; H, 4.71; N, 20.24. Calc. for C11H9N3O: C, 66.32; H, 4.55; N, 21.09. 1H NMR (500 MHz, DMSO): δ 12.21 (s, 1H, OH), 8.72 (dd, J = 2.0, 4.5 Hz, 2H, H2/6 or H9/13), 8.59 (dd, J = 2.0, 4.5 Hz, 2H, H2/6 or H9/13), 7.34 (ddd, J = 2.0, 4.5, 4.5, 4H, H3, H5, H10, H12). 13C NMR (500 MHz, DMSO): δ 151.667 (C7), 150.077 (C6/2 or C9/13) 149.940 (C6/2 or C9/13), 142.314 (C4 or 11), 139.852 (C4 or C11), 123.566 (C3/5 or C10/12), 120.762 (C3/5 or C10/12). ESMS (+ve, ESI) m/z calc. for C11H10N3O 200 [M + H+], found 200. Selected IR/cm−1 1847 (m, br), 1596 (m, sh), 1410 (m, sh), 1011 (s, sh). Colourless crystals of L44·2H2O suitable for X-ray determination were grown by slow evaporation a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of H2O:MeOH.
:1 solution of H2O:MeOH.
3,3′-Dipyridyl ketone oxime, L33
The precursor 3,3′-dipyridyl ketone L1 was prepared by the literature procedure.53 Colourless crystals suitable for X-ray determination were grown by slow evaporation of a MeOH solution of the ketone L1. Hydroxylamine hydrochloride (550 mg, 7.91 mmol) dissolved in water (20 mL) was added to L1 (364 mg, 1.98 mmol) dissolved in EtOH (5 mL). NaOH (324 mg, 8.10 mmol) was added slowly to the solution with vigorous stirring at 0 °C. The solution was warmed to room temperature and heated at reflux overnight. The reaction was allowed to cool to room temperature, diluted with water and extracted with DCM. The organic extracts were combined and dried over anhydrous Na2SO4 and concentrated in vacuo. The product was purified by column chromatography [ethyl acetate (EA)/triethylamine (TEA) 5%] to give the oxime as a white solid (388 mg, 98%). Anal. found: C, 66.22; H, 4.57; N, 20.92. Calc. for C11H9N3O: C, 66.32; H, 4.55; N, 21.09. 1H NMR (500 MHz, DMSO): δ 11.92 (s, 1H, OH), 8.64 (dd, J = 1.7, 4.9 Hz, 1H, H6), 8.60 (dd, J = 0.8, 2.3 Hz, 1H, H9), 8.59 (dd, J = 1.6, 4.8 Hz, 1H, H13), 8.56 (dd, J = 0.9, 2.2 Hz, 1H, H2), 7.79 (dt, J = 2.0, 2.0, 8.0 Hz, 1H, H4), 7.74 (ddd, J = 1.7, 2.2, 8.0 Hz, 1H, H11), 7.52 (ddd, J = 0.9, 4.9, 7.8 Hz, 1H, H5), 7.43 (ddd, J = 0.9, 4.8, 8.0 Hz, 1H, H12). 13C NMR (DMSO): δ 150.749 (C7), 149.904 (C13), 149.693 (C6), 149.394 (C2), 147.640 (C9), 136.737 (C4), 134.330 (C11), 131.966 (C10), 128.369 (C3), 123.611 (C5), 123.537 (C12). HRMS (+ve, ESI) m/z calc. for C11H10N3O 200.2199 [M + H+], found 200.0818. Selected IR (KBr)/cm−1: 3076 (m, sh), 3030 (m, sh), 2983 (m, sh), 2755 (s, sh), 1615 (w, sh), 1595 (m, sh), 1481 (m, sh), 1455 (m, sh), 1417 (m, sh), 1401 (m, sh), 1315 (w, sh), 955 (m, sh), 938 (s, sh), 929 (m, sh), 835 (m, sh), 816 (m, sh), 754 (w, sh), 707 (s, sh). UV-Vis (MeOH)/cm−1 (ε/L mol−1 cm−1): 45 045 (13 846), 40 000 (12 939), 6109 (256), 5194 (118). Colourless X-ray quality crystals of two different morphologies were isolated. Crystals of L33 were obtained by cooling of a 5:1 H2O:MeOH solution and crystals of L33·2H2O were obtained by the slow diffusion of MeCN into a MeOH solution of L33 through a layer of EtOH.
2,4-Dipyridyl ketone oxime, L24
The precursor 2,4-dipyridyl ketone L2 was prepared by the literature procedure.54 Colourless crystals suitable for X-ray determination were grown by slow evaporation of a CDCl3 solution of the ketone L2. Hydroxylamine hydrochloride (357 mg, 5.13 mmol) dissolved in water (16 mL) was added to L2 (236 mg, 1.28 mmol) dissolved in EtOH (5 mL). NaOH (210 mg, 5.26 mmol) was added slowly to the solution with vigorous stirring at 0 °C. The solution was warmed to room temperature and heated at reflux overnight. The reaction was cooled to room temperature, filtered and washed with water to give the isolated isomer A (46.0 mg, 18%). The filtrate was diluted with water and extracted with DCM. The organic extracts were combined and dried over anhydrous Na2SO4 and concentrated in vacuo. The product was purified by column chromatography (EA/TEA 5%) to give a mixture of isomers of the oxime as a cream solid (205 mg, 80%). Anal. found: C, 66.35; H, 4.86; N, 20.93. Calc. for C11H9N3O: C, 66.32; H, 4.55; N, 21.09. 1H NMR (500 MHz, DMSO): single isomer A δ 11.98 (s, 1H, OH), 8.64 (dd, J = 1.6, 4.4 Hz, 2H, H9, H13), 8.47 (dd, J = 1.6, 4.4 Hz, 1H, H6), 7.95 (dt, J = 1.1, 1.1, 7.9 Hz, 1H, H3), 7.87 (td, J = 1.7, 7.8, 7.8 Hz, 1H, H4), 7.38 (ddd, J = 1.2, 4.8, 7.6 Hz, 1H, H5), 7.30 (dd, 1.7, 4.4 Hz, 2H, H10, H12). 13C NMR (500 MHz, DMSO): single isomer A δ 153.789 (C7), 153.709 (C2), 149.156 (C9, C13), 148.795 (C6), 140.808 (C11), 136.826 (C4), 124.084 (C10, C12), 123.825 (C5), 120.962 (C3). 1H NMR (500 MHz, DMSO): mixture: δ 12.07 (s, 1H, OH-b), 11.97 (s, 1H, OH-a), 8.68 (ddd, J = 1.0, 1.5, 5 Hz, 1H, H6b), 8.63 (dd, J = 2.0, 4.5 Hz, 1H, H9, H9a, H13a), 8.55 (dd, J = 1.5, 4.5 Hz, 2H, H9b, H13b), 8.47 (ddd, J = 1.0, 1.5, 5.0 Hz, 0.5H, H6a), 7.94 (m, 1.5H, H3a, H4b), 7.87 (m, 0.5H, H4a), 7.63 (dt, J = 1.0, 7.5 Hz, 1H, H3b), 7.47 (ddd, J = 1.0, 5.0, 7.5 Hz, 1H, H5b), 7.39 (ddd, J = 1.5, 5.0, 7.5 Hz, 0.5H, H5a), 7.31 (m, 2H, H10b, H12b), 7.29 (m, 1H, H10a, H12a). 13C NMR (500 MHz, DMSO): mixture δ 153.749 (C7a), 153.699 (C2a), 152.830 (C7b), 150.867 (C2b), 149.768 (C9b, C13b), 149.497 (C6b), 149.136 (C9a, C13a), 148.776 (C6a), 142.980 (C11b), 140.779 (C11a), 136.821 (C4a), 136.468 (C4b), 125.630 (C3b), 124.057 (C10a, C12a), 123.860 (C5b), 123.815 (C5a), 121.082 (C10b, C12b), 120.940 (C3a). HRMS (+ve, ESI) m/z calc. for C11H10N3O 200.2199 [M + H+], found 200.0818. Selected IR (KBr)/cm−1: 3421 (m, br), 3085 (m, sh), 2999 (s, sh), 1604 (s, sh), 1583 (m, sh), 1563 (m, sh), 1550 (w, sh), 1334 (m, sh), 1007 (s, sh), 993 (s, sh), 958 (s, sh), 836 (m, sh), 790 (m, sh), 741 (w, sh), 684 (s, sh). UV-Vis (MeOH)/cm−1 (ε/L mol−1 cm−1): 43 101 (12 371), 37 594 (11 534), 6365 (179), 6305 (219), 5845 (179). Colourless crystals suitable for X-ray determination were grown by slow evaporation of a H2O:MeOH (1:1) solution of the isolated isomer A.
