Manipulation of DNA damage checkpoint signaling in cancer cells by antioxidant biofactor (AOB)
Naoto
Tatewaki
a,
Hari Narayan
Bhilwade
b,
Hiroshi
Nishida
a,
Yuki
Nakajima
a and
Tetsuya
Konishi
*a
aDepartment of Applied Life Sciences, Niigata University of Pharmacy & Applied Life Sciences, Higashijima 265-1, Niigata 956-8603, Japan. E-mail: konishi@nupals.ac.jp
bRadiation Biology and Health Sciences Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
First published on 4th September 2012
Abstract
Antioxidant biofactor (AOB) is one of the fermented grain food supplements commercially available in Japan and other countries. Herein, we investigated the effect of AOB on the UVC (254 nm) induced DNA damage in A549 cells. Both distilled water and MeOH extracts of AOB did not show any significant cell toxicity. However, the UV (25–75 J m−2) induced cell death was amplified in the presence of these extracts, especially the MeOH extract. When the DNA damage was evaluated by comet assay, the AOB water extract prevented the UV induced DNA damage at the initial stage but significantly inhibited the repair process, especially in the cells exposed to a high dose of UV. The retardation of DNA repair was significantly higher in the presence of the MeOH extract, concentrating such components as caffeine and polyphenols, and thus the damage was enhanced both in the cells irradiated by low and high doses of UV. The DNA damage profile was consistent with the inhibitory profile of ATR, a key kinase of DNA damage checkpoint signaling. The AOB MeOH extract markedly reduced the phosphorylation level of the checkpoint proteins activated by UV, such as p53, SMC1 and Chk1, together with ATR. The inhibitory effect of the AOB water extract was less effective as compared to the MeOH extract, but was dose-dependent both in the cells irradiated with high and low doses of UV. The dual role of AOB as an antioxidant and a checkpoint modulator suggests its beneficial use in complementary medicine as a potential sensitizer of anticancer treatment.
1. Introduction
Recently, the advantageous role of food factors or dietary supplements is expanding in health promotion and the prevention of diseases. Since antioxidant protection is the basic requirement of disease prevention,1 many antioxidant dietary supplements are on the market. Besides disease prevention, the complementary application of dietary supplements in the medical treatment of a disease is an important field covered by food function, especially in cancer treatment. We currently reported Schisandrin B (SchB), an antioxidant lignan isolated from the fruit of Schisandra chinensis and a specific inhibitor of ATR (Ataxia telangiectasia and Rad-3-related), which plays an important role in sensing DNA damage and also in activating checkpoint signaling together with other kinases like ATM (Ataxia telangiectasia mutated) and DNA kinase, suggesting that this functional property of the food factor may be useful as an adjuvant in cancer treating modalities.2DNA is prone to attack by chemicals, ionizing radiation (IR), ultraviolet (UV) radiation, and oxidative stressors. When the DNA damage occurs, cells activate cellular DNA damage responses including cell cycle checkpoint activation. As a result, the cell cycle is arrested at G1, S, G2 and M phase, respectively, for the damage repair or apoptosis induction.3–5 Activation of checkpoint signaling is quite important for the cells to maintain the genomic stability and reduce the incidence of mutation-related disorders such as cancer. Moreover, the checkpoint modulation attracts much attention from the view of sensitizing cancer treatment. Abrogation of checkpoints forces the damaged cancer cell to undergo cell division before the damage is repaired, thus leading to death due to mitotic trouble. Therefore, chemicals and natural products that are capable of modulating DNA damage checkpoint activity are currently attracting much attention because they could be applied for cancer treatment as a sensitizer.2,6,7 An additional requirement for the molecule or compound to exhibit such an adjuvant effect, is low toxicity or a potential to protect against adverse effects induced, such as by oxidative stress. Antioxidant biofactor (AOB) could be one of such dietary supplement to be investigated for its possible complementary role in cancer therapy.
AOB is a commercially available fermented health food in Japan, America and China. It is rich in micronutrients and has potent antioxidant activity.8 The antioxidant properties of AOB have been reported both in vitro and in vivo, for example, free radical scavenging potential,9 inhibition of lipid peroxidation,10 tissue oxidative damage protection,11 and improvement of clastogenic factors in the plasma from Chernobyl liquidators.12 AOB also abrogated the toxicity and tumor promoting effect of iron nitrilotriacetate and served as a chemopreventative agent to suppress oxidant induced tissue injury and tumorigenesis in rats.13 However, no study has been reported on DNA damage protection and repair.
In the present study, we focused our attention on DNA damage protection and modulation of DNA damage checkpoint signaling by AOB in UVC irradiated A549 cancer cells.
2. Materials and methods
2.1. Reagents
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and propidium iodide (PI) were purchased from Dojindo (Kumamoto, Japan). Stock solutions of MTT (0.5%) and PI (1 mg ml−1) were prepared in phosphate-buffered saline (PBS), and stored at 4 °C under protection from light. SYBR green II was purchased from Lonza (Rockland, ME, USA) and stored at −20 °C.2.2. Cell culture
Human adenocarcinoma A549 cells (ATCC: American Tissue Type Collection, VA, USA) were maintained in Dulbecco's modified eagle medium (DMEM) (Sigma Chemical Co., St Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 100 μg ml−1 streptomycin and 100 units per ml penicillin [all were purchased from Invitrogen (Carlsbad, CA, USA)]. All cultures were kept in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. DNA damage of cells was induced by UV irradiation (CL-1000 Ultraviolet Crosslinker) (UVP, INC., CA, USA). The 8 watt UVC lamp (UVP, 34-0007-01) equipped in the illuminator has a sharp 254 nm emission peak and the contribution of UVA and B is negligible. The UV sensor continually measures the UV energy and automatically adjusts the UV dose in the exposure chamber.2.3. Preparation of AOB extracts
