Screening for polymorphs of cocrystals: a case study†
Mark D.
Eddleston
,
Saranja
Sivachelvam
and
William
Jones
*
Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: wj10@cam.ac.uk; Tel: +44 (0) 1223336468
First published on 5th November 2012
Abstract
A comprehensive crystal form screen was performed on the phenazine:mesaconic acid system using a variety of techniques including solution based approaches, dry and liquid assisted grinding, thermal methods and sublimation. A novel approach to preparing pharmaceutical cocrystals involving crystallisation at a solvent–solvent interface was also employed, and produced a new thermodynamically stable polymorph of the anhydrous cocrystal. In total, three anhydrous polymorphs, a monohydrate and a DMSO solvate were obtained from the screen, and the crystal structures of Form II (the thermodynamically stable polymorph) and the hydrate were determined. Traditional solution based approaches to polymorphism screening were found to have limitations when applied to this cocrystal system due to differences in the solubilities of phenazine and mesaconic acid in many solvents. Furthermore, several cocrystal preparation methods were required in order to isolate all of the crystal forms that were identified during the study, confirming the need for a multi-technique approach when screening for polymorphs of cocrystals.
Introduction
It has long been recognised that the solid state behaviour of a compound can be modified by preparing different crystal forms. Cocrystals are solid forms where two or more neutral molecules are present in a crystal lattice and have recently received interest in the pharmaceutical industry as a means of improving the solubility of hydrophobic drug molecules where salt formation is not practical.1,2Currently, cocrystallisation is not an entirely predictable process3–5 as many selected pairs of molecules, for example chosen on the basis of potential synthon formation,6 do not yield a cocrystal, and the propensity for a particular molecule to cocrystallise with a variety of coformers would appear to vary greatly. For example, there are over 40 reported carbamazepine cocrystals in the Cambridge Structural Database (CSD),7 formed with molecules containing a range of functional groups, whereas with other molecules such as artemisinin preparing cocrystals was found to be difficult.8 Much attention has been paid to methods of preparing cocrystals, and although solution based approaches are desirable for scale-up and manufacturing,3 and have been used successfully for cocrystallisation,9 often the cocrystal formers crystallise as separate phases due to the differences in their solubilities.3,10–12 Alternative techniques such as slurrying13,14 and dry/liquid assisted grinding11,15 avoid these solubility issues by reducing the amount of solvent to the point where neither cocrystal former is completely dissolved.10 These methods, however, use crystalline input material and can therefore be prone to seeding effects, whereby individual coformers persist rather than converting to a more stable cocrystal phase. In addition, neither slurrying or grinding is ideal for the large-scale manufacture of a drug substance.5,9 There is, therefore, a need to develop new methods for preparing cocrystals of compounds to overcome the difficulties associated with current approaches.
Investigating the polymorphic behaviour of an active pharmaceutical ingredient (API) is a critical part of the drug development process, and will be equally important with cocrystal forms of APIs. To date, however, detailed screening for polymorphism in pharmaceutical cocrystals has not been extensively studied.16 Initial suggestions were that cocrystals would be less prone to polymorphism than free and salt forms of APIs,17–19 but there is no clear scientific rationale for why this should be the case. A study of the CSD in 2008, not limited to pharmaceutical systems, identified 33 sets of polymorphic cocrystals,20 and there are now also several reported examples of polymorphism in pharmaceutical cocrystals,21–27 including three different polymorphs of the ethenzamide:gentisic acid cocrystal.28 It has been speculated that the lack of extensive polymorphism reported in cocrystal systems may well be the result of difficulties with methods for screening for cocrystals rather than a decreased tendency for cocrystals to be polymorphic.21 Methods of searching for polymorphs of APIs and salt forms of APIs are well established, and are typically based around medium or high-throughput solution crystallisation screens,29,30 but it has yet to be demonstrated that these approaches will be equally applicable to screening for polymorphs of cocrystals. It is likely that investigating cocrystal polymorphism will require special attention due to the difficulties in preparing cocrystals from solution caused by differences in the solubilities of the coformers.