2,3-Dipyridyl ketone oxime, L23
Hydroxylamine hydrochloride (214 mg, 3.08 mmol) dissolved in water (10 mL) was added to 2,3-dipyridyl ketone (142 mg, 0.770 mmol) dissolved in EtOH (5 mL). NaOH (126 mg, 3.16 mmol) was added slowly to the solution with vigorous stirring at 0 °C. The solution was warmed to room temperature and heated at reflux overnight. The reaction was cooled to room temperature, filtered and washed with water to give the isolated isomer A (14.0 mg, 9%). The filtrate was diluted with water and extracted with DCM. The organic extracts were combined and dried over anhydrous Na2SO4 and concentrated in vacuo. The product was purified by column chromatography (EA/TEA 5%) to give a mixture of the oxime isomers as a cream solid (123 mg, 80%). Anal. found: C, 66.54; H, 4.72; N, 20.84. Calc. for C11H9N3O: C, 66.32; H, 4.55; N, 21.09. 1H NMR (500 MHz, DMSO): single isomer A δ 11.94 (s, 1H, OH), 8.56 (dd, J = 1.7, 4.9 Hz, 1H, H13), 8.51 (dd, J = 0.9, 2.2 Hz, 1H, H9), 8.48 (ddd, J = 0.9, 1.8, 4.8 Hz, 1H, H6), 7.97 (dt, J = 1.1, 1.1, 8.0 Hz, 1H, H3), 7.87 (td, J = 1.5, 8.0 Hz, 1H, H4), 7.75 (ddd, J = 2.9, 4.9, 8.0 Hz, 1H, H11), 7.46 (ddd, J = 0.9, 4.9, 7.8 Hz, 1H, H12), 7.39 (ddd, J = 1.1, 4.8, 7.5 Hz, 1H, H5). 13C NMR (500 MHz, DMSO): single isomer A δ 154.132 (C2), 153.195 (C7), 149.678 (C9), 148.859 (C13), 148.751 (C6), 137.098 (C11), 136.804 (C4), 128.643 (C10), 123.763 (C5), 122.954 (C12), 121.077 (C3). 1H NMR (500 MHz, DMSO): mixture: δ 11.93 (s, 0.5H, OH-a), 11.87 (s, 1.0H, OH-b), 8.67 (ddd, J = 1.0, 2.0, 5.0 Hz, 1.0H, H6b), 8.55 (m, 2.5H, H13a, H9b, H13b), 8.49 (m, 1.0H, H6a, H9a), 7.94 (m, 1.5H, H3a, H4b), 7.88 (ddd, J = 1.5, 7.5, 7.5 Hz, 0.5H, H4a), 7.74 (m, 1.5H, H11a, H11b), 7.69 (dt, J = 1.0, 1.0, 8.0 Hz, 1H, H3b), 7.46 (m, 1.5H, H12a, H12b), 7.39 (m, 1.5H, H5a, H5b). 13C NMR (500 MHz, DMSO): mixture δ 154.11 (C2a), 153.172 (C7a), 152.436 (C2b), 151.089 (C7b), 149.650 (C9a), 149.505 (C6b), 149.447 (C13b or 9b), 148.842 (C13a), 148.732 (C6a), 147.742 (C9b or 13b), 137.065 (C11a), 136.790 (C4a), 136.404 (C4b), 134.308 (C11b), 131.824 (C10b), 128.618 (C10a), 125.782 (C3b), 123.821 (C12b), 123.748 (C5a), 123.389 (C5b), 123.935 (C12a), 121.059 (C3a). HRMS (+ve, ESI) m/z calc. for C11H10N3O 200.2199 [M + H+], found 200.0841. Selected IR (KBr)/cm−1: 3159 (s, br), 3049 (s, sh), 1636 (w, sh), 1589 (s, sh), 1498 (s, sh), 1472 (s, sh), 1415 (s, sh), 1311 (w, sh), 998 (s, sh), 963 (m, sh), 941 (s, sh), 812 (s, sh), 790 (m, sh), 743 (m, sh). UV-Vis (MeOH)/cm−1 (ε/L mol−1 cm−1): 44 444 (13 829), 38 168 (11 895), 7037 (222), 5147 (191), 5045 (213). Colourless crystals suitable for X-ray determination were grown by slow evaporation of a MeOH solution of the isolated isomer A.Syntheses of complexes
:1). The solution was allowed to evaporate to give colourless X-ray quality crystals (5 mg, 32.7%). Anal. found: C, 42.35; H, 3.01; N, 12.52; F, 8.73. Calc. for C23H18N6O5AgF3S: C, 42.15; H, 2.77; N, 12.82; F, 8.70. Selected IR (ATR)/cm−1: 3439 (w, br) (L44), 3051–2870 (w, sh) (L44), 1600 (w, sh) (L44), 1424 (w, sh) (L44), 1239 (s, sh) (CF3SO3−), 1220 (vs, sh) (CF3SO3−), 1166 (s, sh) (CF3SO3−), 1021 (vs, sh) (CF3SO3−), 955 (m, sh) (CF3SO3−), 835 (m, sh) (CF3SO3−), 672 (s, sh) (CF3SO3−).
X-ray crystallography
X-ray diffraction data were collected on a Bruker APEX II CCD diffractometer, with graphite monochromated Mo Kα (λ = 0.71073 Å) radiation. Intensities were corrected for Lorentz polarisation effects55,56 and a multiscan absorption correction57 was applied. The structures were solved by direct methods (SHELXS58 or SIR-9759) and refined on F2 using all data by full-matrix least-squares procedures (SHELXL 9760). Non-hydrogen atoms were refined with anisotropic thermal parameters. H atoms were placed in ideal positions except for the H atoms of H2O in L44·2H2O, L33·2H2O, and 3, and the oxime units in L44, L33, L23, L24, 2, 3 and 4 which were located from the Fourier synthesis maps. The absolute configuration of L1 could not be determined because the compound was a weak anomalous scatterer. The data for L24 were de-twinned using the RLATT procedure of SHELXTL.60 In 1 the disordered CF3SO3− anion could not be modelled and the electron density associated with it was removed using the SQUEEZE procedure in PLATON.61 Crystals of 3 were very thin and diffracted weakly at high angles. All calculations were performed using the WinGX interface.62 Detailed analyses of the extended structure were carried out using PLATON63 and MERCURY64,65 (Version 1.4.2). In both L33·2H2O and 2 the oxime units were disordered over two sites having site occupancy factors of 0.69 and 0.31. L44·2H2O and L1 belonged to non-centrosymmetric space groups and as they contained no heavy atoms the assignment of absolute configuration through the Flack parameter was ambiguous.66–68 Crystal data and refinement details are summarised in Table 2.| L44·2H2O | L1 | L33 | L33·2H2O | |
| Empirical formula | C11H13N3O3 | C11H8N2O | C11H9N3O | C11H13N3O3 |
| M | 235.24 | 184.19 | 199.21 | 235.24 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P21 | Pna21 | Pbca | P21/c |
| a/Å | 4.0517(4) | 21.0949(17) | 11.4954(17) | 13.9556(14) |
| b/Å | 13.9045(14) | 10.5185(8) | 10.6744(17) | 10.0763(11) |
| c/Å | 10.2601(11) | 3.8315(4) | 15.506(3) | 8.6383(8) |
| β/° | 94.720(4) | — | — | 101.106(3) |
| V/Å3 | 576.06(10) | 850.16(13) | 1902.5(6) | 1192.0(3) |
| Z | 2 | 4 | 8 | 4 |
| T/K | 90(2) | 90(2) | 90(2) | 90(2) |
| μ/mm−1 | 0.078 | 0.096 | 0.094 | 0.098 |
| Reflections collected | 2430 | 7927 | 6102 | 12 219 |
| Unique reflections (Rint) | 2430 (0.0000) | 1360 (0.0297) | 1708 (0.0289) | 2348 (0.0243) |
| R 1 indices [I > 2σ (I)] | 0.0396 | 0.0299 | 0.0377 | 0.0347 |
| wR2 (all data) | 0.0995 | 0.0759 | 0.0987 | 0.0939 |
| L2 | L24 | L23 | ||
| Empirical formula | C11H8N2O | C11H9N3O | C11H9N3O | |
| M | 184.19 | 199.21 | 199.21 | |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | |
| Space group | C2/c | Pnma | P21/c | |
| a/Å | 21.6741(18) | 14.2517(8) | 9.1149(2) | |
| b/Å | 3.7762(3) | 8.1077(6) | 10.1937(2) | |
| c/Å | 21.1734(18) | 8.9124(6) | 10.1280(2) | |
| β/° | 102.254(5) | — | 91.9285(12) | |
| V/Å3 | 1639.5(2) | 1029.81(12) | 940.51(3) | |
| Z | 8 | 4 | 4 | |
| T/K | 90(2) | 90(2) | 90(2) | |
| μ/mm−1 | 0.096 | 0.087 | 0.095 | |
| Reflections collected | 12 280 | 19954 |
10 203 | |
| Unique reflections (Rint) | 1587 (0.0289) | 859(0.0352) | 1752 (0.0195) | |
| R 1 indices [I > 2σ (I)] | 0.0349 | 0.0384 | 0.0307 | |
| wR2 (all data) | 0.0951 | 0.0952 | 0.0789 | |
| 1 | 2 | 3 | 4 | |
| Empirical formula | C44H36N12O4Ag2 F0S0 | C12H9N3O4F3SAg | C12H11N3O5F3SAg | C13H9N3O7F6S2Ag2 |
| M | 1012.59 | 456.15 | 474.17 | 713.11 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | C2/c | P21/n | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 34.844(3) | 11.3883(12) | 12.0134(6) | 10.2591(8) |
| b/Å | 13.5225(11) | 9.4818(10) | 9.9549(5) | 10.2823(8) |
| c/Å | 6.7362(5) | 15.1208(16) | 13.2918(7) | 11.0872(15) |
| α/° | — | — | — | 96.288(5) |
| β/° | 91.412(3) | 110.395(4) | 90.9902(14) | 103.286(5) |
| γ/° | — | — | — | 116.555(4) |
| V/Å3 | 1029.81(12) | 1530.4(4) | 1589.3(2) | 987.45(18) |
| Z | 2 | 4 | 4 | 2 |
| T/K | 90(2) | 90(2) | 90(2) | 90(2) |
| μ/mm−1 | 0.656 | 1.510 | 1.463 | 2.296 |
| Reflections collected | 10 121 | 25 891 | 5492 | 10 790 |
| Unique reflections (Rint) | 2660 (0.0377) | 2847 (0.0226) | 2578 (0.0250) | 3668 (0.0288) |
| R 1 indices [I > 2σ (I)] | 0.0578 | 0.0291 | 0.0485 | 0.0373 |
| wR2 (all data) | 0.1621 | 0.0725 | 0.1009 | 0.1080 |
Results and discussion
