Oxidative dehydrogenation of ethylbenzene to styrene over alumina: effect of calcination
Christian Nederlofa, Valeriya Zarubinab, Ignacio Melián-Cabrerab, Hero Jan (Eric) Heeresb, Freek Kapteijna and Michiel Makkee*a
aDelft University of Technology, Faculty of Applied Sciences, Chemical Engineering department, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: m.makkee@tudelft.nl; Fax: +31 15 278 5006; Tel: +31 15 278 1391
bUniversity of Groningen, Institute of Technology & Management, Chemical Engineering department, Nijenborg 4, 9747 AG Groningen, The Netherlands
First published on 14th November 2012
Abstract
Commercially available γ-Al2O3 was calcined at temperatures between 500 and 1200 °C and tested for its performance in the oxidative ethylbenzene dehydrogenation (ODH) over a wide range of industrially-relevant conditions. The original γ-Al2O3, as well as η- and α-Al2O3, were tested. A calcination temperature around 1000/1050 °C turned out to be optimal for the ODH performance. Upon calcination the number of acid sites (from 637 to 436 μmol g−1) and surface area (from 272 to 119 m2 g−1) decrease, whereas the acid site density increases (from 1.4 to 2.4 sites per nm2). Less coke, being the active catalyst, is formed during ODH on the Al-1000 sample compared to γ-Al2O3 (30.8 wt% vs. 21.6 wt%), but the coke surface coverage increases. Compared with γ-Al2O3, the EB conversion increased from 36% to 42% and the ST selectivity increased from 83% to 87%. For an optimal ST selectivity the catalyst should contain enough coke to attain full conversion of the limiting reactant oxygen. The reactivity of the coke is changed due to the higher density and strength of the Lewis acid sites that are formed by the high temperature calcination. The Al-1000 sample and all other investigated catalysts lost ODH activity with time on stream. The loss of selectivity towards more COX formation is directly correlated with the amount of coke.
Introduction
Alumina is a widely used catalyst support in the oxidative dehydrogenation (ODH) of hydrocarbons.1–4 Alumina itself is not a selective ODH catalyst; fresh Al2O3 shows about 20% ethylbenzene (EB) conversion with about 70% selectivity to styrene (ST) at a 0.6 O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EB molar feed ratio and 450 °C. In the first hours of the reaction, coke is deposited on the alumina and both EB conversion and ST selectivity increase, up to 36% EB conversion and 83% ST selectivity. The role of the alumina is to enable this essential coke formation. In the literature there are several studies that have investigated this phenomenon. An overview of the characterization data of several alumina samples in the ODH of EB is given in Table 1.
:EB molar feed ratio and 450 °C. In the first hours of the reaction, coke is deposited on the alumina and both EB conversion and ST selectivity increase, up to 36% EB conversion and 83% ST selectivity. The role of the alumina is to enable this essential coke formation. In the literature there are several studies that have investigated this phenomenon. An overview of the characterization data of several alumina samples in the ODH of EB is given in Table 1.
| Sample# | Initial material | Calcination temperature [°C] | SA [m2 g−1] | Number of acid centers [μmol g−1] | Crystalline structure | Cokea 120 min TOS [wt%] | CO2 yielda [%] | Cokea monolayer amount [wt%] |
|---|---|---|---|---|---|---|---|---|
| a These numbers were not determined under the same ODH conditions. Sample # corresponds to the samples of Lisovskii and Aharoni.1 | ||||||||
| 7 | Al(OH)3 | 1500 | 1.5 | 90 | α-phase | |||
| 1 | AlCl3 | 800 | 73 | 299 | Amorphous | 3 | 0.4 | 3 |
| 9 | θ-Al2O3 | — | 73 | — | θ-phase | 6 | 1.0 | 4.5 |
| 6 | Al(OH)3 | 1000 | 120 | 804 | γ-phase, high temperature | 11 | 1.6 | 6.5 |
| 8 | γ-Al2O3 | (commercial) | 152 | 839 | γ-phase | 15 | 1.9 | 8 |
| 3 | Al(OH)3 | 600 | 154 | 789 | γ-phase, low temperature | 14 | 2.0 | 6 |
| 5 | Al(OH)3 | 800 | 157 | 810 | 16 | 2.1 | 5.5 | |
| 4 | Al(OH)3 | 700 | 160 | 888 | 17 | 2.3 | 9 | |
| 2 | Al-isopropoxide | 700 | 235 | 1149 | Amorphous | 22 | 1.3 | 12 |
The acidic nature and specific surface area of the alumina play a crucial role in its activity for ODH. The acidic centres on the surface take part in the formation of coke deposits on the surface, which constitutes the actual catalytic phase for the selective ODH. Once a kind of monolayer of coke is deposited on the alumina, the amount of coke stabilizes, together with the highest yield of styrene and lowest yield of CO2.1,5,6 Several correlations for alumina were found between the amount of coke and:
- The number of acid sites on alumina.
- The amount of CO2 that is produced.
- The specific surface area and “monolayer” coke amount.