AOB was generously provided by AOA Japan Co. Ltd. AOB granules are prepared by the process of roasting and fermentation of a mixture containing extracts from soybean (20%), rice germs (18%), rice bran (15%), wheat (9%), green leaves (Japanese radish leaf, green tea; 10%), sesame (4%), citron juice (2.5%) and malted rice.9 AOB also contains various phenolic compounds, trace elements and vitamins with antioxidant components, such as ascorbic acid, tocopherol and polyphenol.8 In our experiment, both the distilled water extract (AOB DDW) and methanol extract of AOB (AOB MeOH) were prepared for the test samples as described below. MeOH was used to extract the rather lipophilic components that are expected to be the functional factors of AOB, such as polyphenols. AOB granules (2 g) were dissolved in 5 ml of DDW and MeOH, respectively, and vortexed for 20 min at room temperature. Each extract was centrifuged at 5000 rpm for 20 min at 4 °C. The supernatants thus obtained was filtrated through a 0.45 μm ϕ pore size membrane filter for AOB DDW and a 0.20 μm ϕ pore size membrane filter for AOB MeOH, and were stored at −30 °C until use. The concentration of AOB is given as the weight of AOB granules per volume (w/v) in the reaction medium.2.4. Determination of cell viability by a MTT assay
Cell viability was measured by a MTT assay as reported previously.14 A549 cells were seeded at 1 × 104 cells per cm2 in a 96-well plate and grown for 24 h. One hour prior to UV exposure, cells were incubated in the presence or absence of AOB extracts (AOB DDW and AOB MeOH) at the AOB concentrations of 0.0012, 0.004 and 0.012% (w/v). The MeOH concentration in the medium was kept as 0.03% for the duration of the reaction. After removal of the growth medium, the cells were exposed to UV irradiation (25–75 J m−2), followed by the addition of fresh growth medium supplemented with AOB. After incubating for 72 h, the growth medium was removed, and 100 μl of 0.05% MTT solution was added to the cells, which were then incubated for 4 h at 37 °C. After the cells were incubated with 100 μl of lysis buffer [20% SDS, 50% N,N-dimethyl formamide (DMF), pH 4.7], the absorbance was measured by a microplate reader (Bio-Rad Laboratories, CA, USA) at 595 nm. All incubations were carried out at 37 °C in 5% CO2.2.5. Effect of AOB on DNA strand breaks measured by an alkaline comet assay
A549 cells were seeded at 1 × 104 cells per cm2 in a 35 mm ϕ dish and grown for 48 h. One hour prior to UV exposure, cells were pre-incubated with 0.012% (w/v) AOB DDW and AOB MeOH, respectively. After the removal of growth medium, cells were exposed to UV irradiation (25 and 50 J m−2). Immediately after the irradiation, the cells were processed for the alkaline comet assay. The comet assay was carried out by the method described by Bhilwade et al.15 Briefly, cells were mixed with 1.5 ml of 0.8% low melting agarose solution prepared in 0.9% saline at 38 °C and poured onto fully frosted slides. After solidification, the slides were kept in lysis buffer [2.5 M NaCl, 100 mM EDTA (pH 8.0), 10% DMSO and 1% Triton X-100] for 1 h at 4 °C. After lysis treatment, the slides were set in horizontal electrophoresis apparatus (Cleaver Scientific Ltd) and kept in alkaline buffer [300 mM NaOH, 1 mM EDTA (pH 8.0), pH 13.0] at room temperature for 20 min to unwind the DNA strand. Electrophoresis was carried out for 40 min at 25 V, 400 mA. After electrophoresis, the slides were washed gently in the neutralizing buffer (0.4 M Tris–HCl, pH 7.5) to remove the alkaline buffer and detergent, and were stored on wet tissue paper in a closed plastic box at 4 °C until observation. The slides were stained with SYBR green II, and at least 60 cells were captured per slide at 20× magnification using a fluorescence microscope (Olympus (BH2-RFCA), Japan) equipped with green light excitation and 590 nm barrier filter. The images of the comets were analyzed with digital imaging software “CASP”. Two parameters, tail moment (TM) and tail length (TL), were analyzed. TM is the product of the TL and the % DNA in the tail.162.6. Detection of DNA fragmentation by flow cytometry
A549 cells were seeded at 2 × 104 cells per cm2 in a 35 mm ϕ dish and grown for 24 h. One hour prior to UV exposure, the cells were pre-incubated with 0.012% (w/v) AOB extracts (AOB DDW and AOB MeOH). After the removal of growth medium, the cells were exposed to UV irradiation (25 and 50 J m−2). The cells were cultured in a fresh growth medium supplemented with AOB for 1 h, 3 h and 6 h, respectively, and harvested by trypsinization. The cells were washed with ice-cold PBS, fixed with ice-cold 70% ethanol and then stored at −30 °C until use. The cells were centrifuged at 5000 rpm for 5 min at 4 °C, and then washed with 200 μl of ice-cold PBS and incubated for 30 min on ice. Following centrifugation at 5000 rpm for 10 min at 4 °C, the supernatant was removed and then 100 μl of PI solution (50 μg ml−1 propidium iodide, 100 μg ml−1 RNase in PBS) was added to each sample and incubated in the dark for 30 min. DNA fragmentation was detected as the Sub-G1 fraction by FACS (COULTER EPICS® XL-MCL, Beckman Coulter, CA, USA).2.7. Preparation of cell lysates
The cells harvested by scraping after trypsin treatment into ice-cold PBS were centrifuged at 3000 rpm for 5 min at 4 °C to get a pellet. For immunoblot analysis, the protein was extracted from the cell pellet with IP buffer [10 mM Tris, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 0.5% NP-40, 1% Triton X-100, 1 mM phenylmethanesulfonyl fluoride (PMSF), 2 μg ml−1 pepstatin, 2 μg ml−1 aprotinin, 10 mM Sodium fluoride (NaF) and 1 mM dithiothreitol (DTT), pH 7.4] by incubating for 30 min on ice. Following centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min at 4 °C, the supernatant was solubilized in SDS buffer [200 mM Tris–HCl (pH 6.8), 8% SDS, 40% glycerol, 3.4 M 2-mercaptoethanol and bromophenol blue] and stored at −30 °C until use. Protein concentrations were determined using the Bradford protein assay reagent (Bio-Rad Laboratories).
000 rpm for 20 min at 4 °C, the supernatant was solubilized in SDS buffer [200 mM Tris–HCl (pH 6.8), 8% SDS, 40% glycerol, 3.4 M 2-mercaptoethanol and bromophenol blue] and stored at −30 °C until use. Protein concentrations were determined using the Bradford protein assay reagent (Bio-Rad Laboratories).