The isomeric dipyridyl ketone oximes were prepared in a two-step synthesis from the precursor materials, except for the 4,4′-dipyridyl ketone which was commercially available. Methyl nicotinate was reacted with the product of the lithiation of 2- or 3-bromopyridine to give the respective 2,3-dipyridyl or 3,3′-dipyridyl (L1) ketones as solids in moderate yields. Methyl isonicotinate was used to prepare 2,4-dipyridyl ketone (L2) in a similar manner. Subsequent reaction of the respective ketones with hydroxylamine hydrochloride gave the required oximes L44, L23, L33, and L24 in high yield. Isolation of one isomer of each of the non-symmetrical oximes L23 and L24 was achieved by initial recrystallisation. Further work up produced a mixture of the E and Z isomers for L23 (Fig. 1). Symmetrical oximes L33 and L44 were isolated under different conditions; L44 precipitated out of solution on cooling, whereas work up of the solution was required to form L33. These compounds have been briefly mentioned in the literature before, typically as intermediates in pharmaceutical applications. However, details of preparation and characterisation are rather vague.69–71 All four oximes were thoroughly characterised by 1H, 13C and 2-D NMR techniques, microanalysis, mass spectrometry and X-ray structural analysis. In the case of L33 a pseudo-polymorph L33·2H2O was also isolated. In addition, the precursor ketones L1 and L2 were characterised by X-ray crystallography to provide comparisons with the dipyridyl ketone oximes.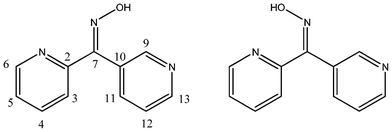 | ||
| Fig. 1 E and Z isomers of L23. | ||
4,4′-Dipyridyl ketone oxime L44·2H2O
X-ray structural analysis of L44·2H2O revealed that it crystallised in the monoclinic space group P21. The asymmetric unit consisted of one complete ligand molecule and two water molecules (Fig. 2). The angle between the plane of each pyridyl rings was 56.66° giving a two-bladed chiral propeller conformation to the ligand.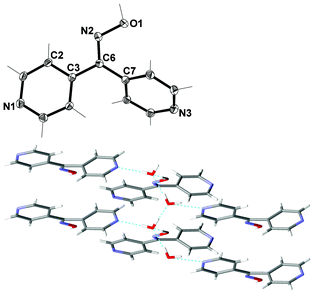 | ||
| Fig. 2 (Top) View of L44 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C(3)–C(6) 1.482(3), C(6)–C(7) 1.497(3), C(6)–N(2) 1.295(3), N(2)–O(1) 1.387(2); C(2)–N(2)–O(1) 114.09(16), C(3)–C(6)–C(7) 121.12(17), C(7)–C(6)–N(2) 124.77(17), C(3)–C(6)–N(2) 114.11(17). (Bottom) View down c axis of double chain showing interaction between water chain and ligands (H-bonds shown in green). | ||
A series of extensive H-bonding was observed in this structure. Individual water molecules were H-bonded to each other forming zigzag chains along the a axis (Fig. 2). The H–O⋯H distances were found to be 1.96 and 1.78 Å respectively, with corresponding O⋯O distances of 2.7740(19) and 2.6821(19) Å respectively. Such chains of water molecules are increasingly being recognised as important in supramolecular systems because they may provide insight into the properties of bulk water and its role in chemical and possibly biological systems.72–78
The first two H-bonding interactions between water molecules and the ligand were between each of the Npy atoms in L44 and two distinct water molecules, generating an infinite chain along the b axis. The Npy⋯H–O distances were found to be 1.93 and 1.95 Å for N1 and N3 respectively, with corresponding N⋯O distances of 2.800(2) and 2.760(2) Å. The third H-bonding interaction between the water chain and ligands was to the oxime unit of L44, generating a chain along the a axis. The O–Hox⋯O distance was found to be 1.70 Å with an O⋯O distance of 2.650(2) Å. Overall these H-bonding interactions generated a 2-D network in the ab plane.
3,3′-Dipyridyl ketone L1
X-ray structural analysis of L1 showed that it crystallised in the orthorhombic space group Pna21. The asymmetric unit consisted of one complete molecule (Fig. 3). The angle between the planes of each of the pyridyl rings was found to be 42.71° giving a two-bladed chiral propeller conformation of the ligand.51 Notably, the 3-substituted pyridyl rings were not arranged in a symmetrical fashion. Analysis of the structure suggested the respective orientation of the pyridyl rings may have been a consequence of the nature of the packing.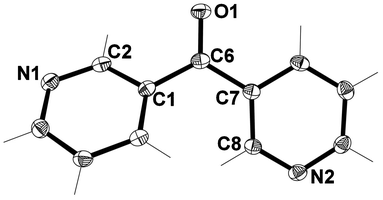 | ||
| Fig. 3 View of the asymmetric unit of L1 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C(1)–C(6) 1.496(2), C(6)–C(7) 1.498(2), C(6)–O(1) 1.228(2); C(1)–C(6)–C(7) 121.73(13). | ||
The packing of L1 involved only non-classical H-bonds which formed an infinite 3-D network. Each molecule within this network was H-bound to four others due to two crystallographically distinct H-bonds (Fig. 4). These H-bonds existed between the Npy donor atoms and nearby H atoms of adjacent pyridyl rings. The Npy⋯H–C bond distances were 2.59 and 2.46 Å, respectively, with corresponding Npy⋯C distances of 3.497(5) and 3.353(5) Å, respectively. The stronger H-bond generates a helix running along the c axis. No other significant interactions existed between individual molecules.
 | ||
| Fig. 4 View of L1 showing the four H-bonds for each molecule and the helix running along the c axis. | ||
3,3′-Dipyridyl ketone oxime L33
The X-ray structure of L33 revealed that it crystallised in the orthorhombic space group Pbca. The asymmetric unit consisted of one complete ligand entity (Fig. 5). The arrangement of the pyridyl rings were such that a two-bladed chiral propeller51 was formed, with the angle between the planes of each ring being 84.93°. This was almost twice the value found for the angle between the planes of the pyridyl rings for the related carbonyl precursor. Once again the Npy atoms were not arranged in a symmetrical fashion.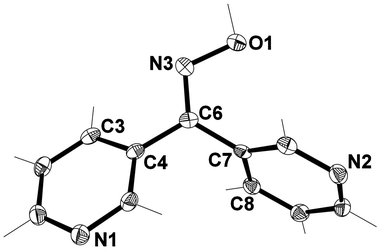 | ||
| Fig. 5 View of asymmetric unit of L33 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C(4)–C(6) 1.476(2), C(6)–C(7) 1.490(2), C(6)–N(3) 1.293(2), N(3)–O(1) 1.3903 (17); C(6)–N(3)–O(1) 111.47(12), C(4)–C(6)–C(7) 118.79(12), C(4)–C(6)–N(3) 116.48(13), C(7)–C(6)–N(3) 124.68(14), C(5)–C(4)–C(6) 120.11(13), C(8)–C(7)–C(6) 120.09(13). | ||
The oxime L33 formed an infinite 1-D zigzag chain running along the a axis. The zigzag chains contained only one enantiomer. However other zigzag chains in the structure were formed with the opposite enantiomer. These chains were formed as a result of a single H-bond between the oxime unit and donor N2 on the pyridyl ring of an adjacent ligand. The Npy⋯H–O distance was found to be 1.77 Å (corresponding to a Npy⋯O distance of 2.737(2) Å). Individual chains were linked into a 2-D sheet consisting of chains of alternating enantiomers by a T-shaped C–H⋯π interaction between H11 and the ring containing N1 (Fig. 6). The C–H⋯ring centroid distance was 2.72 Å.79
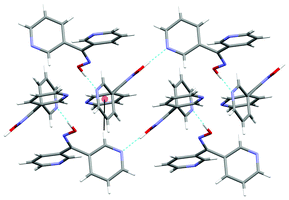 | ||
| Fig. 6 View of L33 in the ab plane of two zigzag chains formed as a result of classical H-bonding interactions (shown in green) and C–H⋯π interactions (shown in black). | ||
3,3′-Dipyridyl ketone oxime L33·2H2O
X-ray structural analysis of L33·2H2O revealed that it crystallised in the monoclinic space group P21/c. The asymmetric unit of this structure contained a complete ligand molecule in which the oxime group was disordered in a 69:31 ratio (Fig. 7) and two water molecules. The disorder arose from the possibility of two conformations of L33 existing in the same crystal (Fig. 1). In the crystal analysed the dominant conformation had the oxime unit pointing towards the pyridyl ring containing N2. The other conformation had the oxime unit pointing towards the pyridyl ring containing N1. Analysis will focus on the main component of disorder. As with the pseudo polymorph L33, the pyridyl rings were arranged in such a way as to form a two-bladed chiral propeller51 with an angle of 69.72° between the planes of each ring. This angle was smaller than that found in L33. Interestingly, the individual Npy atoms were arranged symmetrically in that they were on the same side of the molecule as the oxime functional group [C6, N3, O1].