Here we report a systematic investigation into various techniques for screening for polymorphs of cocrystals using the phenazine:mesaconic acid system. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 phenazine:mesaconic acid cocrystal has been used previously as a model system for investigating cocrystal formation,31,32 and been prepared both from solution and by grinding, with only one polymorph having been isolated prior to this study (Form I).32 The molecular structures of phenazine and mesaconic acid are shown in Fig. 1. In particular, we report an alternative approach to cocrystal preparation, involving crystallisation at the interface of two saturated and immiscible solutions. This approach may provide a facile and process-friendly method for producing pure phases of cocrystals from solution. Crystallisation between layers of immiscible solvents is not a new technique, but to our knowledge has not been applied to the issue of cocrystallisation.33
:1 phenazine:mesaconic acid cocrystal has been used previously as a model system for investigating cocrystal formation,31,32 and been prepared both from solution and by grinding, with only one polymorph having been isolated prior to this study (Form I).32 The molecular structures of phenazine and mesaconic acid are shown in Fig. 1. In particular, we report an alternative approach to cocrystal preparation, involving crystallisation at the interface of two saturated and immiscible solutions. This approach may provide a facile and process-friendly method for producing pure phases of cocrystals from solution. Crystallisation between layers of immiscible solvents is not a new technique, but to our knowledge has not been applied to the issue of cocrystallisation.33
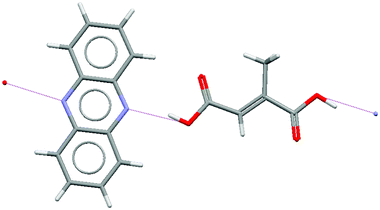 | ||
| Fig. 1 Hydrogen bonding interactions between phenazine and mesaconic acid molecules in Form I of the phenazine:mesaconic acid cocrystal (CSD structure WOQBAF).32 | ||
Experimental
All chemicals were purchased from Sigma-Aldrich and used as received.Powder X-ray diffraction (PXRD) analysis was performed on a Philips X'Pert Diffractometer equipped with an X'celerator RTMS detector using Cu Kα radiation at a wavelength of 1.5406 Å. Typically, 20 mg of solid was used for analysis and pressed gently on a glass slide to give a level surface. PXRD overlays are plotted with an arbitrary intensity scale and were generated using X'Pert Highscore software. Measurements at non-ambient temperature were made using an Anton Paar TK450 heating stage.
Single crystal X-ray data were collected at 180 K on a Nonius Kappa CCD diffractometer equipped with an Oxford Cryosystems cooling device using Mo Kα radiation (λ = 0.71073 Å).
Differential scanning calorimetry (DSC) thermograms were recorded in a nitrogen atmosphere using a Mettler Toledo STARe DSC822e/700 calorimeter using a heating rate of 10 °C min−1. Endotherms are plotted as downward peaks. Samples were prepared in 40 μl aluminum pans which were sealed using a cold weld.
Thermogravimetric analysis (TGA) was performed in air in a Mettler Toledo TGA/SDTA851e/SF/1100 instrument with a heating rate of 10 °C min−1. 5 to 20 mg of sample was analysed in a 100 μl aluminum pan.
Ball milling was performed in a Retsch MM200 grinder for 30 minutes at a frequency of 30 Hz. The grinding was done in a metal vial with two 7 mm diameter metal balls. For liquid assisted grinding 20 μl of liquid was also added. Typically, experiments were performed on a 300 mg scale.