2.8. Immunoblot analysis
For immunoblot analysis, 20 μg of protein (80 μg in case of ATR blot) was loaded and separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE), and the protein bands were transferred to a 0.45 μm pore size polyvinylidene difluoride (PVDF) membrane (Millipore, MA, USA) by electro-blotting apparatus (Invitrogen, CA, USA). The membrane was blocked by 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature. Immunoblotting was performed by incubating the membrane for 16 h at 4 °C with each of the primary antibodies targeting p53 (Cell Signaling Technologies, NJ, USA), phospho-p53 Ser 15 (Cell Signaling Technologies), SMC1 (Bethyl Laboratories, TX, USA), phospho-SMC1 Ser 966 (Bethyl Laboratories), Chk1 (G-4) (Santa Cruz, CA, USA), phospho-Chk1 Ser 317 (Bethyl Laboratories), phospho-Cdc25C Ser 216 (Santa Cruz), ATR (Bethyl Laboratories), phospho-ATR Ser 428 (Cell Signaling Technologies), ATM (Bethyl Laboratories), phospho-ATM Ser 1981 (Cell Signaling Technologies), Tubulin (Cell Signaling Technologies) and GAPDH (Cell Signaling Technologies). Following incubation with secondary antibodies (anti-mouse or anti-rabbit IgG) conjugated with horse radish peroxidase (Cell Signaling) for 1 h at room temperature, the target proteins were visualized using an Immobilon Western Chemiluminescent HRP Substrate (Millipore) and X-ray film (Fujifilm Co., Ltd., Tokyo, Japan). Densitometric analysis of the bands was done using Image Quant 5.2 software (GE Healthcare UK Ltd., UK).2.9. HPLC analysis
AOB DDW and AOB MeOH were analyzed by a Shimadzu HPLC system (Kyoto) equipped with a photodiode array detector (SPD-M10Avp) and a Gemini 5 μm C18 column (150 × 4.6 mm i.d., Phenomenex, Torrance, CA, USA) using a gradient of MeOH in water. The column and auto-sampler tray temperature were kept constant at 40 and 4 °C, respectively, and a flow-rate was 0.7 ml min−1. The gradient program was as follows: from 0% methanol for 0–5 min, 80% methanol for 60–70 min. The sample injection volume was 5 μl. The elution peaks were monitored at 254 nm and 350 nm, respectively.2.10. Statistical analysis
All the data are expressed as mean ± SD. Statistical analysis of the data was carried out by one-way ANOVA using the Tukey–Kramer test. The differences were considered significant at P < 0.05.3. Results
3.1. UV induced cell cytotoxicity and its modulation by AOB (MTT assay)
First, the cytotoxicity of AOB DDW and AOB MeOH was examined in A549 adenocarcinoma cells. Both extracts did not change the cell viability up to 0.012% (w/v) AOB (Fig. 1A). MeOH as the vehicle of AOB MeOH did not show any significant toxicity up to 0.1%, and there was no difference of cell viability in DDW and MeOH even after the UV irradiation (data not shown). Therefore, the MeOH concentration in the reaction medium was kept at 0.03% for all AOB concentrations. The cell death induced by UVC (254 nm) exposure was, however, significantly enhanced in both cells pre-treated with either AOB DDW or AOB MeOH (Fig. 1B). However, it was noted, in the AOB DDW treated cells, the enhancing effect shown by the net enhanced fraction (%) was rather independent of UV dose, but in the AOB MeOH treated cells, it was more marked in the cells irradiated by a higher UV dose (50 J m−2). When the effect of AOB DDW and AOB MeOH was compared at low UV dose (25 J m−2), the enhanced effect was larger in AOB DDW but at higher UV dose (50 J m−2), AOB MeOH showed marked amplification of UV toxicity than AOB DDW (Fig. 1B and C). The effects of AOB extracts, especially of AOB DDW were fairly dependent on the concentration both at low and high doses of UV irradiation.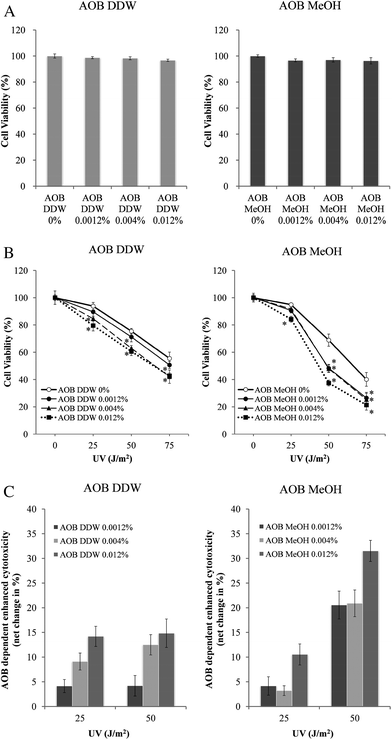 | ||
| Fig. 1 AOB extracts enhanced cytotoxicity of UVC. Cell viability was measured by MTT assay. A549 adenocarcinoma cells were seeded at 1 × 104 cells per cm2 in 96-well plate, and then incubated for 24 h before the experiment. The cells were subjected to 1 h pre-incubation and 72 h post-incubation (A) with AOB extracts (0, 0.0012, 0.004 and 0.012%) after (B) UV irradiation (0, 25, 50 and 75 J m−2). AOB 0% indicates vehicle control (DDW or MeOH). (C) The net increase of cytotoxicity induced by AOB was evaluated from the above data. Each value represents a mean ± S.D. of three independent experiments (*P < 0.05) (vs. AOB extracts 0% cells). P-values were obtained by one-way ANOVA followed by the Tukey–Kramer test. | ||
3.2. UV induced DNA damage and its modulation by AOB
The effect of AOB extracts was examined on the DNA damage caused by UVC in A549 cells. Typical comet shapes are given in Fig. 2 and the quantitative data in Fig. 3 shows the results evaluated by tail length (Fig. 3A) and tail moment (Fig. 3B), respectively. Both AOB DDW and AOB MeOH, themselves did not produce any DNA damage without UV exposure. UV irradiation at doses of 25 and 50 J m−2 significantly produced DNA damage in the control cells (AOB 0%). In the cells exposed to UV 25 J m−2, the damage decreased in a time-dependent manner, indicating the DNA repair progressed, however, the repair process was found to be significantly retarded at UV 50 J m−2 (Fig. 2A and Fig. 3). As both the tail length and tail moment data showed, AOB DDW significantly prevented the initial production of DNA damage at 1 h after UV irradiation with a dose of 25 and 50 J m−2, respectively (Fig. 2B and Fig. 3). However, the AOB DDW effect on the repair process was dependent on the UV doses. In the cells irradiated by a low dose UV (25 J m−2), the repair occurred as well as the control (Fig. 2B and Fig. 3, left graph), but in the cells exposed with a higher dose of UV (50 J m−2), the damage level was rather amplified at 3 h and was maintained even at 6 h, indicating that the repair process was retarded (Fig. 2B and Fig. 3, right graph). This damage enhancing effect was more prominent in AOB MeOH treated cells. In the presence of AOB MeOH, the DNA damage occurred by the low dose of UV (25 J m−2) showed an approximately 30% higher extent than the control, even at 1 h after UV irradiation and the damage was still maintained at a high level at 3 h (Fig. 2C and Fig. 3A, left graph). In the cells heavily damaged with a higher dose of UV (50 J m−2), the damage level was not more than that of control at 1 h after the UV irradiation but was also markedly enhanced at 3 h (Fig. 2C and Fig. 3A, right graph). This trend was more clearly shown when evaluated by the tail moment changes in Fig. 3B.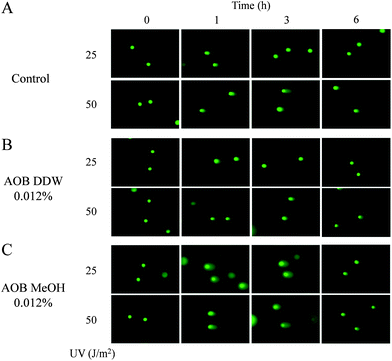 | ||