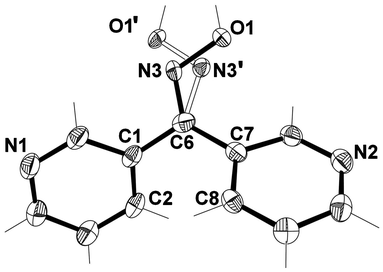 | ||
| Fig. 7 View of asymmetric unit of L33·2H2O with crystallographic numbering, and showing oxime disorder. Thermal ellipsoids are drawn at 50% probability level. Solvent molecules have been omitted for clarity. Selected bond lengths (Å) and angles (°): C(1)–C(6) 1.4833(18), C(6)–C(7) 1.4857(17), C(6)–N(3) 1.300(3), N(3)–O(1) 1.398(2), C(6)–N(3′) 1.360(6), N(3′)–O(1′) 1.388(5); C(6)–N(3)–O(1) 108.6(2), C(1)–C(6)–C(7) 118.61(10), C(1)–C(6)–N(3) 108.85(14), C(7)–C(6)–N(3) 132.54(15), C(2)–C(1)–C(6) 121.91(11), C(8)–C(7)–C(6) 120.32(10), C(6)–N(3′)–O(1′) 105.5(5), C(1)–C(6)–N(3′) 139.9(3), C(7)–C(6)–N(3′) 101.2(3). | ||
In this structure an extensive series of H-bonding interactions was observed. The individual water molecules were H-bound to each other to form zigzag chains (Fig. 8) in a manner similar to L44·2H2O. These chains ran along the c axis and lay in the cavities between individual ligand entities. The H–O⋯H distances were found to be 1.91 and 1.80 Å, respectively, with corresponding O⋯O distances of 2.751(2) and 2.639(2) Å, respectively. These chains of water molecules were also H-bound to individual ligand entities through three separate H-bonding interactions to generate an infinite 3-D network (Fig. 8). The first two H-bonding interactions were between two water molecules and each of the Npy atoms of L33. The Npy⋯H–O distances were found to be 1.93 and 1.98 Å for N1 and N2, respectively, with corresponding N⋯O distances of 2.791(3) and 2.807(2) Å, respectively. The third H-bonding interaction was between one water molecule and the oxime unit of L33. The O–Hox⋯O distance was found to be 1.85 Å with an O⋯O distance of 2.657(4) Å. One weak π–π interaction, with a centroid–centroid distance of 3.82 Å, was observed between two N2 containing rings on adjacent ligands. No other significant interactions were observed between individual ligand entities.
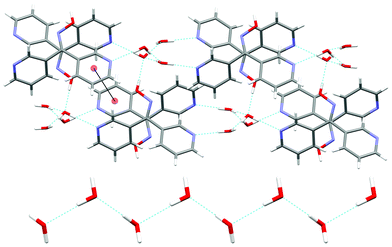 | ||
| Fig. 8 (Top) View of the network formed in the ab plane as a result of H-bonding between L33 and individual water molecules with one example of the weak π–π interaction shown by the solid line between two N2 pyridyl rings. View is along the c axis. (Bottom) Zigzag chain of water molecules formed as a result of two H-bonding interactions (shown in green). View is along the b axis. | ||
2,4-Dipyridyl ketone L2
X-ray structural analysis revealed that L2 crystallised in the monoclinic space group C2/c. The asymmetric unit consisted of one complete molecule (Fig. 9). Each pyridyl ring was twisted relative to the other giving rise to an angle of 48.41° between them. The N2 atom was arranged such that it was in a trans relationship to the ketone functional group [C6, O1].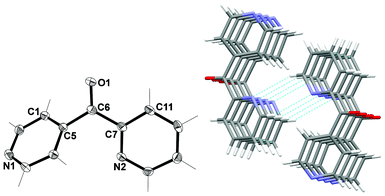 | ||
| Fig. 9 (Left) View of the asymmetric unit of L2 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C(5)–C(6) 1.5038(16), C(6)–C(7) 1.5015(16), C(6)–O(1) 1.2200(14); C(5)–C(6)–C(7) 120.01(10). (Right) Stacked nature of 1-D chains formed as a result of π–π interactions and intermolecular H-bonds (shown in green). View is in the bc plane. | ||
Infinite 1-D π-stacked chains of the same enantiomer were formed as a result of π–π interactions between identical rings of adjacent ligand entities. The centroid–centroid distances were 3.78 Å. Each π-stacked chain was linked to one other chain, containing the opposite enantiomer to form a double chain as a result of two complementary non-classical H-bonds between N2 of the 2-substituted pyridyl ring and H8 of adjacent ligands (Fig. 9). The Npy⋯H–C distance was found to be 2.71 Å (corresponding to a Npy⋯C distance of 3.537(2) Å). As a further consequence of these H-bonds, the carbonyl units were positioned on the outside edges of each double chain and therefore pointed in opposite directions.
2,4-Dipyridyl ketone oxime L24
X-ray structural analysis of L24 revealed that it crystalised in the orthorhombic space group Pnma. The asymmetric unit consisted of one half of a ligand molecule (Fig. 10). The ligand had a mirror plane of symmetry such that the oxime unit and one ring (N1) lay in the plane while the other ring (N2) was bisected by the plane. Given this imposed crystallographic symmetry L24 cannot act as a two bladed chiral propeller like the other ligands. The N2 atom was in a Z relationship to the oxime functional group while the N1 atom was in an E relationship.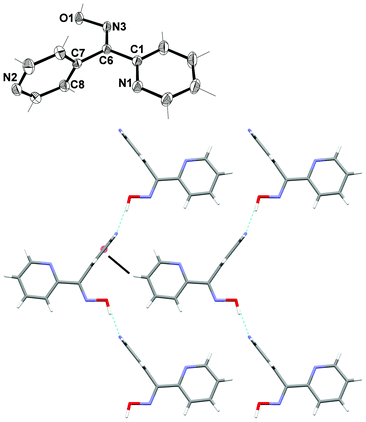 | ||
| Fig. 10 (Top) View of L24 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C(1)–C(6) 1.480(3), C(6)–C(7) 1.495(3), C(6)–N(3) 1.283(2), N(3)–O(1) 1.390(2); C(6)–N(3)–O(1) 112.83(19), C(7)–C(6)–C(1) 121.59(15), C(7)–C(6)–N(3) 122.22(16), C(1)–C(6)–N(3) 116.67(16), C(8)–C(7)–C(6) 120.98(9), N(1)–C(1)–C(6) 115.90(16). (Bottom) View along b axis showing the 1-D chain (H-bonds shown in green) and T shaped C–H⋯π interaction (black) linking chains together. | ||
Individual ligand molecules were held together with H-bonding forming an infinite 1-D chain running parallel to the a axis (Fig. 10). The H-bonding interaction occurred between the H atom of the oxime group and the N2 of the 4-substituted ring. The Npy⋯H–O distance was found to be 1.77(3) Å with a corresponding N⋯O distance of 2.695(2) Å. These individual chains were then further linked into a 2-D sheet in the ac plane by a T shaped C–H⋯π interaction between H4 and the N2 ring. The C–H⋯ring distance was 2.77 Å, corresponding to a C⋯ring distance of 3.586(3).79
2,3-Dipyridyl ketone oxime L23
L23 crystallised in the monoclinic space group P21/c. The asymmetric unit consisted of one complete ligand molecule (Fig. 11). The angle between the plane of each of the pyridyl rings was 59.94° giving rise to the formation of a two-bladed chiral propeller.51 The N2 atom of the 2-substituted pyridyl ring was in a trans relationship to the oxime functional group while the N1 atom of the 3-substituted pyridyl ring was in a cis relationship.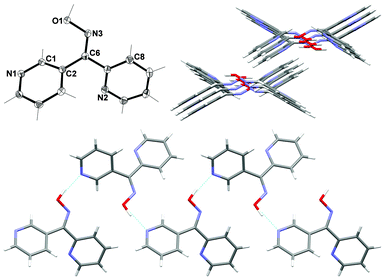 | ||
| Fig. 11 (Top left) View of asymmetric unit of L23 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): C(2)–C(6) 1.4891(15), C(6)–C(7) 1.4919(15), C(6)–N(3) 1.2873(10), N(3)–O(1) 1.3960(12); C(6)–N(3)–O(1) 114.59(9), C(2)–C(6)–C(7) 119.46(10), C(2)–C(6)–N(3) 127.18(10), C(7)–C(6)–N(3) 113.35(10), C(3)–C(2)–C(6) 120.62(10), N(2)–C(7)–C(6) 116.35(10). (Top right) View down c axis of the 1-D double chain showing the chains offset from one another. (Bottom) View of 1-D double chains running parallel to the c axis (H-bonds shown in green). | ||
Individual ligand molecules were held together through one main H-bonding interaction to form an infinite 1-D double chain running parallel to the c axis (Fig. 11). Each side of the double chain contained only one enantiomer. This key H-bonding interaction occurred between the H atom of the oxime unit and N1 of the 3-substituted pyridyl ring. The Npy⋯H–O distance was found to be 1.79 Å (corresponding to a N⋯O distance of 2.7281(12) Å). Individual chains were offset from one another and no significant interactions existed between them (Fig. 11).