Results
Cocrystal polymorph screening by solution crystallisation
A typical approach for investigating the polymorphic behaviour of single component and salt forms of a compound is to perform solution crystallisations using a range of solvents chosen to have diverse properties in order to maximise the chances of isolating different solid forms. Here, a set of 12 solvents with a range of polarities (measured by dielectric constant) and functional groups was selected for solution crystallisations with phenazine and mesaconic acid (see Table 1). Equimolar amounts of phenazine and mesaconic acid were dissolved in each of these solvents and then crystallised by evaporation to dryness. With four of the solvents the solubility of either phenazine (water) or mesaconic acid (dichloromethane, chlorobenzene and cyclohexane) was found to be poor (<2 mg ml−1) and a solid remained undissolved. In these cases, the mixture was slurried in the solvent for one week before being isolated by filtration. The solutions resulting from these filtrations were then allowed to evaporate to yield a second crop of crystals. All material that was generated during the solution crystallisation screen was analysed by PXRD to identify the crystal form or forms obtained. Observations are summarised in Table 1.:1 molar ratio of phenazine and mesaconic acid from 12 different solvents
| Solvent | Dielectric constant34 | Crystal forms identified by PXRD (in order of composition) |
|---|---|---|
| a Solid obtained from filtering a slurry. b Solid obtained after evaporation of the solution resulting from this filtration. c Dimethylsulphoxide. d N,N-Dimethylformamide. e t-Butylmethylether. | ||
| Watera | 80.1 | Phenazine α, phenazine dihydrate |
| Waterb | 80.1 | Mesaconic acid |
| DMSOc | 47.2 | Phenazine β, phenazine α, cocrystal Form I |
| DMFd | 36.6 | Phenazine α, phenazine β |
| Acetonitrile | 38.3 | Cocrystal Form I |
| Methanol | 33.0 | Phenazine α, cocrystal Form I, mesaconic acid |
| Acetone | 21.0 | Cocrystal Form I, phenazine α, mesaconic acid, phenazine β |
| 1-Butanol | 17.8 | Cocrystal Form I, phenazine α |
| Dichloromethanea | 8.9 | Mesaconic acid |
| Dichloromethaneb | 8.9 | Phenazine α, cocrystal Form I |
| Chlorobenzenea | 5.7 | Mesaconic acid |
| Chlorobenzeneb | 5.7 | Phenazine β, phenazine α |
| Diisopropyl ether | 3.8 | Cocrystal Form I |
| TBMEe | 3.1 | Cocrystal Form I, phenazine α |
| Cyclohexanea | 2.0 | Mesaconic acid, cocrystal Form I |
| Cyclohexaneb | 2.0 | Phenazine α |
Cocrystallisation did occur during this solution based polymorph screen, but only the known Form I was identified. Importantly, in 7 of the 12 experiments, Form I was either obtained as a minor component in a mixture with unconverted phenazine and/or mesaconic acid or not observed at all, making the screen highly inefficient. The likely reason for this inefficiency is the very different solubility profiles of phenazine and mesaconic acid, with the hydrophilicity of mesaconic acid giving it a high solubility in polar solvents and low solubility in non-polar solvents, whereas phenazine is strongly hydrophobic and has opposite solubility properties. As might be expected, the cocrystal was obtained as a majority phase only when the solubility of both compounds in the solvent used for crystallisation was similar. Importantly, the cocrystal was not obtained from solvents at either end of the polarity scale, which will have had the effect of reducing the experimental space that was probed during the solution polymorph screen.
Cocrystallisation at the interface between two immiscible solvents
An alternative cocrystallisation method which exploits the differences in solubility of the components was therefore employed in which two saturated solutions, one of mesaconic acid in a polar solvent, and one of phenazine in a non-polar solvent immiscible with that used for the mesaconic acid solution were brought into contact. It was expected that cocrystallisation would occur at the solvent interface if the cocrystal of the two compounds is thermodynamically more stable, and hence less soluble, than either of the individual components. A key advantage of this technique is that by starting with solutions of the coformers that are not supersaturated, the possibility of crystallising either compound as a single component phase is avoided.Saturated solutions of phenazine in xylene and mesaconic acid in water were prepared, and the phenazine solution was layered onto the aqueous mesaconic acid solution in a 2.5 cm diameter glass vial. A precipitate formed at the solvent interface within a few seconds and was isolated on a filter paper. The resulting needle-shaped crystals were found to be a monohydrate of a 1:1 phenazine:mesaconic acid cocrystal and a full single crystal X-ray structure of this phase was obtained from interface grown crystals. The monohydrate crystallises with an orthorhombic Pna21 structure having unit cell parameters of a = 27.495 Å, b = 14.043 Å and c = 4.0898 Å. Water molecules form columns which are bridged in the perpendicular plane by a molecule of phenazine and a molecule of mesaconic acid which interact via an OH⋯N hydrogen bond (Fig. 2). A comparison of simulated and experimental PXRD traces showed that the single crystal used to obtain the crystal structure was representative of the bulk of the sample. This monohydrate was found to dehydrate to Form I of the phenazine:mesaconic acid cocrystal during 10 days of storage under ambient conditions.