After a monolayer deposition of coke the coke formation still continues and results in a constant deactivation with less ST and more CO2 formation.
Reducing the acidity by adding KOH to the alumina reduces the amount of coke that is deposited. Also the addition of NH3 to the feed decreases the formation of coke. Increasing the acidity by adding H3BO3 to alumina increases the amount of coke that is deposited. The addition of mineral acids like H3BO3 or H3PO4 has another side effect, the composition of the coke deposits is changed. This resulted in a more active and selective catalyst.1
Another way to change the acidity of the alumina could be by a high temperature calcination. A heat treatment will, first of all, reduce the amount of bound water on the alumina surface. Two hydroxyl groups on the alumina surface that act as Brønsted acid sites can form a Lewis acid site upon release of a water molecule. This takes place at temperatures above 300 °C. Two-thirds of the surface hydroxyls can be removed without affecting the local order, when more hydroxyls are removed oxygen vacancies will be formed that behave as strong Lewis acid sites. Even at temperatures as high as 800–1000 °C and under vacuum, still a few tenths of a per cent of surface hydroxyls exist.7
By applying a heat treatment to γ-Al2O3, other crystalline phases can be obtained. At a calcination temperature of 900–1000 °C, it is converted into δ- and θ-Al2O3 crystalline phases. Above 1100 °C, it changes into α-Al2O3. The oxygen packing of γ-Al2O3 and η-Al2O3 is cubic close packing, which is transformed into hexagonal close packing for α-Al2O3. In γ-Al2O3 the (110) plane is predominant and in η-Al2O3 it is the (111) plane. The (111) plane has a higher density of Al3+ tetrahedral sites, making η-Al2O3 more acidic than γ-Al2O3.8 The transformation into α-Al2O3 takes place by a nucleation and growth process, whereby the pores rapidly grow in size, especially in the transformation stage from the θ to α phase (around 1000 °C).9,10
In this paper the effect of the calcination temperature on a commercially available γ-Al2O3 is reported with regard to the catalyst performance relationships (EB conversion, ST selectivity, stability, coke amount, acidity, and surface area) in the ODH reaction of EB to ST. The samples were tested using a screening protocol including two O2 :EB feed ratios and the use of two diluents (N2 and CO2).
Results
Catalyst characterization
Nitrogen adsorption analysis was done for all fresh samples. Ammonia TPD, XRD, and coke amount determination were done for selected fresh samples. Thermogravimetric analysis was done for all spent samples to determine the amount of coke. The data are given in Table 2. These results show that with increasing calcination temperature the surface area, pore volume, acidity, and amount of coke decrease. The ODH experiments were performed with a constant bed volume of catalyst (0.8 ml). The increasing sample mass reflects, therefore, the increasing sample density with increasing calcination temperature.| Sample name | XRD crystalline phase | Calcination temperature [°C] | Sample massa [mg] | SA [m2 g−1] | VP [ml g−1] | Dp [Å] | Number of acid sites [μmol g−1] | Coke 62 h TOS [wt%] | Tmax TGA [°C] |
|---|---|---|---|---|---|---|---|---|---|
| a The given sample mass is equivalent to 0.8 ml of catalyst volume. | |||||||||
| α-Al2O3 | α | 1432 | 10 | 0.50 | >1000 | 37.4 | 555 | ||
| boehmite | 500 | 601 | 24.8 | 504 | |||||
| η-Al2O3 | η | 590 | 386 | 0.20 | 21 | 23.5 | 494 | ||
| γ-Al2O3 | γ | 487 | 272 | 0.64 | 94 | 637 | 30.8 | ||
| Al-500 | γ | 500 | 532 | 271 | 0.65 | 96 | 32.7 | 512 | |
| Al-600 | γ | 600 | 554 | 255 | 0.64 | 100 | 31.3 | 512 | |
| Al-700 | γ | 700 | 506 | 239 | 0.64 | 107 | 24.9 | 504 | |
| Al-800 | γ | 800 | 549 | 214 | 0.64 | 120 | 29.0 | 513 | |
| Al-900 | δ | 900 | 622 | 179 | 0.61 | 136 | 540 | 25.8 | 520 |
| Al-1000 | θ | 1000 | 707 | 119 | 0.49 | 165 | 436 | 21.6 | 522 |
| Al-1050 | θ | 1050 | 572 | 101 | 0.46 | 182 | 398 | 20.8 | 522 |
| Al-1100 | θ | 1100 | 804 | 54 | 0.35 | 260 | 244 | 10.6 | 522 |
| Al-1150 | α | 1150 | 763 | 20 | 0.17 | 340 | 1.3 | ||
| Al-1200 | α | 1200 | 981 | 16 | 0.12 | 300 | 20 | 1.1 | |
The TGA profiles of the coked catalyst samples shown in Fig. 1 show the maximum in the oxidation rate of coke around 500 °C (oxidation temperature) for all the samples. Coke on the commercial η-Al2O3 is the most reactive with a maximum oxidation rate at 494 °C, coke on the α-Al2O3 is the least reactive with a maximum oxidation rate at 555 °C. The amount of coke on the calcined samples ranges from 10.6 wt% for Al-1100 up to 32.7 wt% for Al-500, all measured after 62 hours time on stream. Also a shift in the temperature of maximum coke combustion is observed for the calcined samples going from 512 °C for Al-500 to 522 °C for Al-1100.