:1 molar ratio of L44 and AgCF3SO3, respectively, in a 1:1 mixture of MeCN and MeOH. Microanalysis of the crystalline sample was consistent with a 2:1 ligand-to-metal ratio. Despite many attempts, ligand to metal ratios of 1:1 and 1:2 only gave the 2:1 product upon crystallisation. Analysis of the IR spectrum confirmed the presence of L44 and the CF3SO3− (Experimental section) and suggested that the symmetry of the CF3SO3− anion was unchanged upon formation of the complex.
X-ray structural analysis of 1 showed that it crystallised in the monoclinic C2/c space group as a discrete cation forming a 2-D hydrogen-bonded network. The asymmetric unit comprises of one complete L44 ligand, half an Ag(I) cation and half a very disordered CF3SO3− anion (Fig. 12). The disordered CF3SO3− anion could not be appropriately modelled and the electron density associated with the CF3SO3− anion was removed using the SQUEEZE procedure in PLATON.61 However, the presence of the CF3SO3− anion was confirmed from the microanalysis and the IR spectrum (Experimental section). The Ag(I) ion was two-coordinate and formed a centrosymmetric dimer through binding to two symmetry related ligands through the Npy donors of the N1 atom with a N–Ag distance of 2.134(4) Å. The ligand acted as a chiral two bladed propeller with an angle of 72.65° with the dimer consisting of both enantiomers. Hydrogen bonding between the N(2) and O(1) formed an infinite planar 2-D honey-comb network. The O–H⋯N distance was found to be 1.87 Å (corresponding to an O⋯N distance of 2.677(5) Å). This extended into an infinite 3-D network via π–π stacking between the N1 rings of two ligands with a centroid distance of 3.575(3) Å. This interaction was reinforced by weak argentophilic interactions with an Ag⋯Ag distance of 3.3680 Å. The overall stacking was further reinforced by π–π stacking of N2 rings with distance of 3.948(3) Å between the centroids (Fig. 13). The 2-D sheets were relatively well aligned generating channels running along c axis. This left no place for the CF3SO3− anion to bind or interact with the complex, as all significant functional groups were already involved in some type of interaction. This essentially left the CF3SO3− anion to fill the space in the channels, causing it to be highly disordered.
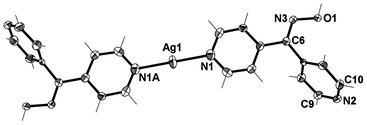 | ||
| Fig. 12 View of the cationic complex 1 with crystallographic numbering. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): N(1)–Ag(1) 2.134(4), C(6)–N(3) 1.276(6), N(3)–O(1) 1.384(5); N(1)–Ag(1)–N(1A) 180.00, C(6)–N(3)–O(1) 112.1(4). (Symmetry code: A − x, −y, 1 − z). | ||
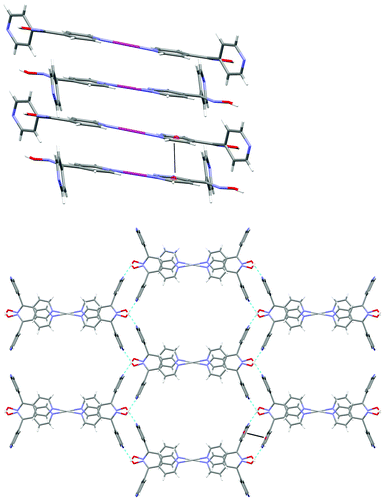 | ||
| Fig. 13 (Top) view of 1 along b axis showing π–π stacking of N1 rings and argentophilic interactions. (Bottom) View along c axis showing infinite planar 2D honeycomb in ab plane (H-bonding shown in green, π–π stacking shown in black). | ||
:1 molar ratio produced a tan coloured precipitate in low yield and microanalysis was consistent with a 1:1 metal-to-ligand ratio. Analysis of the IR spectrum confirmed the presence of L33 in the bulk material and peaks corresponding to the CF3SO3− anion suggested the symmetry of the anion remained unchanged upon formation of the complex (Experimental section).80 Colourless X-ray quality crystals were grown by the slow diffusion of L33 in MeOH into a solution of AgCF3SO3 in MeCN.
X-ray structural analysis of 2 showed that it crystallised in the monoclinic space group P21/n and formed a tightly wound 1-D helical polymer (Fig. 14) encapsulating very weakly interacting CF3SO3− anions. The asymmetric unit contained a Ag(I) cation, a CF3SO3− anion and a complete L33 ligand disordered around the oxime group in a 69:31 ratio in a similar fashion to the free ligand, vide supra (Fig. 14). The main component of the disordered oxime was used in the structural analysis of 2. The Ag(I) was bound to two symmetry related ligands through the Npy donors and adopted an essentially linear arrangement with a N–Ag–N angle of 174.84(9)°. A CF3SO3− anion sat at a distance of 2.745(3) Å from the Ag(I) ion. This distance was well above the mean for Ag–OSO2CF3 distances (Table 1). The small deviation from linearity in the N–Ag–N bond angle can be attributed to the very weak interaction of the CF3SO3− anion.4
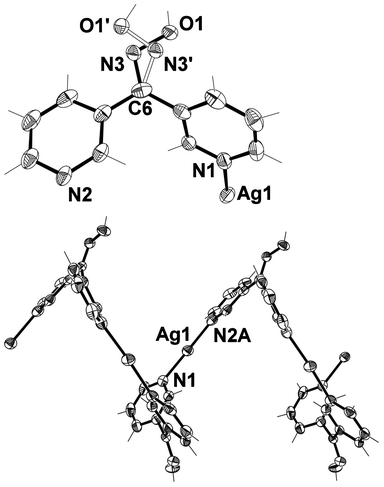 | ||
| Fig. 14 (Top) View of asymmetric unit of 2 showing disorder of the oxime unit (crystallographic numbering). The CF3SO3− anion is omitted for clarity. Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): N(1)–Ag(1) 2.150(3), N(2)–Ag(1) 2.142(3), C(6)–N(3) 1.276(6), C(6)–N(3′) 1.404(13), N(3)–O(1) 1.390(6), N(3′)–O(1′) 1.377(14); N(1)–Ag(1)–N(2) 174.84(9), C(6)–N(3)–O(1) 109.5(6), C(6)–N(3′)–O(1′) 101.9(11). (Bottom) View showing the coordination environment of the Ag(I) ion within the 1-D helical polymer chain (crystallographic numbering) (symmetry code: A ½ − x, −½ + y, ½ − z). | ||
The L33 ligand was bound to two symmetry related Ag(I) ions through the 3-substituted Npy donor atoms and thus acted as a bridge between individual Ag(I) ions. It adopted a conformation in which each ring was approximately orthogonal to the other at 71.12°. As a consequence the ligand acted as a two-bladed chiral propeller forming the 1-D helical polymer. The centrosymmetric space group meant that both M- and P-helices were present within the structure. The 1-D helix ran parallel to the crystallographic b axis and had a helical pitch of 9.4818(10) Å, corresponding to the length of the b axis.