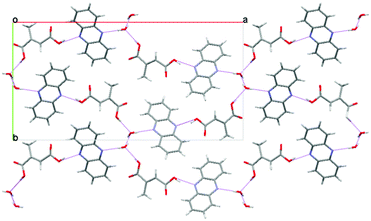 | ||
| Fig. 2 The crystal structure of the monohydrate of the 1:1 phenazine:mesaconic acid cocrystal viewed down the c-axis. | ||
After removing the precipitate of the cocrystal monohydrate from the solvent interface, the vial was re-sealed and stored under ambient conditions. A second crop of precipitate formed slowly at the interface over several days. The PXRD trace of this sample was different to those of both the monohydrate and Form I of the phenazine:mesaconic acid cocrystal, indicating that another new crystal form had been obtained. Crystals from this second crop were suitable for single crystal X-ray structure determination, revealing that it was a second anhydrous polymorph of the cocrystal (Form II). Form II crystallises with an orthorhombic Pna21 structure having unit cell dimensions of a = 16.5230 Å, b = 15.9952 Å and c = 5.4792 Å. Phenazine and mesaconic acid molecules form hydrogen bonded chains (Fig. 3), as in Form I, but there is a different 3-dimensional packing arrangement of the chains. A comparison of simulated and experimental PXRD traces showed that the single crystal used to obtain the crystal structure was representative of the bulk sample.
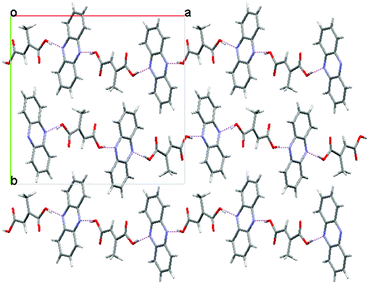 | ||
| Fig. 3 The crystal structure of Form II of the 1:1 phenazine:mesaconic acid cocrystal viewed down the c-axis. | ||
Cocrystal polymorph screening by grinding
Phenazine and mesaconic acid were ground in equimolar amounts in a ball mill, both dry and with the addition of a small volume of liquid (liquid assisted grinding)15 to promote cocrystal formation. Dry grinding phenazine with mesaconic acid for 30 minutes resulted in conversion to Form I of the phenazine:mesaconic acid cocrystal, a result which matches previous findings reported by Nguyen et al.31 Liquid assisted grinding was performed using each of the solvents selected for the solution cocrystallisation screen (in each case 20 μl of liquid was added). The resulting solids were analysed by PXRD and the observations are summarised in Table 2. Both Form I and Form II of the cocrystal were isolated from this cocrystallisation screen, and it was found that grinding with polar liquids (high dielectric constant) tended to favour generation of Form II, whereas non-polar liquids gave Form I. The main exception to this trend was grinding with water, where Form I was obtained despite the existence of a monohydrate of the cocrystal and water being a liquid with a high dielectric constant.:1 molar ratio of phenazine and mesaconic acid with 12 different liquids
| Liquid | Dielectric constant34 | Cocrystal form as identified by PXRD |
|---|---|---|
| a 100 μl of solvent was used in this experiment. | ||
| Water | 80.1 | Form I |
| DMSOa | 47.2 | DMSO cocrystal solvate |
| DMF | 36.6 | Form II |
| Acetonitrile | 38.3 | Form II |
| Methanol | 33.0 | Form I |
| Acetone | 21.0 | Form I |
| 1-Butanol | 17.8 | Form I |
| Dichloromethane | 8.9 | Form I |
| Chlorobenzene | 5.7 | Form I |
| Diisopropyl ether | 3.8 | Form I |
| TBME | 3.1 | Form I |
| Cyclohexane | 2.0 | Form I |
In the case of liquid assisted grinding with DMSO evidence of a new crystal form was obtained with small additional peaks appearing in the PXRD trace. A second grinding experiment was conducted with a greater volume (100 μl) of DMSO. The PXRD trace of this sample was different to those of the known forms of phenazine and mesaconic acid, indicating that a new crystal form had been isolated (Fig. 4). This form converted to Form II of the cocrystal during heating at 90 °C for three hours. In addition, during TGA analysis there was a weight loss of 17.0% between 25 and 150 °C. This weight decrease occurs below the point at which sublimation of the cocrystal was observed (170 °C), and so is assumed to be solvent loss corresponding to 0.82 moles of DMSO, indicating that the form is a mono DMSO solvate. To date it has not been possible to obtain the crystal structure of the DMSO solvate since upon attempting to crystallise phenazine and mesaconic acid from DMSO phenazine crystallises from solution in preference to the cocrystal.