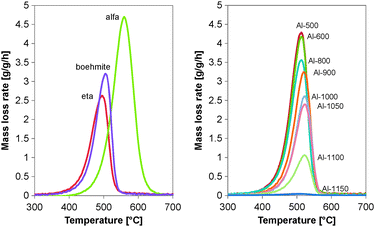 | ||
| Fig. 1 TGA profiles of coked commercial and calcined Al2O3 samples after 62 h TOS. | ||
The pore size (Table 2) shows a shift to larger pore sizes for the samples with higher calcination temperatures. The most dominant pore diameter shifts from 90–100 Å for the commercial γ-Al2O3 to 136 Å for the 900 °C sample. Samples calcined at higher temperatures show a further increase of the pore size, going up to 260 Å for the 1100 °C sample.
The NH3-TPD measurements show a clear decrease in the amount of NH3 desorbing from samples that are calcined at higher temperatures. The shape of the curves is similar with two desorption steps. One step around 200–350 °C and one step around 500–700 °C. At the same time the acid density or the number of acidic sites increase from 2.34 (μmol m−2 for bare γ-alumina) to 4.52 (Al-1100) or 1.4 to 2.4 (sites per nm2), respectively.
XRD analysis of some of the samples agrees well with the expected phases of transitional aluminas.9,10
Texture data from N2 adsorption on selected spent and coked samples are given in Table 3, together with the data of the fresh samples. For the commercial γ-Al2O3, one-third of the surface area and two-thirds of the pore volume is lost due to the coke deposition. The specific surface areas of the calcined samples hardly change, but about half of the pore volume is lost due to coke deposits on the samples.
| Sample | Fresh | Spent | ||
|---|---|---|---|---|
| SA [m2 g−1] | VP [ml g−1] | SA [m2 g−1] | VP [ml g−1] | |
| γ-Al2O3 | 272 | 0.64 | 173 | 0.25 |
| 900 °C | 179 | 0.61 | 152 | 0.28 |
| 1000 °C | 119 | 0.49 | 119 | 0.28 |
ODH experiments
The performance with time on stream (TOS) of all samples tested according to the screening protocol in the ODH reaction of EB to ST is shown in Fig. 2. To facilitate a comparison, the performance as a function of the calcination temperature is presented in Fig. 3. All oxygen was completely converted, unless stated otherwise. The role of oxygen is three-fold, namely: (1) to assist in the formation of carbon deposits (oxidation of adsorbed organics that polymerize at the surface of the alumina support); (2) to perform the oxidative dehydrogenation; and (3) to oxidize the deposited carbon to CO and CO2.1,3,4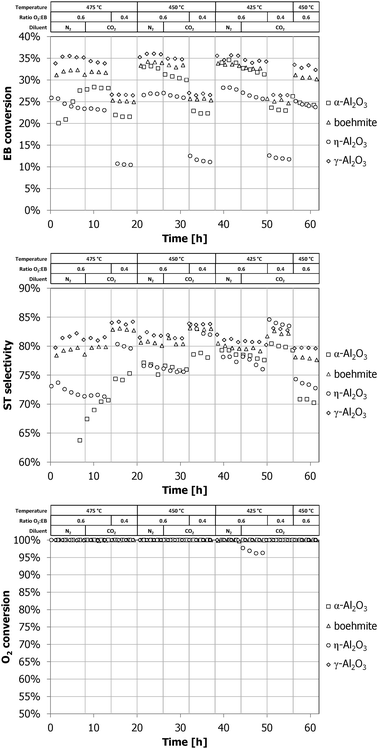 | ||
| Fig. 2 EB conversion, ST selectivity and O2 conversion of commercial alumina samples with time on stream. | ||
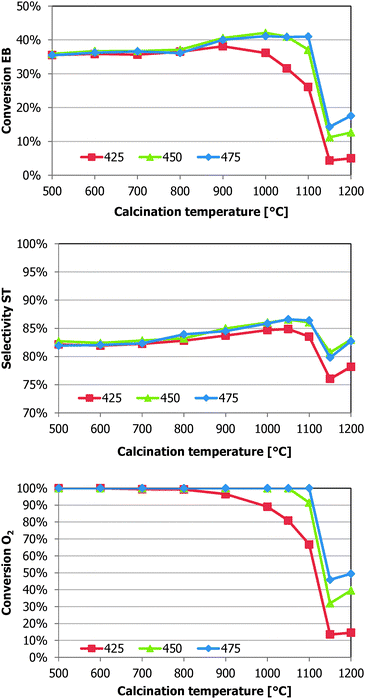 | ||
| Fig. 3 Styrene selectivity and conversions of EB and O2 for the alumina samples calcined at different temperatures. The data are time averages for the conditions 1 (475 °C, 4–8 h TOS), 4 (450 °C, 20–26 h TOS), and 7 (425 °C, 38–44 h TOS). All at a 0.6 O2:EB molar feed ratio. | ||
At 475 °C (0–14 h TOS), the samples Al-1100, Al-1050, and Al-1000 have the best performance. Their 42% EB conversion and 86% ST selectivity is an improvement over the 36% EB conversion and 83% ST selectivity for the reference γ-Al2O3. At 450 °C (20–32 h TOS), the samples Al-1050, Al-1000, and Al-900 have the best performance. The EB conversion of the Al-1100 sample decreased compared with operation at 475 °C. The available oxygen is not completely converted over the Al-1100 sample anymore. At 425 °C (38–50 h TOS), the Al-900 sample has the highest ST yield of 32%, with 38% EB conversion and 83% ST selectivity. Both the EB conversion and O2 conversion of the samples Al-900, Al-1000, and Al-1050 decreased 5%, 10%, and 20%, respectively, compared with operation at 450 °C. The 85% ST selectivity of the Al-1000 and Al-1050 samples is the highest of the series at 425 °C. Full oxygen conversion was not always attained at 425 °C. Samples calcined at higher temperatures had lower O2 conversions. No full oxygen conversion was reached over the Al-1150 and Al-1200 samples under all investigated reaction conditions. An incomplete oxygen conversion results in a lower selectivity due to the increased formation of partly oxygenated aromatics (tars) and an increased COX formation.