The CF3SO3− anion sat within the helix (Fig. 15) by virtue of the very weak Ag(I)⋯OSO2CF3 interaction. Also holding it in place was a weak H-bonding interaction between an O atom of the CF3SO3− anion and a pyridyl H atom of the helix. The C–H⋯O distance was found to be 2.54 Å (corresponding to a C⋯O distance of 3.219(5) Å).
 | ||
| Fig. 15 View showing how the CF3SO3− anions sit within the 1-D helices running along the crystallographic b axis. | ||
The strongest interaction between individual helices was an H-bonding interaction between an O atom of a CF3SO3− encapsulated in one helix and the oxime OH group of an adjacent helix. The O–H⋯O distance was found to be 1.94 Å (corresponding to a O⋯O distance of 2.683(7) Å). As a result of this H-bonding interaction the helices involved were slightly interdigitated and in this way chirality was transferred between adjacent oxime H-bonded helices. This gave rise to H-bonded sheets composed of helices with the same chirality. Each helix was surrounded by six others (Fig. 16).
 | ||
| Fig. 16 View in the ac plane showing the chirality of adjacent helices. | ||
:1 molar ratio afforded off-white precipitates in low yield which gave analyses consistent with 1:1 metal-to-ligand ratios. Analysis of the IR spectrum confirmed the presence of both CF3SO3− and L24 in the complex (Experimental section). The peaks corresponding to the CF3SO3− anion were consistent with the symmetry of CF3SO3− remaining essentially unchanged upon formation of the complex.80 Colourless X-ray quality crystals were grown by the slow diffusion of L24 in MeOH into a solution of AgCF3SO3 in MeCN.
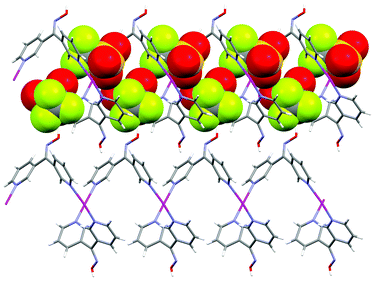
X-ray structural analysis of 3 revealed the complex to be a 1-D polymer chain decorated on one side with weakly bound CF3SO3− anions (Fig. 17). The complex crystallised in the monoclinic space group P21/n. The asymmetric unit contained one Ag(I) ion, one ligand, one CF3SO3− counterion and a H2O molecule of crystallisation. The Ag(I) ion was in a distorted trigonal pyramidal arrangement. The trigonal plane was formed with one chelating ligand involving 2-substituted Npy and Nox donors and a 4-substituted Npy donor from an adjacent symmetry related ligand. The remaining site was occupied by a weakly bound CF3SO3− anion at a Ag–O distance of 2.698(4) Å. This distance was near the limit of what would be considered a Ag–OSO2CF3 bond (Table 1). The Ag(I) ion was 0.108 Å below the plane and pointing away from the CF3SO3− anion. The ligand L24 adopted a conformation in which the 4-substituted pyridine ring was approximately orthogonal to the remainder of the ligand at 70.49°. This twist allowed L24 to behave as a two-bladed chiral molecular propeller.
![View of a 1-D chain of 3 (crystallographic numbering) running along the [1 0 1] direction with H2O molecules omitted for clarity (50% probability ellipsoids). Selected bond lengths (Å) and angles (°): Ag1–N1 2.174(5), Ag1–N2A 2.247(5), Ag1–N3A 2.467(5), Ag1–O11 2.698(4); N1–Ag1–N2A 160.12(18), N1–Ag1–N3A 128.42(19), N2A–Ag1–N3A 70.31(17), N1–Ag1–O11 88.56(15), N2A–Ag1–O11 84.60(15), N3A–Ag1–O11 89.74(14) (symmetry code: A ½ + x, ½ − y, −½ + z).](/image/article/2013/CE/c2ce26449h/c2ce26449h-f17.gif) | ||
| Fig. 17 View of a 1-D chain of 3 (crystallographic numbering) running along the [1 0 1] direction with H2O molecules omitted for clarity (50% probability ellipsoids). Selected bond lengths (Å) and angles (°): Ag1–N1 2.174(5), Ag1–N2A 2.247(5), Ag1–N3A 2.467(5), Ag1–O11 2.698(4); N1–Ag1–N2A 160.12(18), N1–Ag1–N3A 128.42(19), N2A–Ag1–N3A 70.31(17), N1–Ag1–O11 88.56(15), N2A–Ag1–O11 84.60(15), N3A–Ag1–O11 89.74(14) (symmetry code: A ½ + x, ½ − y, −½ + z). | ||
The complex formed a 1-D chain running parallel to the [1 0 1] diagonal axis. All the ligands pointed in one direction along the chain and adjacent ligands were oriented at 113.7° with respect to each other forming an L-shaped chain. The CF3SO3− anions sat within the bend of the L shaped chain and point outwards. The ligands and CF3SO3− anions, respectively, were related by a glide plane which bisected the dihedral angle of the L-shaped chain. As a consequence of the glide plane the chirality of individual ligands alternated along the chain giving rise to an overall achiral structure. Each L-shaped chain interacted with two adjacent chains forming a zigzag pattern through a series of H-bonding interactions. The H-bonding interactions were responsible for forming a series of 18-membered rings (Fig. 18). These rings involved Ag1–N3–O1H1A⋯O2H2A⋯O12–S1–O11–Ag1–N3–O1H1A⋯O2H2A⋯O12–S1–O11 at H-bonding distances of 1.72 and 1.85 Å, respectively [corresponding to an O1⋯O2 separation of 2.614(5) and a O2⋯O12 separation of 2.806(7) Å, respectively]. The chains were arranged in such a way that the chelated oxime unit of one chain registered with the monodentate 4-pyridyl unit of an adjacent chain. These adjacent chains were bowed such that the Ag⋯Npy and Ag⋯Ag distances were 3.157(6) and 3.3236(18) Å, respectively (Fig. 18). This bowing appeared to arise as a consequence of steric distortion of the chains rather than any weak bonding interactions involving the Ag(I) ions. Similarly the influence of π-bonding interactions was ruled out as they were too weak at 4.04 Å. Therefore, the steric distortion was caused by the presence of the 18-membered rings pulling adjacent chains into close proximity resulting in the formation of an overall 3-D network. Interactions within this network structure were augmented by inter-chain π–π interactions at 3.78 Å.
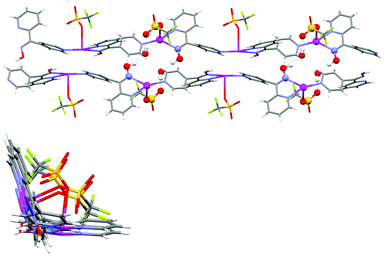 | ||
| Fig. 18 (Left) View down the diagonal ac axis showing the L-shaped nature of the chain. (Right) View of two adjacent 1-D chains of 3 showing the 18-membered H-bonded rings and the bowing of the chains. | ||
[Ag2L23(CF3SO3)2]∞4
Colourless X-ray quality crystals of [Ag2L23(CF3SO3)2]∞4 were grown by the slow diffusion of a solution of AgCF3SO3 in MeCN into a solution of L23 in MeOH through blank layers of EtOH and MeCN. Microanalysis of the crystalline sample was consistent with a 1:2 ligand-to-metal ratio. Analysis of the IR spectrum confirmed the presence of L23 and the CF3SO3− (Experimental Section).
A bulk reaction of L23 with AgCF3SO3 was consequently carried out with a 1:2 ligand-to-metal ratio and a pale cream precipitate was obtained in good yield. Microanalysis of this solid was consistent with a 1:1 ligand-to-metal ratio. The solid was found to be insoluble in all common organic solvents. X-ray quality crystals of the 1:1 product could not be obtained, despite many attempts at growing crystals of this material. The bulk reaction was repeated a number of times using different molar ratios. However the same result of a 1:1 metal-to-ligand complex was always obtained.
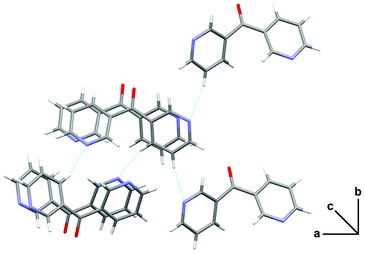
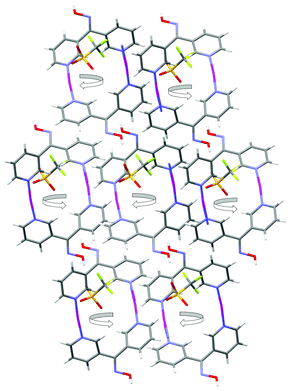
X-ray analysis of 4 revealed it to be an infinite neutral 2-D network lying in the ab plane. The complex crystallised in the triclinic space group P and the asymmetric unit consisted of one L23 entity, two Ag(I) cations and two CF3SO3− anions (Fig. 19). The Ag1 cation was bound to L23 through the chelating Npy, Nox donor set and through O atoms from three different CF3SO3− anions and adopted a distorted square-based pyramidal geometry with a τ5 value of 0.28.81 The Ag1 ion sat 0.15 Å above the basal plane (N1, N2, O2, O4B). The second Ag2 cation was in a distorted tetrahedral coordination environment being bound to L23 through the 3-substituted Npy donor atom and through O atoms from three different CF3SO3− anions. The τ4 value was found to be 0.8782 as the bond angles around Ag2 varied from 88.11° to 149.67°. The Ag–O distances of each of the Ag(I) ions were found to be in the range of 2.239(4) to 2.584(3) Å (Table 1). The ligand itself acted as a two-bladed chiral propeller with an angle of 74.47° between the plane of each ring. The centrosymmetric nature of the space group meant that both enantiomers of L23 were present within the 2-D network and were found to alternate along the ligand ribbons (vide infra).