| Fig. 2 Typical comets images from damaged A549 adenocarcinoma cells stained with SYBR green II. Comets induced by UV 25 and 50 J m−2 with or without AOB extracts (0.012%) at 0, 1, 3 and 6 h. (A) Control, (B) pre-treated with AOB DDW, (C) pre-treated with AOB MeOH. | ||
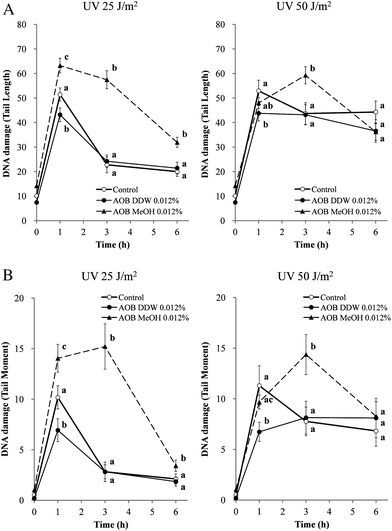 | ||
| Fig. 3 Effect of AOB extracts on UVC-induced DNA damage in A549 adenocarcinoma cells. Cells were pre-incubated with or without AOB extract (0.012%) for 1 h prior to UV exposure. Cells were subjected to 25 or 50 J m−2 of UV irradiation and the DNA damages were examined by the comet assay as described in the Materials and methods section. Damage is expressed as (A) tail length and (B) tail moment at different time intervals. Each value is the mean ± S.D. of over 60 randomly selected cells obtained from three independent experiments. Values with different superscripts are significantly different (P < 0.05) to each other. P-Values were obtained by one-way ANOVA followed by the Tukey–Kramer test. | ||
3.3. Effect of AOB on UV induced DNA fragmentation
FACS analysis was performed to determine the Sub-G1 fraction as the indices of apoptosis occurred in individual cells (Fig. 4). Cells in the Sub-G1 phase increased in a time-dependent manner after UVC irradiation (25 and 50 J m−2) in the A549 cells. AOB DDW did not change the profile of the Sub-G1 fraction in the low dose UV (25 J m−2) treated cells as compared to the control cells (AOB 0%), even at 6 h, but at the higher dose (50 J m−2) a significant increase of Sub-G1 cells was observed. On the other hand, AOB MeOH significantly increased the Sub-G1 cell population as compared to the control at 6 h at both the 25 and 50 J m−2 UV irradiation doses.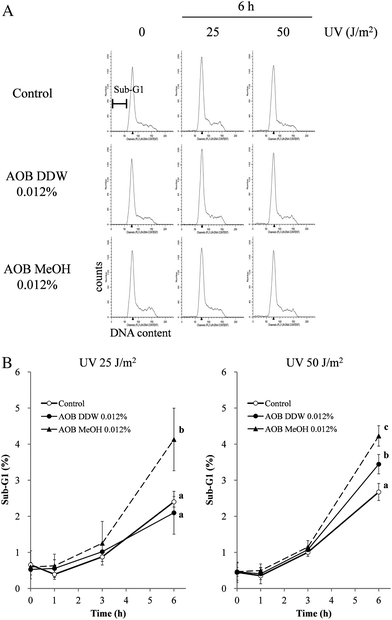 | ||
| Fig. 4 Effect of AOB extracts on DNA fragmentation induced by UVC in A549 adenocarcinoma cells. DNA fragmentation was analyzed by FACS after 1 h, 3 h and 6 h of UV exposure, followed by data analysis using ModFit LT 3.2 software. (A) The cells treated with or without AOB extract (0.012%) and/or UV (25 or 50 J m−2) were harvested and fixed before staining with propidium iodide. Red fluorescence intensity was measured and histograms of DNA content generated. (B) Data are expressed as the percentage of Sub-G1 DNA fragmentation. Values are expressed as the mean ± SD of three independent experiments. Values with different superscripts are significantly different (P < 0.05) to each other. P-Values were obtained by one-way ANOVA followed by the Tukey–Kramer test. | ||
3.4. AOB inhibits the phosphorylation of checkpoint protein
To investigate whether AOB directly modulated the DNA damage response, especially the ATM- or ATR-related signal transduction pathways, we studied the phosphorylation of p53 at serine 15 (Ser 15), a direct target of ATM/ATR at 3 h after UV irradiation using specific antibodies for the phosphorylation site. As expected, the phosphorylation level of p53 at Ser 15 was significantly enhanced by UV irradiation (25 and 50 J m−2) in the control cells (Fig. 5A and B, lane 2). It was revealed that AOB MeOH inhibited p53 phosphorylation induced by UV for the same extent in both cells irradiated by 25 and 50 J m−2 UV (Fig. 5A and B, lanes 6–8). On the other hand, the inhibitory activity of AOB DDW was weak but dose-dependent, especially in the cells irradiated at 25 J m−2. However, the inhibition level was almost the same as for AOB MeOH in the cells irradiated at 50 J m−2 UV (Fig. 5B, lanes 4 and 5).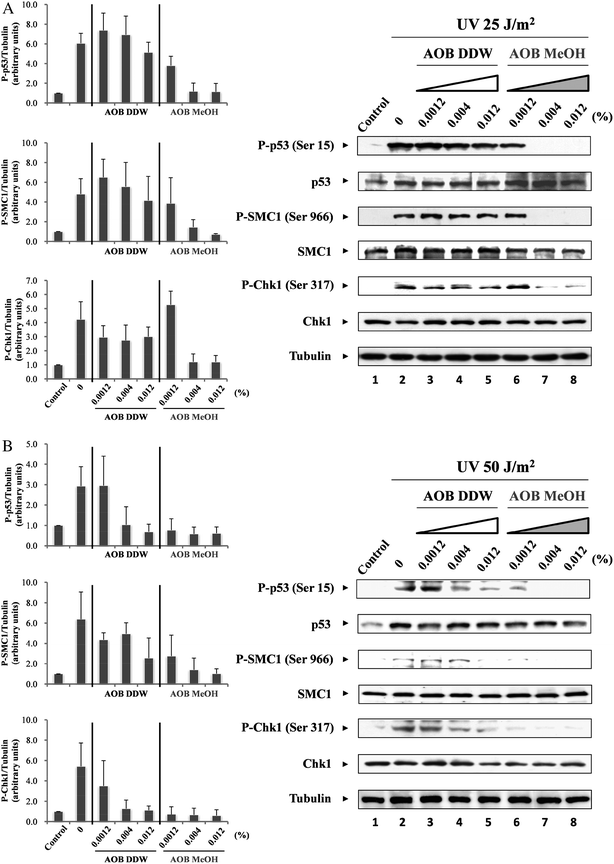 | ||
| Fig. 5 Effect of AOB extracts on checkpoint proteins. A549 adenocarcinoma cells were pre-incubated with or without AOB extracts (0.0012, 0.004 and 0.012%) for 1 h prior to induction of DNA damage. After pre-incubation, cells were irradiated with UV (25 or 50 J m−2), and then cultured at 37 °C for 3 h. Dose-dependent effects of AOB extracts on the phosphorylation level of p53, SMC1 and Chk1 were observed by (A) 25 J m−2 or (B) 50 J m−2 of UV irradiation. Tubulin was used as a loading control. | ||
Next, we examined the phosphorylation of SMC1 at serine 966 (Ser 966) as the marker of the intra-S phase checkpoint. The phosphorylation level of SMC1 was enhanced by UV (25 and 50 J m−2) irradiation in the control cells but the AOB extracts inhibited the phosphorylation in a similar fashion to the p53 phosphorylation case. AOB MeOH significantly inhibited the phosphorylation of SMC1 in both cells exposed to 25 and 50 J m−2 UV irradiation (Fig. 5A, lanes 7 and 8, and Fig. 5B, lanes 6–8). AOB DDW also inhibited the phosphorylation, but the inhibitory effect was weaker than AOB MeOH, especially in the cells exposed with a low dose of UV (25 J m−2) (Fig. 5A, lane 5). The inhibitory profile of AOB extracts on Chk1 (Ser 317) phosphorylation was essentially the same as those in p53 and SMC1. AOB MeOH dose-dependently inhibited Chk1 phosphorylation in the cells irradiated both by high and low doses UV (Fig. 5A, lanes 7 and 8, and Fig. 5B, lanes 6–8). Similarly, AOB DDW inhibited Chk1 phosphorylation, however, the inhibitory effect on Chk1 was found to be more marked than on other checkpoint molecules, p53 and SMC1, either in the cells exposed to low dose UV (25 J m−2) or high dose UV (50 J m−2) (Fig. 5A, lanes 3–5).