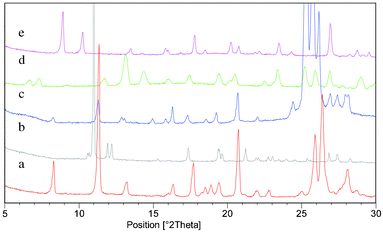 | ||
| Fig. 4 PXRD overlay of the five forms of the 1:1 phenazine:mesaconic acid cocrystal that were isolated from the polymorphism screen: (a) Form I, (b) Form II, (c) Form III (measured at 175 °C), (d) monohydrate and (e) DMSO solvate. | ||
Cocrystal polymorph screening by sublimation
A sample containing equimolar amounts of phenazine and mesaconic acid (as cocrystal) was placed on a glass slide and heated to 175 °C while a second, cooler, glass slide was held in place above the sample. Solid which sublimed and crystallised on the second glass slide was analysed by PXRD and found to be Form I of the phenazine:mesaconic acid cocrystal.Cocrystal polymorph screening by thermal methods
Polymorphism of the phenazine:mesaconic acid cocrystal was investigated both by crystallisation of a melt phase of phenazine and mesaconic acid and by screening for form changes that occur on heating the cocrystal. Firstly, a 1:1 physical mixture of phenazine and mesaconic acid was heated to 195 °C, causing both compounds to melt. Crystallisation occurred on cooling to ambient temperature, and PXRD analysis showed that Form I of the cocrystal had been obtained. Secondly, a sample of Form I of the phenazine:mesaconic acid was heated by DSC (Fig. 5). A small endotherm was observed at 161 °C, below the melting point of the sample at 183 °C, suggesting that a form change occurs prior to melting. In a second DSC experiment, the cocrystal was heated to 170 °C and then cooled back to ambient temperature. During the cooling phase, an exotherm was observed, and PXRD analysis of the solid resulting from this experiment showed it to be Form I cocrystal, demonstrating that the form change is reversible.
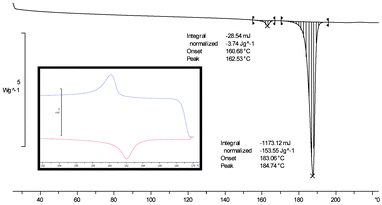 | ||
| Fig. 5 DSC thermogram of Form I of the 1:1 phenazine:mesaconic acid cocrystal. The inset shows the reversible nature of the event that occurs below the melting point of the cocrystal. | ||
Variable temperature PXRD (VT-PXRD) measurements were made on Form I of the cocrystal to investigate the form change further. The sample was held for two minutes at each temperature to equilibrate prior to PXRD analysis. Clear differences in PXRD traces recorded at 145 °C and 175 °C showed that a form change occurs between these temperatures (Fig. 6). This is a solid–solid transition from Form I of the phenazine:mesaconic acid cocrystal to a third polymorph of the cocrystal (Form III). As the form change is reversible, Form III must be a metastable, high temperature polymorph, with Forms I and III related enantiotropically.
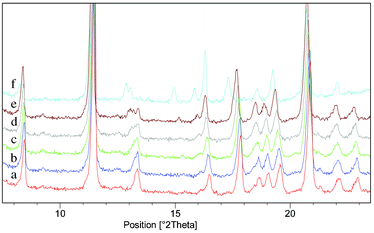 | ||
| Fig. 6 VT-PXRD analysis of a sample which was initially Form I of the phenazine:mesaconic acid cocrystal: (a) 25 °C. (b) 55 °C. (c) 85 °C. (d) 115 °C. (e) 145 °C. (f) 175 °C. A form change from Form I to Form III occurred between 145 °C and 175 °C. | ||
Discussion
The various different types of polymorph screening experiments that were performed on the phenazine:mesaconic acid cocrystal generated two new polymorphic forms of this cocrystal as well as a monohydrate and a DMSO solvate. An overlay of PXRD traces of all five forms of the cocrystal is shown in Fig. 4. Form II is the thermodynamically stable form under ambient conditions (during a competitive slurry experiment conducted at room temperature in diisopropyl ether a 1:1 mixture of Forms I and II fully converted to Form II within 4 days), but Form I can also be retained under ambient conditions for several months without conversion to Form II.
Table 3 summarises the different polymorph screening approaches that generated each form of the cocrystal. It could be concluded that liquid assisted grinding was the most important single method used in the screen as the greatest number of crystal forms were isolated using this approach, but perhaps the more important observation is that there was no single approach which yielded all five forms of the cocrystal.