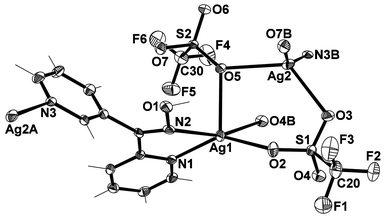 | ||
| Fig. 19 View of the coordination environment of each unique Ag(I) ion in 4 (crystallographic numbering). Thermal ellipsoids are drawn at 50% probability level. Selected bond lengths (Å) and angles (°): Ag(1)–N(1) 2.298(4), Ag(1)–N(2) 2.433(4), Ag(1)–O(2) 2.344(3), Ag(1)–O(4B) 2.447(3), Ag(1)–O(5) 2.584(3), Ag(2)–N(3B) 2.239(4), Ag(2)–O(3) 2.495(3), Ag(2)–O(5) 2.561(3), Ag(2)–O(7B) 2.350(3), N(2)–O(1) 1.387(5); N(1)–Ag(1)–O(2) 142.95(12), N(1)–Ag(1)–O(4B) 112.47(12), N(1)–Ag(1)–O(5) 113.79(11), N(1)–Ag(1)–N(2) 70.06(12), N(2)–Ag(1)–O(2) 108.54(12), N(2)–Ag(1)–O(4B) 159.82(11), N(2)–Ag(1)–O(5) 86.23(11), O(2)–Ag(1)–O(4B) 81.76(11), O(2)–Ag(1)–O(5) 102.85(11), O(4B)–Ag(1)–O(5) 74.38(10), N(3B)–Ag(2)–O(3) 99.70(12), N(3B)–Ag(2)–O(5) 109.60(12), N(3B)–Ag(2)–O(7B) 149.67(12), O(3)–Ag(2)–O(7B) 89.99(11), O(3)–Ag(2)–O(5) 123.04(10), O(5)–Ag(2)–O(7B) 88.11(11) (symmetry codes: A 1 + x, 1 + y, z; B −1+ x, −1 + y, z). | ||
The infinite 2-D network was formed as a result of the bonding interactions observed between the O atoms of the CF3SO3− anions and the Ag(I) ions. These interactions played a key role in the formation of the overall structure. The first CF3SO3− anion was bound to two Ag(I) ions, one in a monodentate fashion and the other in a bis-monodentate fashion. The second CF3SO3− anion was bound to two Ag(I) ions in a tris-monodentate fashion. Thus each acted as a bridge between two Ag(I) ions in differing binding modes. There are only a few examples of AgCF3SO3 complexes where the CF3SO3− anion is bound in a bridging fashion through a single O atom (Table 1). The presence of these bonding interactions resulted in the formation of AgCF3SO3 units which then assembled to form threads along the a axis (Fig. 20). The L23 ligand then acted as a bridge between each Ag(I) ion within the threads and in this way linked the individual AgCF3SO3 threads to each other. The only H-bonding interaction present within the structure was an H-bond between the oxime OH and an O atom of the CF3SO3− anion. The O–H⋯O distance was found to be 1.81 Å (corresponding to an O⋯O distance of 2.629(4) Å).
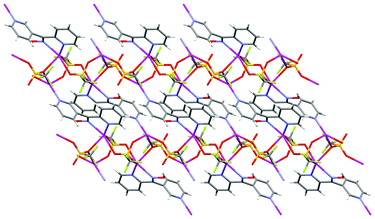 | ||
| Fig. 20 View of the ab plane showing how the L23 and AgCF3SO3 threads run parallel to each other along the a axis. | ||
Comparison
The resulting polymeric Ag(I) structures were a subtle balance between the influences of the CF3SO3− anion and the Npy substitution pattern of the isomeric dipyridyl ketone oxime ligands. This balance appeared to dictate the stoichiometry in the complexes formed. The network of 1 was formed from the Ag(I) ion binding exclusively to one pyridyl ring of the ligand. This entity then essentially formed an infinite 2-D structure of closed circles through H-bonding between the remaining pyridyl ring and the oxime of an adjacent ligand. An infinite 3-D honeycomb network with large channels throughout the structure was generated by π–π stacking combined with weak argentophilic interactions. The CF3SO3− anion was seriously disordered as no significant functional groups remained to interact with thus leaving it to occupy the channels in a random fashion.The helical nature of 2 arose in part from the 3,3′-substitution pattern of L33 and the propeller twist of pyridyl rings relative to each other. Crucial to the formation of a helix was the need to link these chiral propellers through a linear Ag(I) ion. Such a coordination environment precluded any strong interaction with the counterion. Thus the CF3SO3− anion remained unbound and by virtue of a very weak interaction was encapsulated, essentially filling space, within the tightly wound helical polymer chain. A strong H-bonding interaction between the encapsulated CF3SO3− anion of one chain and the oxime OH group of an adjacent chain assisted with the transfer of chirality to the neighbouring chain.
In contrast, the achiral L-shaped nature of the 1-D chain in 3 arose from the essentially linear-linking nature of L24. Formation of a chelate ring with Ag(I) locked one pyridyl ring into a set conformation and, although the other pyridyl ring remained free to rotate, the N donor atom of the 4-substituted pyridyl ring essentially remained static as the ring rotated about its axis. In addition, the monodentate binding by the CF3SO3− anion to the Ag(I) ion limited the possible structural outcomes with a trigonal pyramidal coordination environment being adopted by the Ag(I) ion and as a consequence the L24 ligands all pointed in the same direction along the chain. Positioning the CF3SO3− anion in the bend of the L-shaped chain allowed H-bonding interactions which were responsible for the formation of 18-membered rings between adjacent 1-D polymer chains. These 18-membered rings contained solvent H2O molecules and were approximately orthogonal giving rise to a 3-D H-bonding network.
Finally, in 4 a 2-D network was formed as a consequence of the CF3SO3− anions strongly binding to the two different Ag(I) ions through two rare bridging modes. These bridging interactions in 4 were facilitated by a metal rich 2:1 metal-to-ligand ratio, compared to the 1:1 ratio in 2 and 3. Again formation of a 5-membered chelate ring by L23 restricts the possible ways this ligand can contribute to a structural outcome for this polymer.
The twisting of the pyridyl rings in the complex arrays relative to each other was about the same at 72°. In contrast, the twists observed in the free ligands and related precursor ketones varied markedly. This suggested there was a reasonable degree of conformational freedom in the ligand structures which was probably influenced by crystal packing forces. The approximate uniformity of the twist in each of the complexes suggested that the conformational freedom inherent in the Cpy–Cox bond was not being fully utilised in these structures. These effects were compounded in 2 and 3 by the fact that in 2 each polymer chain was composed of only one enantiomeric form of L33 whereas in 3 the handedness of the enantiomers alternate along the chain.
The strongly coordinating and structurally complex role of the CF3SO3− anion in 4 necessitated such a high metal salt ratio and generated the final 2-D structure. In 4 the two Ag(I) ions each took on different coordination geometries, namely distorted tetrahedral and distorted square-based pyramidal. In particular the varying Npy substitution patterns and the ability of L23 and L24 to form chelate rings, have an effect on the nature of the final polymer formed. Formation of a chelate ring in both L23 and L24 locks one pyridyl ring into a set conformation upon binding to a Ag(I) ion. However, given that one pyridyl ring remains free to rotate, sufficient conformational freedom remains allowing the Ag(I) ions together with the anion to have an impact on the overall structure.
Acknowledgements
We acknowledge the Tertiary Education Commission New Zealand for the award of a Top Achiever Doctoral Scholarship (VJA) and thank the University of Otago Research Committee (VJA) and the University of Otago for financial support. We thank Dr Scott A. Cameron for assistance with the twinning in the ligand structure L24.References
- S. Natarajan and P. Mahata, Chem. Soc. Rev., 2009, 38, 2304 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- M. A. M. Abu-Youssef, V. Langer and L. Oehrstroem, Dalton Trans., 2006, 2542 RSC.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688 CrossRef CAS.
- D. G. Mantero, A. Neels and H. Stoeckli-Evans, Inorg. Chem., 2006, 45, 3287 CrossRef CAS.
- A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127 CrossRef CAS.
- N. S. Oxtoby, A. J. Blake, N. R. Champness and C. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4905 CrossRef CAS.
- C. Janiak, Dalton Trans., 2003, 2781 RSC.