3.5. Effect of AOB on UV-induced ATM and ATR activation
ATM and ATR protein kinases are key regulators of cell cycle checkpoints, and these kinases operate cooperatively after DNA damage occurs. Since the DNA damage caused by UVC preferentially activates ATR,17–19 the inhibitory effects of the AOB extracts were further examined for ATR. The results revealed that AOB MeOH markedly inhibited ATR phosphorylation (Ser 428) both in 25 and 50 J m−2 UV irradiated cells, and the effect was dose-dependent, especially in the cells exposed to a low dose of UV (Fig. 6A and B, lanes 6–8). AOB DDW also inhibited ATR phosphorylation, but the extent was several times weaker than AOB MeOH, both in the cells exposed with high and low doses of UV. In the cells exposed with a low dose of UV, no significant inhibition was observed, especially at lower concentrations of AOB DDW (Fig. 6A and B, lanes 3–5). AOB also inhibited phosphorylation of p53, SMC1 and Chk1. It seemed that the reduced phosphorylation level in the checkpoint proteins, p53, SMC1 and Chk1 was resulted from the ATR inhibition by AOB extracts.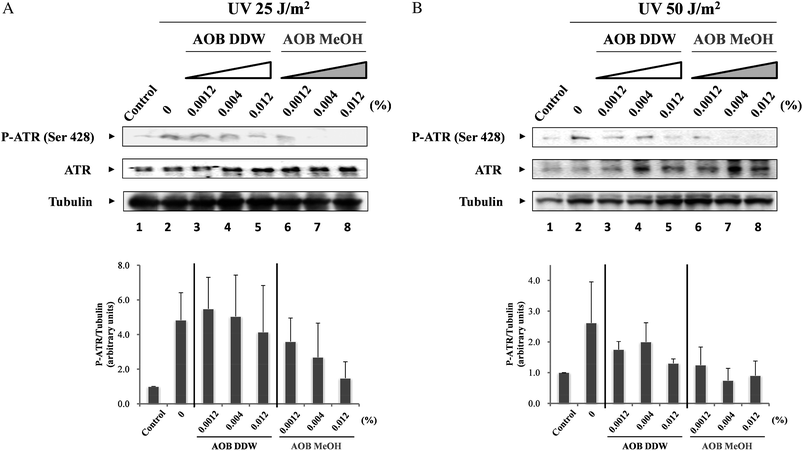 | ||
| Fig. 6 Effect of AOB extracts on UVC-induced ATR activity. A549 adenocarcinoma cells were pre-incubated with or without AOB extracts (0.0012, 0.004 and 0.012%) for 1 h prior to induction of DNA damage. After pre-incubation, cells were irradiated with UV (25 or 50 J m−2), and then cultured at 37 °C for 3 h. Dose-dependent effects of the AOB extracts on the phosphorylation level of ATR was observed by (A) 25 J m−2 or (B) 50 J m−2 of UV irradiation. Tubulin was used as a loading control. | ||
We further examined the effect of AOB on UV-induced ATM activation. The phosphorylation level of ATM at Ser 1981 was significantly enhanced in control cells after UV exposure (25 and 50 J m−2) in a time-dependent manner, although UV damage is reported to activate ATR rather than ATM4,5,20 (Fig. 7A and B, lanes 2 and 3). AOB DDW significantly inhibited ATM phosphorylation, especially at 1 h after the UV irradiation (25 and 50 J m−2). In the cells exposed to a higher dose of UV, the phosphorylation level was slightly higher at 3 h after UV than at 1 h, as observed in the control cells (Fig. 7A and B, lanes 4 and 5). The effect of AOB MeOH differed from that of AOB DDW. In the cells exposed with a low dose of UV, it also inhibited the ATM phosphorylation at 1 h after the irradiation but the phosphorylation level was markedly enhanced at 3 h after UV irradiation as compared to the control cells (Fig. 7A, lane 7). In the cells exposed to higher dose of UV, however, AOB MeOH inhibited ATM phosphorylation to almost the same extent as the AOB DDW extract, both at 1h and 3 h after UV exposure (Fig. 7B, lanes 6 and 7). The Chk1 phosphorylation profile observed along with ATM activation was consistent with the profiles shown in Fig. 5, where the phosphorylation level was determined at 3 h after UV irradiation (25 and 50 J m−2), that is, both AOB extracts significantly inhibited the phosphorylation level of Chk1. As was shown in the control cells, Chk1 activation was also time-dependent . Phosphorylation of Chk1 was prominent shortly after UV exposure, that is, at 1 h and then gradually dephosphorylated (Fig. 7A and B, lanes 2 and 3). A marked inhibition of Chk1 phosphorylation was observed in AOB MeOH treated cells at both 1 h and 3 h after UV irradiation at both doses (25 and 50 J m−2) (Fig. 7A and B, lanes 6 and 7). However, the phosphorylation was not significantly inhibited by AOB DDW at 1 h, but was significantly inhibited to the same level given by AOB MeOH at 3 h (Fig. 7A and B, lanes 4 and 5). Further, the effect of AOB extracts was examined on the G2/M checkpoint regulatory protein Cdc25C. It was found the Cdc25C phosphorylation profile was almost the same as that of Chk1 in the control and AOB treated cells.