:1 phenazine:mesaconic acid cocrystal
| Cocrystal form | Cocrystallisation methods which have yielded this form |
|---|---|
| Form I | Solution crystallisation, dry grinding, liquid assisted grinding, sublimation, thermal methods, desolvation of the monohydrate |
| Form II | Cocrystallisation at the interface between two saturated solutions, liquid assisted grinding, desolvation of the DMSO solvate |
| Form III | Thermal methods |
| Monohydrate | Cocrystallisation at the interface between two saturated solutions |
| DMSO solvate | Liquid assisted grinding |
It is noteworthy that the solution crystallisation screen yielded only one of the five cocrystal forms, Form I. While Form III is unstable under ambient conditions, and would not be expected to crystallise from solution, Form II was obtained reproducibly by liquid assisted grinding, desolvation and interfacial growth and so it is perhaps surprising that it was not obtained from solution crystallisations, especially as it is the thermodynamically stable form at ambient temperature. It is also surprising that neither the monohydrate nor the DMSO solvate of the cocrystal were obtained during crystallisations from water and DMSO respectively, but this observation can be rationalised in terms of the much greater solubility of mesaconic acid than phenazine in these solvents. These findings highlight the danger of using solution crystallisation based approaches in isolation when screening for polymorphs of cocrystals, as is common in screens on single compounds, as an incorrect understanding of the propensity of a cocrystal to form different polymorphs may be obtained.
In contrast, with cocrystallisation at the interface between two immiscible solvents not only were two new crystal forms of the phenazine:mesaconic acid cocrystal isolated, but both phases were obtained as pure forms containing no unconverted phenazine or mesaconic acid, and with a crystal size suitable for crystal structure determination. This is a significant improvement over the more conventional solution crystallisation experiments where mixtures of phases were obtained. Given that cocrystallisation at the interface between two immiscible solvents is a facile, solution based technique it could be an important way of producing cocrystals on a larger scale than used here, but would be equally amenable to dealing with small amounts of material.
Conclusions
Polymorph screening on the phenazine:mesaconic acid cocrystal yielded five cocrystal forms including three anhydrous polymorphs, a monohydrate and a DMSO solvate.Solution crystallisation experiments, however, gave no indication of this rich polymorphic behaviour, primarily we believe because of the large solubility differences between phenazine and mesaconic acid, meaning that coformers crystallised separately in preference to forming a cocrystal. In general, solution based polymorph screening may be less effective for cocrystals than it is for single component phases as the range of solvents that can be employed will be limited to those in which the two cocrystal formers have approximately equal solubility.36 Wherever large solubility differences between coformers exist, such as with pharmaceutical cocrystals where highly soluble coformers are crystallised with hydrophobic APIs to improve solubility, this could be especially problematic. There is also indication that during solution based crystallisation screens new cocrystal forms might in some cases be obtained as minor components in mixtures, and analytical techniques such as transmission electron microscopy, which are ideal for characterising such samples,35 may become important.
Cocrystallisation at the interface between two immiscible solutions was found to be a simple method of preparing the phenazine:mesaconic acid cocrystal in a pure form, where the likelihood of contamination from unconverted phenazine or mesaconic acid is removed, while retaining the benefits of a solution based approach. This cocrystallisation method produced the thermodynamically stable polymorph, Form II.
At least three different cocrystallisation methods were needed to isolate all of the identified crystal forms of the phenazine:mesaconic acid cocrystal, suggesting that when screening for polymorphs of cocrystals a multi-technique approach should be employed.
Acknowledgements
The authors thank Dr. John E. Davies for the crystal structure determinations, and acknowledge the EU INTERREG IVA 2 Mers Seas Zeeën Cross-border Cooperation Programme, the EPSRC and the Old Bancroftians Association Trust Fund for financial support.References
- J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R. Guzman and O. Almarsson, J. Am. Ceram. Soc., 2003, 125, 8456–8457 CAS.
- N. J. Babu and A. Nangia, Cryst. Growth Des., 2011, 11, 2662–2679 CAS.
- A. M. Thayer, Chem. Eng. News, 2007, 17–30 Search PubMed.
- M. L. Peterson, M. B. Hickey, M. J. Zaworotko and O. Almarsson, J. Am. Pharm. Assoc., 2006, 9, 317–326 CrossRef.