- J. J. M. Amoore, C. A. Black, L. R. Hanton and M. D. Spicer, Cryst. Growth Des., 2005, 5, 1255 CAS.
- M. O. Awaleh, A. Badia and F. Brisse, Inorg. Chem., 2005, 44, 7833 CrossRef CAS.
- X.-H. Bu, W. Chen, W.-F. Hou, M. Du, R.-H. Zhang and F. Brisse, Inorg. Chem., 2002, 41, 3477 CrossRef CAS.
- J. M. Knaust and S. W. Keller, CrystEngComm, 2003, 5, 459 RSC.
- X.-H. Bu, Y.-B. Xie, J.-R. Li and R.-H. Zhang, Inorg. Chem., 2003, 42, 7422 CrossRef CAS.
- N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161 CrossRef CAS.
- M.-Y. He, S.-C. Chen, Z.-H. Zhang, K.-L. Huang, F.-H. Yin and Q. Chen, Inorg. Chim. Acta, 2009, 362, 2569 CrossRef CAS.
- A. J. Blake, N. R. Champness, P. A. Cooke and J. E. B. Nicolson, J. Chem. Soc., Dalton Trans., 2000, 3811 RSC.
- L. R. Hanton and K. Lee, J. Chem. Soc., Dalton Trans., 2000, 1161 RSC.
- L. Mimassi, C. Guyard-Duhayon, L. Raehm and H. Amouri, Eur. J. Inorg. Chem., 2002, 9, 2453 CrossRef.
- K. A. Hirsch, S. R. Wilson and J. S. Moore, Inorg. Chem., 1997, 36, 2960 CrossRef CAS.
- C.-Q. Wan, Z.-J. Wang, G. Wang, H. Liu, Y.-H. Deng and Q.-H. Jin, Cryst. Growth Des., 2012, 12, 376 CAS.
- F. Gschwind and K. M. Fromm, CrystEngComm, 2012, 14, 4008 RSC.
- I. Bassanetti, F. Mezzadri, A. Comotti, P. Sozzani, M. Gennari, G. Calestani and L. Marchio, J. Am. Chem. Soc., 2012, 134, 9142 CrossRef CAS.
- J. Ni, K.-J. Wei, Y. Liu, X.-C. Huang and D. Li, Cryst. Growth Des., 2010, 10, 3964 CAS.
- M. O. Awaleh, A. Badia, F. Brisse and X.-H. Bu, Inorg. Chem., 2006, 45, 1560 CrossRef CAS.
- J. W. Lee, E. A. Kim, Y. J. Kim, Y.-A. Lee, Y. Pak and O.-S. Jung, Inorg. Chem., 2005, 44, 3151 CrossRef CAS.
- O.-S. Jung, Y. J. Kim, Y.-A. Lee, K.-M. Park and S. S. Lee, Inorg. Chem., 2003, 42, 844 CrossRef CAS.
- O.-S. Jung, Y. J. Kim, Y.-A. Lee, J. K. Park and H. K. Chae, J. Am. Chem. Soc., 2000, 122, 9921 CrossRef CAS.
- O.-S. Jung, Y. J. Kim, Y.-A. Lee, H. K. Chae, H. G. Jang and J. Hong, Inorg. Chem., 2001, 40, 2105 CrossRef CAS.
- O.-S. Jung, Y.-A. Lee, Y. J. Kim and J. Hong, CrystEngComm, 2002, 2, 497 CAS.
- X.-D. Chen and T. C. W. Mak, Dalton Trans., 2005, 3646 RSC.
- D. Pogozhev, S. A. Baudron and M. W. Hosseini, Dalton Trans., 2011, 40, 437 RSC.
- D. Pocic, J.-M. Planeix, N. Kyritsakas, A. Jouaiti and M. W. Hosseini, CrystEngComm, 2005, 7, 624 RSC.
- K. K. Bisht, A. C. Kathalikkattil and E. Suresh, RSC Adv., 2012, 2, 8421 RSC.
- N. C. Di, Effendy, F. Marchetti, C. Nervi, C. Pettinari, W. T. Robinson, A. N. Sobolev and A. H. White, Dalton Trans., 2010, 39, 908 RSC.
- X.-D. Chen and T. C. W. Mak, Chem. Commun., 2005, 3529 RSC.
- C.-L. Chen, B.-S. Kang and C.-Y. Su, Aust. J. Chem., 2006, 59, 3 CrossRef CAS.
- O.-S. Jung, Y. J. Kim, J. Y. Park and S. N. Choi, J. Mol. Struct., 2003, 657, 207 CrossRef CAS.
- M. Bardaji, O. Crespo, A. Laguna and A. K. Fischer, Inorg. Chim. Acta, 2000, 304, 7 CrossRef CAS.
- R. Terroba, M. B. Hursthouse, M. Laguna and A. Mendia, Polyhedron, 1999, 18, 807 CrossRef CAS.
- X. L. Lu, W. K. Leong, T. S. A. Hor and L. Y. Goh, J. Organomet. Chem., 2004, 689, 1746 CrossRef CAS.
- A. Bacchi, E. Bosetti, M. Carcelli, P. Pelagatti and D. Rogolino, Eur. J. Inorg. Chem., 2004, 1985 CrossRef CAS.
- A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schroder, Coord. Chem. Rev., 2001, 222, 155 CrossRef CAS.
- H.-P. Wu, C. Janiak, G. Rheinwald and H. Lang, J. Chem. Soc., Dalton Trans., 1999, 183 RSC.
- C. Janiak, L. Uehlin, H.-P. Wu, P. Klufers, H. Piotrowski and T. G. Scharmann, J. Chem. Soc., Dalton Trans., 1999, 3121 RSC.
- M. Munakata, L. P. Wu and T. Kuroda-Sowa, Adv. Inorg. Chem., 1998, 46, 173 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- F. H. Allen, J. E. Davies, J. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G. Watson, J. Chem. Inf. Model., 1991, 31, 187 CrossRef CAS.
- A. B. Caballero, J. K. MacLaren, A. Rodriguez-Dieguez, I. Vidal, J. A. Dobado, J. M. Salas and C. Janiak, Dalton Trans., 2011, 40, 11845 RSC.
- C. J. Milios, T. C. Stamatatos and S. P. Perlepes, Polyhedron, 2006, 25, 134 CrossRef CAS.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, John Wiley and Sons Inc., New York, 1994 Search PubMed.
- F. F. Blicke and C. E. Maxwell, J. Am. Chem. Soc., 1939, 61, 1780–1782 CrossRef CAS.
- X.-D. Chen and T. C. W. Mak, J. Mol. Struct., 2005, 743, 1 CrossRef CAS.
- X.-D. Chen and T. C. W. Mak, Inorg. Chem. Commun., 2005, 8, 393 CrossRef CAS.
- Z. Otwinowski and W. Minor, Processing of X-Ray Diffraction Data Collected in Oscillation Mode, Academic Press, New York, 1st edn, 1997 Search PubMed.
- SAINT-V4, Area Detector and Integration Software, Siemens Analytical X-Ray Systems Inc., Madison, WI, 1996 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
- A. L. Speck, J. Appl. Crystallogr., 2003, 36, 7 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- H. D. Flack, M. Sadki, A. L. Thompson and D. J. Watkin, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 21 CAS.
- H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55, 908 CrossRef.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681 CrossRef CAS.
- J. Tirouflet and E. Laviron, Comptes Rendus, 1958, 246, 274 CAS.
- E. Niemers and R. Hiltmann, Synthesis, 1976, 593 CrossRef.
- K. Niewiadomski, Acta Poloniae Pharm., 1976, 33, 307 CAS.
- H.-D. Xian, H.-Q. Li, X. Shi, J.-F. Liu and G.-L. Zhao, Inorg. Chem. Commun., 2009, 12, 177 CrossRef CAS.
- S.-P. Chen, M. Li, Y. Xiao, Y.-x. Yuan, L.-L. Pan and L.-J. Yuan, CrystEngComm, 2008, 10, 1227 RSC.
- R. Carballo, A. Castiñeiras, B. Covelo, a. B. Lago and E. M. V. López, Z. Anorg. Allg. Chem., 2007, 633, 687 CrossRef CAS.
- C.-K. Xia, C.-Z. Lu, X.-Y. Wu, L.-J. Chen, Q.-Z. Zhang, J.-J. Zhang and D.-M. Wu, Inorg. Chim. Acta, 2006, 359, 4639 CrossRef CAS.
- F. Li, T.-H. Li, W. Su, S.-Y. Gao and R. Cao, Eur. J. Inorg. Chem., 2006, 8, 1582 CrossRef.
- D. L. Reger, R. F. Semeniuc, C. Pettinari, F. Luna-giles and M. D. Smith, Cryst. Growth Des., 2006, 6, 1068 CAS.
- X.-L. Zhang, B.-H. Ye and X.-M. Chen, Cryst. Growth Des., 2005, 5, 1609 CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, 3rd edn, 2001 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
Footnote |
| † CCDC reference numbers 899902–899912. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ce26449h |
| This journal is © The Royal Society of Chemistry 2013 |