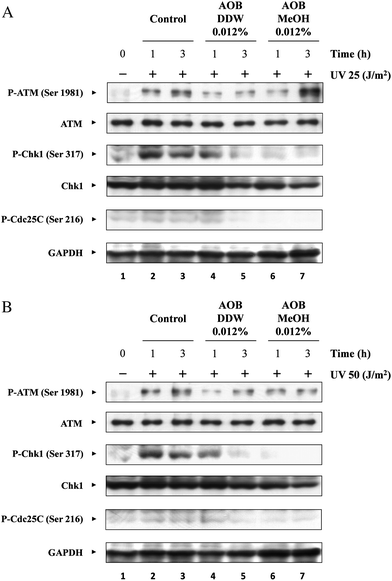 | ||
| Fig. 7 Effect of AOB extracts on the checkpoint protein activation of different times. A549 adenocarcinoma cells were pre-incubated with or without AOB extracts (0.012%) for 1 h prior to induction of DNA damage. After pre-incubation, cells were irradiated with UV (25 or 50 J m−2), and then cultured at 37 °C for 1 h and 3 h. Effect of AOB extracts on the phosphorylation level of ATM, Chk1 and Cdc25C were observed by (A) 25 J m−2 or (B) 50 J m−2 of UV irradiation. GAPDH was used as a loading control. | ||
3.6. Analysis of AOB ingredients by HPLC
Components of AOB DDW and AOB MeOH, were separated and analyzed by HPLC. Typical HPLC chromatograms of AOB DDW and AOB MeOH monitored at 254 and 350 nm are given in Fig. 8A and B. The peaks appeared at 28.9 and 34.6 min were assigned as caffeine and (−)-epigallocatechin gallate (EGCG), respectively, and 43.5 min as rutin using respective authentic references. The peak profiles of AOB DDW and AOB MeOH were quite different. The hydrophilic peaks with a retention time (Rt) shorter than 22 min were absent in the AOB MeOH. Other moderately lipophilic peaks including caffeine and EGCG were present in both extract, however, their contents in AOB DDW were low and were no more than 30% of those in AOB MeOH.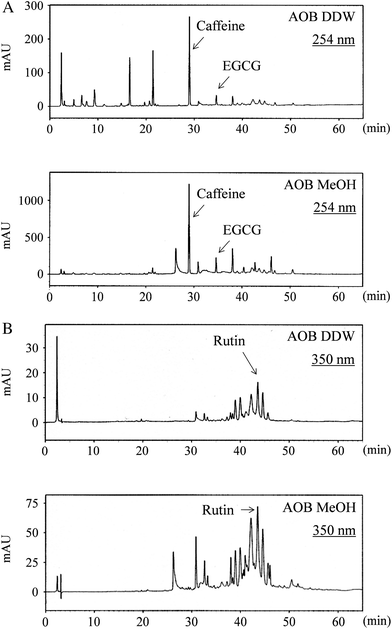 | ||
| Fig. 8 HPLC chromatogram of AOB extracts (AOB DDW and AOB MeOH) monitored at (A) 254 nm and (B) 350 nm with a linear gradient of methanol in water. Caffeine, EGCG and rutin appeared at (A) 28.9 min, 34.6 min and (B) 43.5 min, respectively. | ||
4. Discussion
Currently, the beneficial health effects of foods or food factors are attracting much attention and thus many studies are focused on the pharmacological or physiological functions of food factors, for example, immune modulation by soy isoflavones and genotoxicity protection by squalene.21,22 Under the circumstances, functional foods and dietary supplements are gaining social status as beneficial modalities for improving the quality of life. For further understanding of the beneficial role of food and food factors in human health, the underlying mechanisms of their action need to be extensively studied at biochemical and molecular levels either for a crude mixture or a specific isolated ingredient.AOB is one of the commercially available health food supplements made from fermented mixed grain and vegetables, and has been used for daily health promotion because of its high potential as an antioxidant. In the present study, the effect of AOB on the DNA checkpoint signaling was precisely examined. It is shown in Fig. 1, that the AOB extracts enhanced UVC (254 nm) toxicity in cancer cell line, as was previously demonstrated by SchB.2 This indicated that the AOB extracts, especially the MeOH extract modulates DNA damage checkpoint signaling.
Indeed, the UVC dependent phosphorylation of checkpoint molecules was significantly inhibited by AOB, especially in the AOB MeOH treated cells (Fig. 5–7). For example, the phosphorylation levels of p53 at Ser 15 and SMC1 at Ser 966 were significantly reduced by AOB MeOH treatment in the cells exposed to both low (25 J m−2) and high (50 J m−2) doses of UV, as well as ATR phosphorylation. The Chk1 phosphorylation at Ser 317, a specific and direct phosphorylation site of ATR and also of ATM, was considerably suppressed by AOB MeOH in a dose-dependent manner. This indicated that AOB essentially contains component(s) for modulating DNA damage checkpoint signaling. Indeed, HPLC analysis revealed that the MeOH extract was concentrated with such components as caffeine, EGCG and rutin (Fig. 8), those which are reported to have DNA damage response modulating activity.23–25 AOB DDW, that might reflect the form of daily intake, did not inhibit p53 and SMC1 at its lower concentration range in the cells exposed to a low dose of UV, but in the high dose UV exposed cells, a dose-dependent inhibition was observed. The Chk1 phosphorylation at Ser 317 was not significantly modulated by AOB DDW in the low dose UV exposed cells, but was markedly inhibited by AOB MeOH in the cells exposed to high and low doses of UV (Fig. 5A and B and Fig. 6).
It is known that the activation of Chk1 by ATR follows the activation of Cdc25C at Ser 216, which leads to G2/M checkpoint activation.20,26–28 In the present study, AOB, especially, the MeOH extract, inhibited Cdc25C phosphorylation, as shown in Fig. 7. This supports the notion that AOB, especially the components concentrated in the MeOH extract, inhibited ATR related checkpoint signaling.
In the present study, UVC irradiation activated both ATR and ATM at 3 h after irradiation although UVC is known to preferentially activate ATR.17–19 The effect of AOB on these kinases was slightly different for AOB DDW and AOB MeOH. As was expected from the phosphorylation profiles of the downstream checkpoint molecules, AOB DDW did not show any significant inhibition of the ATR activity, even in cells irradiated at a high dose of UV, except at its higher concentration (Fig. 6B). However, AOB MeOH clearly inhibited the ATR activity in a dose-dependent manner in the low dose UV treated cells and almost completely inhibited in the high dose UV treated cells. On the other hand, ATM phosphorylation was also considerably inhibited by the DDW and MeOH extracts, respectively. AOB DDW showed rather stronger inhibition at 1 h after UV irradiation than AOB MeOH. It was noted that the ATM phosphorylation level was rather amplified at 3 h after UV irradiation, both in the cells treated by AOB DDW and AOB MeOH. This enhanced phosphorylation of ATM was more marked by AOB MeOH in the low dose UV treated cells than by AOB DDW in the high dose UV treated cells (Fig. 7). It was expected that ATM is activated in compensation for ATR inactivation by AOB. This also indicated AOB primarily modulates ATR activity.
AOB DDW primarily tended to protect UV dependent DNA damage production but retarded the repair process, especially in the cells irradiated with a high dose of UV (Fig. 3). The retardation of the repair process was more markedly observed in the presence of AOB MeOH after UV irradiation, both at 25 and 50 J m−2. DNA repair was studied by monitoring the changes in tail length and tail moment in cells exposed to UV radiation in the presence or absence of AOB extracts.
ATR is mainly activated by the single strand break (SSB) type DNA damage produced, such as by UV exposure. When the polymerase reaction is stalled at damaged sites on DNA during replication, helicase will continue to unwind the DNA, thus leading to the production of long stretches of single-stranded DNA.17–19 Indeed, the comet tail elongation was observed in the UV damaged cells in the presence of AOB (Fig. 2). The ATR/Chk1 pathway plays a critical role in repairing this type of damage since the single stranded DNA is the common intermediate responsible for activation of ATR. This might explain why the observed DNA damage assessed by the comet assay was enhanced by AOB at 3 h, since AOB, especially AOB MeOH, inhibits ATR/Chk1 to prevent repair. This also explains the enhanced toxicity of UV towards A549 cells. Under the checkpoint abrogation, the cells carrying damaged DNA are forced to progress the cell cycle and thus undergo mitotic death.7
On the other hand, the damage enhancement due to the retardation of DNA repair observed at 3 h after UV exposure was well correlated with the increase in Sub-G1 cell population at 6 h after UV exposure as assessed by FACS in Fig. 4. In addition, the damaged DNA was significantly repaired after 6 h in the AOB treated cells as shown in Fig. 2B and C and Fig. 3. These observations indicate the checkpoint is operational.