Of the commercial samples shown in Fig. 2, α-Al2O3 takes longest (10 h TOS) to become active and selective. At 425 °C it reaches its highest EB conversion (34%) and ST selectivity (79%), only slightly lower than that of γ-Al2O3. The γ-Al2O3 material is used as the basis for the calcined samples. The EB conversion (36%) and ST selectivity (82%) of the commercial γ-Al2O3 is not significantly different from the Al-500, Al-600, and Al-700 samples. The boehmite sample, a precursor for γ-Al2O3, gives an EB conversion (34%) and ST selectivity (81%) that is only a little lower than that of the commercial γ-Al2O3. The η-Al2O3 sample neither becomes very active nor selective at 450 °C after 20 h TOS. Both its EB conversion (27%) and ST selectivity (77%) are lower than those of the other commercial samples.
The stability in performance of the samples, calculated as the ratio of the ST yield at the 10th condition (56–62 h TOS) and the yield at the 5th condition (26–32 h TOS), is shown in Fig. 4, together with the ST yields under these conditions. These two reaction conditions are identical and optimal for our reference γ-Al2O3 sample: 450 °C, molar feed ratio O2 :EB = 0.6 and CO2 :EB = 5. It shows that the calcined γ-Al2O3 samples all have a very similar stability performance, they deactivate about 7% during this period of operation. The η-Al2O3 and boehmite samples are less stable and deactivate about 11%. The α-Al2O3 sample deactivates the most, the yield decreased 25%. The samples that do not show full oxygen conversion at 450 °C (Al-1150 and Al-1200) show the highest stability and only deactivate 5% or less, although their ST yields are the lowest for all measured samples.
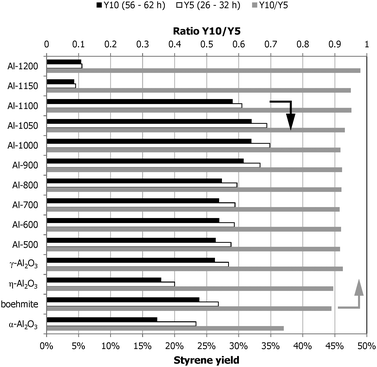 | ||
| Fig. 4 ST yields and performance stability (grey) as the ratio of the styrene yield at the 5th (white, 26–32 h TOS) and 10th (black, 56–62 h TOS) condition, both at 450 °C and a 0.6 O2:EB molar feed ratio. | ||
Discussion
The high temperature calcination of alumina (900–1100 °C) is favourable for both EB conversion and ST selectivity in the oxidative dehydrogenation reaction. By calcination of γ-Al2O3, the samples are dehydrated, the surfaces anneal and they are sintered, while other crystalline phases are being formed. As expected, the δ-phase is formed at 900 °C, and the θ-phase is formed at 1000 °C and higher (Table 1). With increasing calcination temperature, the specific surface area decreases. Also, the mass based acidity, specific surface area, and pore volume of the alumina are reduced. The Al2O3 density increases by the sintering process and more sample is loaded in the reactor for a constant testing volume. Actually, in this study it appears that these four characterization parameters (pore volume, acidity, surface area, and coke) are closely related to each other. As shown in Fig. 5, all these follow a similar trend. Their absolute amounts present in the reactor are almost constant for calcination temperatures up to 900 °C. The loss in the weight based properties (Table 2) is compensated by the density increase. The specific pore volume, surface area, and the amount of coke on the catalyst decrease up to this calcination temperature. Around 900 °C there seems to be a tipping point, all of the parameters decrease more pronounced above this temperature. At this point the crystalline phase of the alumina starts to change from δ to θ. Only the decrease in specific surface area seems to change a little faster than the other parameters.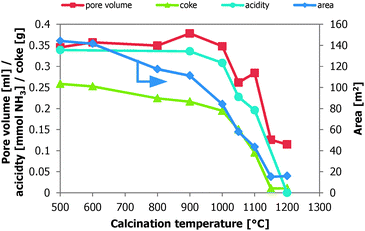 | ||
| Fig. 5 The absolute amounts of pore volume (■), acidity (●), coke (▲), and surface area (◆) in the reactor as a function of the calcination temperature of the alumina samples. | ||
As was expected, less acidic alumina forms less coke deposits,1 but high temperature calcination also gives a large improvement in the ODH performance of EB to ST (Fig. 3). This can be the result of three effects:
(1) the type of coke formed is different and is more active for dehydrogenation;
(2) the production of COX is proportional to the amount of coke;
(3) the accessibility of the coke is improved.