- Z. Li, B.-S. Yang, M. Jiang, M. Eriksson, E. Spinelli, N. Yee and C. Senanayake, Org. Process Res. Dev., 2009, 13, 1307–1314 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- S. L. Childs, P. A. Wood, N. Rodriguez-Hornedo, L. S. Reddy and K. I. Hardcastle, Cryst. Growth Des., 2009, 9, 1869–1888 CAS.
- S. Karki, T. Friscic, L. Fabian and W. Jones, CrystEngComm, 2010, 12, 4038–4041 RSC.
- A. Y. Sheikh, S. Abd. Rahim, R. B. Hammond and K. J. Roberts, CrystEngComm, 2009, 11, 501–509 RSC.
- R. A. Chiarella, R. J. Davey and M. L. Peterson, Cryst. Growth Des., 2007, 7, 1223–1226 CAS.
- T. Friscic and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CAS.
- S. Karki, L. Fabian, T. Friscic and W. Jones, Org. Lett., 2007, 9, 3133–3136 CrossRef CAS.
- G. G. Z. Zhang, R. F. Henry, T. B. Borchardt and X. Lou, J. Pharm. Sci., 2007, 96, 990–995 CrossRef CAS.
- D. K. Bucar, R. F. Henry, X. C. Lou, T. B. Borchardt and G. G. Z. Zhang, Chem. Commun., 2007, 525–527 CAS.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, Cryst. Growth Des., 2010, 10, 2229–2238 CAS.
- O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- P. Vishweshwar, A. M. Jennifer, L. P. Matthew, B. H. Magali, R. S. Tanise and J. Z. Michael, Chem. Commun., 2005, 4601–4603 RSC.
- M. B. Hickey, M. L. Peterson, L. A. Scoppettuolo, S. L. Morrisette, A. Vetter, H. Guzman, J. F. Remenar, Z. Zhang, M. D. Tawa, S. Haley, M. J. Zaworotko and O. Almarsson, Eur. J. Pharm. Biopharm., 2007, 67, 112–119 CrossRef CAS.
- N. J. Babu, L. S. Reddy, S. Aitipamula and A. Nangia, Chem.–Asian J., 2008, 3, 1122–1133 CrossRef CAS.
- W. W. Porter III, S. C. Elie and A. J. Matzger, Cryst. Growth Des., 2008, 8, 14–16 Search PubMed.
- J. H. ter Horst and P. W. Cains, Cryst. Growth Des., 2008, 8, 2537–2542 CAS.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2009, 11, 889–895 RSC.
- N. Schultheiss, M. Roe and S. X. M. Boerrigter, CrystEngComm, 2011, 13, 611–619 RSC.
- A. N. Sokolov, D. C. Swenson and L. R. MacGillivray, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1794–1797 CrossRef CAS.
- D. Braga, G. Palladino, M. Polito, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2008, 14, 10149–10159 CrossRef CAS.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2009, 11, 1823–1827 RSC.
- J. M. Miller, N. Rodriguez-Hornedo, A. C. Blackburn, D. Macikenas and B. M. Collman, Biotechnol.: Pharm. Aspects, 2007, 6, 53–109 CrossRef CAS.
- S. L. Morissette, O. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef CAS.
- K. L. Nguyen, T. Friscic, G. M. Day, L. F. Gladden and W. Jones, Nat. Mater., 2007, 6, 206–209 CrossRef CAS.
- E. Batchelor, J. Klinowski and W. Jones, J. Mater. Chem., 2000, 10, 839–848 RSC.
- S. Childs, Cocrystallization methods for active agents and guest molecules. WO Patent, 2006-US37509, 2007038524, 2007 Search PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 92nd Edition, 2011–2012. http://www.hbcpnetbase.com/ (Accessed March 2012) Search PubMed.
- M. D. Eddleston, E. G. Bithell and W. Jones, J. Pharm. Sci., 2010, 99, 4072–4083 CAS.
- K. Fucke, S. A. Myz, T. P. Shakhtshneider, E. V. Boldyreva and U. J. Griesser, New J. Chem., 2012, 36, 1969–1977 RSC.
Footnote |
| † CCDC reference numbers 901279 and 901280. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ce26496j |
| This journal is © The Royal Society of Chemistry 2013 |