It has been reported that ATM activates p38 kinase through the thousand and one amino acid (TAO) kinases activation.29 TAO kinases are one of the multiple MAPKKK that can activate MEK3/6 (also referred to as MKK3/6). It is demonstrated that p38 promotes the phosphorylation of Cdc25B/C indirectly through the activation of MAPKAP kinase-2 (MK2) after UVC irradiation.30 Regulation of Cdc25B/C activation by p38 is critical for working of the G2/M checkpoint after UVC irradiation. It is thus indicated that DNA damage repair and the increase of apoptosis-like cells were mediated by G2/M checkpoint activation through the ATM-TAO-MKK3/6-p38-MK2-Cdc25B-Cdc2 pathway. Thus it is indicated that the enhanced cell toxicity observed in Fig. 1 results from two competitive reactions, that is, the increased mitotic death due to the inhibition of ATR related checkpoint abrogation and an increased apoptosis mediated by the ATM-TAO-MKK3/6-p38-Cdc2 pathway. In the latter pathway, the compensative ATM activation might play a role and AOB possibly modulates this process. This kind of synergistic modulation of checkpoint might be a characteristic feature of an antioxidant-based multi targeted formula like AOB.
To understand the differential effects of AOB DDW and AOB MeOH we analyzed the chromatographic profile of the components in AOB DDW and AOB MeOH using HPLC (Fig. 8). The contents of the hydrophobic ingredients, including rutin and EGCG, were far less in AOB DDW than in the AOB MeOH fraction. The caffeine peak at 28.9 min appeared in both AOB DDW and AOB MeOH but the content in AOB MeOH was approximately three times larger than in AOB DDW. The diverse component profile of AOB DDW and AOB MeOH suggests that the hydrophilic components in AOB are mainly active as an antioxidant to prevent UV damage of cells and plays a role in DNA damage protection, whereas the hydrophobic ingredients play a role in ATR inhibition. The caffeine present in AOB might be one of the components involved in the latter reaction since caffeine was first reported to inhibit ATM/ATR.31
Inhibition of DNA damage checkpoints lead to chemo- and radio-sensitization of cancer cells6,20,32 and thus the beneficial use of checkpoint inhibitors currently attracts attention for cancer therapy. Caffeine is one of the earliest inhibitors known to sensitize tumor cells to ionizing radiation and other genotoxic agents by inhibiting both ATR and ATM.31 Although several ATM inhibitors have been reported,6,33–36 there are few ATR-specific inhibitors reported except SchB.37,38 Targeting ATM and ATR is a promising approach to modulate the checkpoint signals including G2/M checkpoint. Because in many cancer cells, p53 is mutated or defected39,40 and thus the G1 checkpoint function is not always operative and thus abrogation of the G2/M checkpoint attracts attention as a more appropriate target for sensitizing cancer chemo- or radio-therapy.6,41,42
There would be some argument whether the AOB doses (12–120 μg ml−1) examined in the present study is reliable. Usually, 2–4 packages of AOB (6–12 g) are taken a day as a dietary supplement, and thus the dose is calculated to be approximately 0.1–0.2 g kg−1 body weight supposing the body weight is 60 kg. Even though the active ingredients were reasonably concentrated in the MeOH extract, the data obtained in the present study suggests a certain feasibility of the clinical application of AOB in complementary cancer therapy. Although we need further studies on the practical dose of AOB in clinical use, the present finding that AOB inhibits the G2/M checkpoint is novel and suggests a possible application of AOB in complementary cancer therapy.
5. Conclusion
The present study showed that antioxidant biofactor (AOB) prevents DNA damage production by UVC (254 nm), especially in the cells damaged by a low dose of UV, but amplified the DNA damage produced by UV in a dose-dependent manner when the cellular DNA was heavily damaged. This dual effect of AOB DDW was well explained by the roles of hydrophilic and hydrophobic components present in AOB. The hydrophilic components are involved in the damage protection, but the hydrophobic components concentrated in AOB MeOH play a critical role in DNA damage enhancement through the inhibition of checkpoint signaling resulting from the inhibition of key kinase ATR. AOB thus could be beneficial for the complementary use in cancer chemo- or radio-therapies in addition to the chemopreventative effect in the cells undergoing DNA damage.Acknowledgements
We thank AOA Japan Co., Ltd. for kindly providing AOB. We also thank Dr Sandur Santosh Kumar at BHABHA atomic research centre (India) for critically reading the manuscript.References
- S. R. Maxwell, Prospects for the use of antioxidant therapies, Drugs, 1995, 49, 345–361 CrossRef CAS.
- H. Nishida, N. Tatewaki, Y. Nakajima, T. Magara, K. M. Ko, Y. Hamamori and T. Konishi, Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response, Nucleic Acids Res., 2009, 37, 5678–5689 CrossRef CAS.
- Y. Shiloh, ATM and ATR: networking cellular responses to DNA damage, Curr. Opin. Genet. Dev., 2001, 11, 71–77 CrossRef CAS.
- R. T. Abraham, Cell cycle checkpoint signaling through the ATM and ATR kinases, Genes Dev., 2001, 15, 2177–2196 CrossRef CAS.
- Y. Shiloh, ATM and related protein kinases: safeguarding genome integrity, Nat. Rev. Cancer, 2003, 3, 155–168 CrossRef CAS.
- T. Kawabe, G2 checkpoint abrogators as anticancer drugs, Mol. Cancer Ther., 2004, 3, 513–519 CAS.
- N. Bucher and C. D. Britten, G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer, Br. J. Cancer, 2008, 98, 523–528 CrossRef CAS.
- Y. Minamiyama, S. Takemura, T. Yoshikawa and S. Okada, Fermented grain products, production, properties and benefits to health, Pathophysiology, 2003, 9, 221–227 CrossRef CAS.
- Y. Minamiyama, T. Yoshikawa, T. Tanigawa, S. Takahashi, Y. Naito, H. Ichikawa and M. Kondo, Antioxidative effects of a processed grain food, J. Nutr. Sci. Vitaminol., 1994, 40, 467–477 Search PubMed.
- Y. Minamiyama, S. Takemura, S. Toyokuni, Y. Tanimoto, E. F. Sato and M. Inoue, A processed grain food inhibits hepatic injury in endotoxemic rats, J. Nutr. Sci. Vitaminol., 1998, 44, 547–559 Search PubMed.
- Y. Minamiyama, E. Sato, S. Takemura, M. Inoue and T. Yoshikawa, Oxidative stress and antioxidant biofactor (AOB), in Food and Free Radicals, ed. M. Hiramatsu, T. Yoshikawa and M. Inoue, Plenum Press, New York, 1997, pp. 137–140 Search PubMed.
- I. Emerit, N. Oganesian, R. Arutyunian, A. Pogossian, T. Sarkisian, L. Cernjavski, A. Levy and J. Feingold, Oxidative stress-related clastogenic factors in plasma from Chernobyl liquidators: protective effects of antioxidant plant phenols, vitamins and oligoelements, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1997, 377, 239–246 CrossRef CAS.
- A. Mizote, Y. Okazaki, M. Iqbal and S. Okada, Antioxidant biofactor, a processed grain food, inhibits iron nitrilotriacetate-induced renal tumorigenesis, hyperproliferative response, and oxidative damage, Hum. Exp. Toxicol., 2008, 27, 207–214 CrossRef CAS.