The first effect was also reported when mineral acids (H3BO3 and H3PO4) were added, the C:H ratio and the C:O ratio of the coke increased, as well as the amount of coke.1,11 The type of coke, however, also changes with time on stream and the operating temperature (aging).1 The TGA profiles of the calcined alumina samples shift to higher temperatures for higher calcination temperatures (Fig. 1) and indicate that the reactivity of the coke is different. However, after 62 h time on stream and 10 different operational conditions, it will be very hard to explicitly identify which one of these parameters is responsible for the change in the ODH performance.
The second effect is more straightforward. By lowering the acidity and the surface area, the steady-state amount of coke is reduced. The samples with high surface area and acidity (calcined <900 °C) form much more coke than is necessary for the dehydrogenation reaction, resulting in relatively more gasification of coke and thus loss of selectivity to COX. The samples that have the lowest surface areas have built up about enough coke. This is nicely depicted by the Al-1100 sample shown in Fig. 3. At 475 °C it has an almost identical performance as the samples of lower calcination temperature, while at 450 °C and 425 °C its O2 and EB conversion decrease, as well as its selectivity. The other samples still have enough coke to compensate the lower activity due to the decrease in the reaction temperature. This lower EB conversion with incomplete consumption of oxygen will also result in a loss in ST selectivity due to the higher COX production.
The third effect has been described to explain differences in the ODH performance between several samples with varying pore size distributions. As a result of the sample properties and the coke deposition, some micropores/mesopores could still be accessible to O2 only (molecular diameter 0.35 nm) and not to EB (molecular diameter 0.7 nm). The oxidation of coke can continue in these regions, deteriorating the ODH performance, until EB has access again.6,12 Within the alumina series there is also a clear change in the pore volume for calcination at 1000 °C and above. This accessibility effect can play a role in the observed ODH performance.
Another phenomenon that is observed in this work is the loss of selectivity at an incomplete oxygen conversion (Fig. 3). More COX and heavy condensates are formed. These are both undesired products that do not represent any value and moreover the heavy condensates can cause fouling issues downstream and result in additional operation costs in the downstream equipment. The residual oxygen can also lead to unwanted process conditions in a commercial operation. This emphasises the need to attain complete oxygen conversion. At full oxygen conversion, the improvements in styrene selectivity and ethylbenzene conversion are clearly coupled. In Fig. 3 it can be seen that between 800 and 1100 °C, both go up simultaneously as a higher styrene selectivity implies that less O2 is spent on COX formation, leaving more O2 for ethylbenzene conversion into styrene, resulting in a higher EB conversion and an enhanced ST selectivity.
When the styrene yield is plotted as a function of the amount of coke after 62 h TOS (Fig. 6), these trends become clear. Note that only the last performance data (after 56–62 h TOS) will correspond to the measured amounts of coke. It should be noted that for all our investigated samples after an activation time interval a continuous deactivation with time on stream is observed due to excessive coke deposition, accompanied by a loss in styrene selectivity and a gain in CO2 selectivity. This observation is in contrast to the literature where it is claimed that the amount of coke is reported to reach a steady-state level.1 With low amounts of coke in the reactor at 450 °C (less than 0.15 g), not all oxygen is converted and styrene yields are lower. At 475 °C, an amount of 0.10 g of coke is sufficient to convert all the oxygen and the performance is best at this point. At 475 °C, the samples that have higher amounts of coke exhibit a lower styrene yield. With 0.15–0.20 g coke, also all oxygen is converted at 450 °C and this is the optimum as shown in Fig. 6. In the range of 0.15–0.20 g, the differences are small, and this is the optimal region to operate under the applied conditions. For higher temperatures the range becomes lower, 0.10–0.15 g. Above 0.20 g of coke the loss of ST yield due to increased COX formation becomes clearly visible for both temperatures. Fig. 6 also shows the deactivation of the samples with time on stream. This deactivation can be caused by two effects: increasing amounts of coke or an aging effect of the coke. More data on the coke with time on stream are required, however, to clarify this aspect.
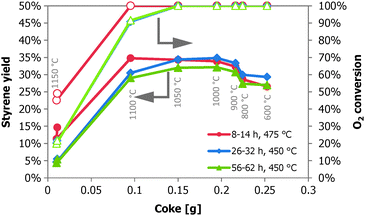 | ||
| Fig. 6 Styrene yield (◆▲●) and oxygen conversion (◇△○) as a function of the amount of coke on the catalyst in the reactor (62 h TOS) for the performance at 450 °C/56–62 h (▲△), at 450 °C/26–32 h (◆◇) and at 475 °C/8–14 h (●○). Other conditions: 0.6 O2:EB molar feed ratio, and 5 CO2:EB molar feed ratio. | ||
Another way to look at the available characterization data is to relate it to the specific surface area and correlate it with the ODH performance (Fig. 7). The average number of coke layers are calculated using the theoretical coverage of graphene (0.76 mg m−2). The acid site density is in the same order of magnitude as determined in the literature (max. 9.3–14.3 sites per nm2).13 The number of coke layers and the acid site density increase with the calcination temperature, because the surface area decreases faster than the other parameters. The NH3-TPD analysis reveals that the ST yield increases with increasing acid site density (from 1.4 to 2.7 sites per nm2) and coke coverage (from 2.4 to 3.4 layers). The measured coke coverages are well above the amounts that are reported for a monolayer of coke (0.54–0.76 mg m−2), similar to the data in the column ‘coke 120 min TOS’1 in Table 1 which corresponds to coverages up to 1.5 layers. This can have the following implications: (1) the catalysts are in the process for further deactivation and the presented data after 62 h TOS, possibly due to very high coke coverage, making these coke data not representative to compare with the ST yield; (2) more of the available catalyst surface is used for the ODH reaction instead of the unselective gasification/oxidation of the deposited coke, resulting in a higher coke coverage, the data are the average of the whole catalyst bed; and (3) a higher coke coverage is required for an improved ODH performance.
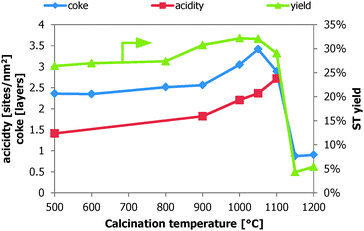 | ||
| Fig. 7 Acid site density, # coke layers and ST yield over the calcined alumina as a function of the calcination temperature. | ||
The η-Al2O3 sample should have the highest acidity (not measured) and, therefore, should produce a lot of coke. The TGA analysis shows that the amount of coke after 62 h TOS is less than for the γ-Al2O3, but in spite of this its ODH performance is worse. It is hardly better than the performance of un-coked or fresh γ-Al2O3. Based on the TGA profile, the type of coke is different and is easier to combust (25 °C lower than that of the high temperature calcined samples (Al-1000 and Al-1050), see Fig. 1 and Table 2). In the ODH reaction this can result in higher selectivity to COX, possibly amplified by diffusional resistance in its smaller pores compared to the γ-Al2O3. When coke is formed faster, but at the same time is gasified/oxidized faster too in these small pores, it will result in a less coke deposition, but a higher selectivity to CO/CO2.
An NH3-TPD analysis cannot differentiate between Brønsted and Lewis acid sites. It only distinguishes the weak from strong acid sites. Pyridine-IR-spectroscopy is needed for this. However, it is well-known that high temperature calcination will lead to the removal of the hydroxyl group from the surface and, thereby, decreasing the number of Brønsted acid sites and creating Lewis acid sites. In our case we observed some decrease in the number of acid sites on a mass basis, but at the same time the BET surface area was decreasing to a larger extent, leading to a higher density of acidic sites per surface area and of a higher strength. Taking these two effects together one can rationalize that the strength and density of Lewis sites increases by the high temperature calcination. It can be hypothesized that these properties are favourable for the selectivity and activity of the coke that is formed. The coke is more difficult to combust, leading to less COX and more ST. The coke that forms on the Brønsted and weak Lewis acid sites on the η- and γ-Al2O3 surface is apparently more reactive for combustion and, implicitly, less selective for ST formation.
The development of a more active and selective catalyst for EB ODH is apparently a complex challenge. Coke is required for activity and selectivity, but there should be an optimum amount of coke. The activity and the ST selectivity are coupled because full conversion of the limiting reactant oxygen is required in this operation. To enable an improvement in ST selectivity, less O2 has to be consumed to produce COX, which is related to the amount of coke, the type of coke, and its accessibility. Too much coke is, therefore, undesired, but too little as well because sufficient coke, the catalyst in this process, is required to achieve the full and selective conversion of oxygen. It will be clear that the optimal ST yield requires a fine tuning of the operational conditions and Lewis acidity of the catalyst.