- H. Nishida, M. Kushida, Y. Nakajima, Y. Ogawa, N. Tatewaki, S. Sato and T. Konishi, Amyloid-beta-induced cytotoxicity of PC-12 cell was attenuated by Shengmai-san through redox regulation and outgrowth induction, J. Pharmacol. Sci., 2007, 104, 73–81 CAS.
- H. N. Bhilwade, N. Tatewaki, V. V. Giridharan, H. Nishida and T. Konishi, Modulation of doxorubicin-induced genotoxicity by squalene in Balb/c mice, Food Funct., 2010, 1, 174–179 RSC.
- K. Konca, A. Lankoff, A. Banasik, H. Lisowka, T. Kuszewski, S. Gozdz, Z. Koza and A. Wojcik, Across platform public domain PC image analysis programme for the comet assay, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2003, 534, 15–20 CrossRef CAS.
- M. J. O'Connell and K. A. Cimprich, G2 damage checkpoints: what is the turn-on?, J. Cell Sci., 2005, 118, 1–6 CrossRef CAS.
- L. Zou and S. J. Elledge, Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes, Science, 2003, 300, 1542–1548 CrossRef CAS.
- J. M. Wagner and S. H. Kaufmann, Prospects for the use of ATR inhibitors to treat cancer, Pharmaceuticals, 2010, 3, 1311–1334 Search PubMed.
- B. B. Zhou and J. Bartek, Targeting the checkpoint kinases: chemosensitization versus chemoprotection, Nat. Rev. Cancer, 2004, 4, 216–225 CrossRef CAS.
- T. Sakai and M. Kogiso, Soy isoflavones and immunity, J. Med. Invest., 2008, 55, 167–173 Search PubMed.
- H. N. Bhilwade, N. Tatewaki, H. Nishida and T. Konishi, Squalene as novel food factor, Curr. Pharm. Biotechnol., 2010, 11, 875–880 CAS.
- G. C. Yen, J. W. Ju and C. H. Wu, Modulation of tea and tea polyphenols on benzo(a)pyrene-induced DNA damage in Chang liver cells, Free Radical Res., 2004, 38, 193–200 CrossRef CAS.
- H. Celik and E. Arinç, Evaluation of the protective effects of quercetin, rutin, naringenin, resveratrol and trolox against idarubicin-induced DNA damage, J. Pharm. Pharm. Sci., 2010, 13, 231–241 CAS.
- K. Kawakami, H. Nishida, N. Tatewaki, Y. Nakajima, T. Konishi and M. Hirayama, Persimmon leaf extract inhibits the ATM activity during DNA damage response induced by Doxorubicin in A549 lung adenocarcinoma cells, Biosci., Biotechnol., Biochem., 2011, 75, 650–655 CrossRef CAS.
- Y. Sanchez, C. Wong, R. S. Thoma, R. Richman, Z. Wu, H. Piwnica-Worms and S. J. Elledge, Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25, Science, 1997, 277, 1497–1501 CrossRef CAS.
- C. Y. Peng, P. R. Graves, R. S. Thoma, Z. Wu, A. S. Shaw and H. Piwnica-Worms, Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216, Science, 1997, 277, 1501–1505 CrossRef CAS.
- H. Zhao and H. Piwnica-Worms, ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1, Mol. Cell. Biol., 2001, 21, 4129–4139 CrossRef CAS.
- M. Raman, S. Earnest, K. Zhang, Y. Zhao and M. H. Cobb, TAO kinases mediate activation of p38 in response to DNA damage, EMBO J., 2007, 26, 2005–2014 CrossRef CAS.
- I. A. Manke, A. Nguyen, D. Lim, M. Q. Stewart, A. E. Elia and M. B. Yaffe, MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation, Mol. Cell, 2005, 17, 37–48 CrossRef CAS.
- J. N. Sarkaria, E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz and R. T. Abraham, Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine, Cancer Res., 1999, 59, 4375–4382 CAS.
- S. J. Collis, M. J. Swartz, W. G. Nelson and T. L. DeWeese, Enhanced radiation and chemotherapy-mediated cell killing of human cancer cells by small inhibitory RNA silencing of DNA repair factors, Cancer Res., 2003, 63, 550–554.
- I. Hickson, Y. Zhao, C. J. Richardson, S. J. Green, N. M. Martin, A. I. Orr, P. M. Reaper, S. P. Jackson, N. J. Curtin and G. C. Smith, Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM, Cancer Res., 2004, 64, 9152–9159 CrossRef CAS.
- S. E. Golding, E. Rosenberg, N. Valerie, I. Hussaini, M. Frigerio, X. F. Cockcroft, W. Y. Chong, M. Hummersone, L. Rigoreau, K. A. Menear, M. J. O'Connor, L. F. Povirk, T. van Meter and K. Valerie, Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion, Mol. Cancer Ther., 2009, 8, 2894–2902 CrossRef CAS.
- J. Won, M. Kim, N. Kim, J. H. Ahn, W. G. Lee, S. S. Kim, K. Y. Chang, Y. W. Yi and T. K. Kim, Small molecule-based reversible reprogramming of cellular lifespan, Nat. Chem. Biol., 2006, 2, 369–374 CrossRef CAS.
- M. D. Rainey, M. E. Charlton, R. V. Stanton and M. B. Kastan, Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation, Cancer Res., 2008, 68, 7466–7474 CrossRef CAS.
- L. I. Toledo, M. Murga, R. Zur, R. Soria, A. Rodriguez, S. Martinez, J. Oyarzabal, J. Pastor, J. R. Bischoff and O. Fernandez-Capetillo, A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations, Nat. Struct. Mol. Biol., 2011, 18, 721–727 CrossRef CAS.
- A. Peasland, L. Z. Wang, E. Rowling, S. Kyle, T. Chen, A. Hopkins, W. A. Cliby, J. Sarkaria, G. Beale, R. J. Edmondson and N. J. Curtin, Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines, Br. J. Cancer, 2011, 105, 372–381 CrossRef CAS.
- M. Hollstein, D. Sidransky, B. Vogelstein and C. C. Harris, p53 mutations in human cancers, Science, 1991, 253, 49–53 CrossRef CAS.
- A. J. Levine, J. Momand and C. A. Finlay, The p53 tumour suppressor gene, Nature, 1991, 351, 453–456 CrossRef CAS.
- S. N. Powell, J. S. DeFrank, P. Connell, M. Eogan, F. Preffer, D. Dombkowski, W. Tang and S. Friend, Differential sensitivity of p53(−) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay, Cancer Res., 1995, 55, 1643–1648 CAS.
- Y. Luo, S. K. Rockow-Magnone, M. K. Joseph, J. Bradner, C. C. Butler, S. K. Tahir, E. K. Han, S. C. Ng, J. M. Severin, E. J. Gubbins, R. M. Reilly, A. Rueter, R. L. Simmer, T. F. Holzman and V. L. Giranda, Abrogation of G2 checkpoint specifically sensitize p53 defective cells to cancer chemotherapeutic agents, Anticancer Res., 2001, 21, 23–28 CAS.
| This journal is © The Royal Society of Chemistry 2013 |