Experimental
Catalyst preparation
The γ-Al2O3 extrudates (Albemarle, 272 m2 g−1, 0.64 ml g−1) were crushed and sieved to 212–425 μm. Each time, 2 g of sample was put in a ceramic cup and calcined for 8 h in a static air calcination oven. The maximum calcination temperature was 1200 °C, the temperature safety limit of this oven. Other calcination temperatures were 500, 600, 700, 800, 900, 1000, 1050, 1100, and 1150 °C. The heating rate was set at 4 °C min−1. The suffixes in the names of the samples correspond to their calcination temperatures. Other alumina samples that were used in this paper are mentioned in Table 4.| Name | SA [m2 g−1] | VP [ml g−1] | Other details |
|---|---|---|---|
| γ-Al2O3 (Albemarle) | 272 | 0.64 | Albemarle |
| η-Al2O3 | 386 | 0.20 | ETA-EXTRUDATE 1.8/360 Sasol Germany GmbH |
| α-Al2O3 | ∼10 | 0.5 | Engelhard Al-4196E 1/12 |
| Boehmite | Sasol Catapal B alumina (boehmite) |
Catalyst testing ODH of EB
The oxidative dehydrogenation experiments are performed in a parallel fixed bed reactor setup with six quartz reactors with an inner diameter of 4 mm, in down-flow operation. In a typical experiment a catalyst bed height of 65 mm is used (0.80 ml). The reactors are loaded – from top to bottom – with a quartz wool plug, 10 cm glass pearls (0.5 mm diameter), the catalyst, 10 cm glass pearls (0.5 mm diameter), and a quartz wool plug. A quartz reactor tube, filled only with glass beads and quartz wool plugs, only showed up to 3% EB conversion under all applied conditions. The catalyst bed is positioned within the 20 cm isothermal zone of the reactor. The reactor gas feed is a mixture that can consist of CO2, N2, and air. To each reactor, the gas-flow rate is 36 ml (NTP) per min, the liquid EB feed flow rate is 1 g h−1 (3.6 ml (NTP) per min vapour), resulting in a 1:10 molar ratio of ethylbenzene and gas with an EB GHSV (gas hourly space velocity) of 3000 l l−1 h−1. The ethylbenzene liquid is fed into a heated α-Al2O3 filled tube, where it is evaporated in a concurrent flow with the gas feed. This evaporator is located in an oven, where the reactor furnace is also installed.The reactors are heated up under nitrogen flow with 5 °C min−1. The temperature is allowed to stabilize for another 15 minutes when the reaction temperature is reached. The EB flow is started and only after 1–5 minutes oxygen is added to the gas mixture in order to prevent any pre-oxidation of the catalyst samples. The reaction starts when oxygen is added to the reactors. The pressure at the setup outlet is atmospheric, the pressure drop over the reactors is typically about 0.2–0.3 bar.
A testing protocol of 10 conditions is used to test the catalyst samples. The protocol consists of three temperatures, going from high to low, because this is the optimal way for the activation of the catalyst (initial coking of the catalyst under reaction conditions). Two O2:EB molar feed ratios are used, and two CO2:EB molar feed ratios to see the effect of these gases on the reaction. One condition is tested twice to see the aging effect of the catalyst (the 5th and 10th condition). An experiment takes 62 hours. The testing protocol is presented in Table 5.
| Condition | Time on stream [h] | Temp. [°C] | O2:EB | CO2:EB |
|---|---|---|---|---|
| 1. | 0–8 | 475 | 0.6 | 0 |
| 2. | 8–14 | 475 | 0.6 | 5 |
| 3. | 14–20 | 475 | 0.4 | 5 |
| 4. | 20–26 | 450 | 0.6 | 0 |
| 5. | 26–32 | 450 | 0.6 | 5 |
| 6. | 32–38 | 450 | 0.4 | 5 |
| 7. | 38–44 | 425 | 0.6 | 0 |
| 8. | 44–50 | 425 | 0.6 | 5 |
| 9. | 50–56 | 425 | 0.4 | 5 |
| 10. | 56–62 | 450 | 0.6 | 5 |
During the ODH experiments, the concentrations of the reactor effluent are measured online by a two channel gas chromatograph with a TCD (columns: 0.3 m Hayesep Q 80–100 mesh with back-flush, 25 m × 0.53 mm Porabond Q, and 15 m × 0.53 mm molsieve 5A with bypass option for CO2 and H2O, all in series) for permanent gas analysis (CO2, H2, N2, O2, CO) and a FID (column: 30 m × 0.53 mm, Df = 3 μm, RTX-1) for the hydrocarbons analysis (methane, ethane, ethene, benzene, toluene, ethylbenzene, styrene, heavy aromatics). The total analysis time takes 15 minutes. Six reactors and one reference gas analysis gives that each reactor is analysed every 1¾ h in order to follow the catalyst performance with time on stream. A constant flow of nitrogen is used as the internal standard. The internal standard was fed just before the GC. The lines from the reactor to the GC are heat-traced at 175 °C to prevent condensation of vapours. The EB conversion, ST selectivity, ST yield, and O2 conversion are based on moles of ethylbenzene, and calculated according to eqn (1)–(4). The overall carbon balance is >99%, only the carbon that is deposited on the catalyst as coke is missing from the carbon balance by the GC analysis. The overall oxygen balance is closed.
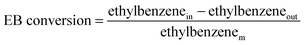 | (1) |
 | (2) |
| ST yield = EB conversion × ST selectivity | (3) |
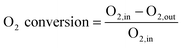 | (4) |
Catalyst characterization
The amount of coke on the spent catalyst samples was determined by thermogravimetric analysis (TGA) in synthetic air using a Mettler-Toledo analyzer (TGA/SDTA851e). Alpha alumina crucibles (90 μl) were filled with 5–10 mg of sample and the weight loss was monitored in a flow of air of 100 ml min−1. The temperature was increased from room temperature up to 900 °C at a rate of 10 °C min−1. A blank experiment is performed employing an empty crucible to correct for physical artefacts; this mass profile is subtracted from the measured weight automatically. The derivative of the TGA signal was used to quantify the rate of mass loss.Nitrogen adsorption was used to determine the porosity characteristics of the catalyst and catalyst supports. Nitrogen adsorption at −196 °C was carried out in a Micromeritics 2420. The samples were pre-treated in N2 at 250 °C for 10 h for fresh catalyst or catalyst supports while 200 °C was applied for the spent catalysts. No appreciable weight loss was detected. The textural parameters were derived from the BET model.14 The total pore volume (Vp) is directly measured in the desorption branch at a relatively pressure of ca. 0.987 as recommended by Micromeritics. The geometrical pore (Dp) size was calculated as 4Vp/SBET.
NH3-TPD experiments were carried out in a Micromeritics AutoChem II system equipped with a thermal conductivity detector. The sample (ca. 30 mg) was pre-treated by heating it up to 500 °C in He at 10 °C min−1. The sample was cooled to 120 °C at a similar cooling rate, and then exposed to 1 vol% NH3–He (25 ml min−1) for 30 min. Subsequently, a flow of He (25 ml min−1) was passed through the reactor for 60 min to remove weakly adsorbed NH3 from the sample’s surface. After the baseline stabilization, the desorption of NH3 was monitored in the range of 120–1000 °C using a heating rate of 10 °C min−1.
The crystal structure of selected materials was analysed by powder X-ray diffraction. XRD patterns were recorded in a Philips PW1840 using a Cu-Kα radiation source. The scanning angle was varied from 10°–100° with a scanning rate of 0.2° per minute. The identification of the different crystalline phases was performed by the comparison with the corresponding JCPDS diffraction data cards.
Conclusions
The performance of γ-Al2O3 in the ODH reaction of EB to ST can be improved by calcination at high temperature. The acid site density and strength increase with the calcination temperature. With increasing calcination temperature more coke is deposited per unit surface area during time on stream, it is less reactive in O2, more selective for the ODH reaction, and is better accessible for both reactants due to the modified porous texture. Full conversion of the limiting reactant oxygen is required for optimal selectivity, setting the lower limit for the amount of coke depending on the reaction temperature. Under the applied conditions, the optimal calcination temperature is around 1000 °C, resulting in about 3 layers of coke on the alumina after 62 h TOS. Too much coke results in a loss in ST selectivity and ST yield due to more COX formation.References
- A. E. Lisovskii and C. Aharoni, Catal. Rev.: Sci. Eng., 1994, 36, 25–74 CAS.
- G. E. Vrieland, J. Catal., 1988, 111, 1–13 CrossRef CAS.
- P. G. Menon, J. Mol. Catal., 1990, 59, 207–220 CrossRef CAS.
- E. Echiogoya, H. Sano and M. Tanaka, 8th Int. Congr. Catal., Berlin, 1984, vol. 623–633 Search PubMed.
- F. Cavani and F. Trifiro, Appl. Catal., A, 1995, 133, 219–239 CrossRef CAS.
- G. Emig and H. Hofmann, J. Catal., 1983, 84, 15–26 CrossRef CAS.
- B. C. Lippens and J. J. Steggerda, in Physical and Chemical Aspects of Adsorbents and Catalysts, ed. B. G. Linsen, J. M. H. Fortuin, C. Okkerse and J. J. Steggerda, Academic Press Inc., London, 1970, pp. 171–211 Search PubMed.
- E. B. M. Doesburg, K. P. De Jong and J. H. C. van Hooff, in Catalysis: An Integrated Approach, ed. R. A. van Santen, P. W. N. M. van Leeuwen, J. A. Moulijn and B. A. Averill, Elsevier, Amsterdam, 2000, pp. 433–484 Search PubMed.
- P. A. Badkar and J. E. Bailey, J. Mater. Sci., 1976, 11, 1794–1806 CrossRef CAS.
- J. L. Mcardle and G. L. Messing, J. Am. Ceram. Soc., 1993, 76, 214–222 CrossRef CAS.
- R. Fiedorow, W. Przystajko and M. Sopa, J. Catal., 1981, 68, 33–41 CrossRef CAS.
- L. E. Cadus, L. A. Arrua, O. F. Gorriz and J. B. Rivarola, Ind. Eng. Chem. Res., 1988, 27, 2241–2246 CrossRef CAS.
- H. Knozinger and P. Ratnasamy, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |