Selective oxidation of alcohols and aldehydes over supported metal nanoparticles
Sara E. Davis, Matthew S. Ide and Robert J. Davis*
Department of Chemical Engineering, University of Virginia, 102 Engineers Way, Charlottesville, VA 22904-4741, USA. E-mail: rjd4f@virginia.edu; Fax: +1 434-982-2658; Tel: +1 434-924-6284
First published on 12th October 2012
Abstract
Oxidation is a key reaction in organic synthesis and will likely play a significant role in the development of value-added chemicals from biomass. The application of heterogeneous catalysis and molecular oxygen to oxidation reactions offers a green alternative to traditional, toxic chemical oxidants. However, making comparisons of catalyst performance (reaction rate, product selectivity) between reports in the literature is difficult because of inconsistencies in the ways results are reported. Herein, we examine the literature on supported metal catalysts for the oxidation of molecules of interest in biomass conversion (primary alcohols, polyols, 5-hydroxymethylfurfural, and various sugars). Reaction rates are calculated and compared in a consistent manner and recommendations for avoiding common pitfalls in kinetic investigations are made.
1. Introduction
The diminishing supply of oil has heightened interest in the development of more sustainable alternatives to petroleum-derived fuels and chemicals. Although much attention is paid to the transportation and energy sectors of the economy, 5% of current consumption of crude oil is used for the production of chemicals.1 Renewable resources such as solar and wind power can meet some of the world's energy demands, but replacements for petroleum-derived chemicals require a source of carbon atoms, such as biomass. The U.S. National Renewable Energy Laboratory (NREL) describes a biorefinery for the conversion of biomass to produce fuels, power, and chemicals that is analogous to the contemporary petroleum refinery.2 A major challenge to the development of a biorefinery, and thus the biorenewable chemicals industry, is identifying key intermediates and platform chemicals. While today's petroleum refinery benefits from years of optimization and a well-defined set of platform chemicals for the production of value-added chemicals, the implementation of the biorefinery is still under development.In 2004, the US Department of Energy (DOE) sought to identify the molecules with the greatest potential for use as value added chemicals from biomass.3,4 In the years since the DOE report, the literature involving the production and transformation of these chemicals has grown substantially. While a consensus on the key intermediates that will be utilized in a biorefinery has yet to be reached, common reactions such as oxidation, reduction and dehydration will undoubtedly play a role in their development. As the oxidation of alcohols to their corresponding ketones, aldehydes, and/or carboxylic acids is one of the most important transformations in organic synthesis, reviews of the transformations of alcohols by biocatalysts5 and heterogeneous photocatalysts6 were recently published. The use of Au catalysts for selective oxidation reactions of sugars and alcohols was also recently discussed.7 In the current review, we focus on the selective oxidation of alcohols (and aldehydes) over supported metal catalysts, because of the ease of recovery of heterogeneous catalysts from the products and their potential for recyclability. In particular, this critical review aims to classify reaction rates on a common basis (the turnover frequency, TOF) so that catalyst performance (rate and selectivity) can be compared in a consistent manner.
We have chosen to examine the oxidation of several classes of alcohols, including monoalcohols, 5-hydroxymethylfurfural (HMF), and polyols. The monoalcohols ethanol, octanol, benzyl alcohol, and cinnamyl alcohol, have been used primarily as model alcohols to test for oxidative activity and selectivity over a wide variety of supported metals and reaction conditions. The oxidation of HMF to 2,5-furan dicarboxylic acid (FDCA) was highlighted by the DOE as a target reaction to produce a monomer for use in polyethyleneterephthalate (PET)-like plastics because HMF can be produced from glucose.4 The polyol glycerol is a byproduct of the transesterification of vegetable oil and animal fats into bio-diesel. While glycerol can be utilized in pharmaceutical and cosmetic applications, bio-diesel production provides low purity glycerol. The oxidation of glycerol to fine chemicals, such as dihydroxyacetone (DHA), glyceric acid (GA), and tartronic acid (TA), might also help offset the cost of bio-diesel production.8 Finally, the direct oxidation of glucose and other simple sugars to acids for use as additives in food and beverages was examined. The oxidation of sugars is also interesting because, in aqueous solution, sugars exist in equilibrium between their ring-opened aldose form and their ring-closed cyclic form.9
As the depth of the literature on selective oxidation grows, the wide variety of reaction conditions, catalyst characteristics, and methods for rate calculations make comparing the conversion, selectivity, and kinetic results difficult. Perhaps the most problematic area involves the rate, as the literature generally does not report turnover frequency calculations in a consistent way. It is our belief that the most useful form of the turnover frequency (TOF) is reported at a low level of conversion, before substantial deactivation of the catalyst takes place, and is based on the amount of surface metal available to the substrate (eqn (1)).
 | (1) |
Therefore, in this review, we discuss the literature that has reported turnover frequency values in this way, or has reported enough information for us to calculate the turnover frequency ourselves, so that various catalysts for oxidation reactions can be compared. Because many of the reactions included in this review are not simple one step oxidations but are sequential reactions, we emphasize the importance of comparing product selectivity at equal levels of conversion.
2. Alcohol oxidation mechanism
The oxidation of a primary alcohol proceeds first to an aldehyde and subsequently to a carboxylic acid, as outlined in Fig. 1. The oxidation of an alcohol to an aldehyde over a heterogeneous catalyst likely occurs in three steps: first, the alcohol adsorbs on the metal surface, producing an adsorbed metal alkoxide. Second, β-hydride elimination occurs to produce a carbonyl species and a metal hydride. Last, the metal–hydride is oxidized by dioxygen to regenerate the metal surface. The oxidation of an aldehyde to carboxylic acid is believed to proceed through a geminal diol intermediate.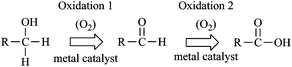 | ||
| Fig. 1 General oxidation scheme for primary alcohol oxidation to acid. | ||
2.1 Metal–alkoxide formation
As mentioned above, the mechanism of primary alcohol oxidation to aldehyde over a supported metal catalyst likely begins with the formation of a metal alkoxide;10–15 however, the nature of the metal or the nature of substrates adsorbed on the metal may influence its formation. In the case of benzyl alcohol and Ru/Al2O3 or Ru(OH)x/Fe3O4 catalysts, a Ru–alkoxide has been hypothesized to form via ligand exchange between Ru–OH and the alcohol, producing a Ru–alkoxide and a water molecule.16 In other work with Ru and Pd catalysts, activation of the O–H bond in benzyl alcohol on the metal surface is proposed, yielding a metal–alkoxide and a metal–hydride.11,12,15,17The selective oxidation of primary alcohols over secondary alcohols was demonstrated for benzyl alcohol and phenylethanol oxidation over Ru catalysts.10,14 These results are consistent with formation of a metal alkoxide intermediate, as the formation of this species is well known for the selective oxidation of primary alcohols.10,16,18,19 In a seeming contradiction, Mori et al. report similar reaction rates for benzyl alcohol and 1-phenylethanol oxidation over a Pd catalyst, yet propose the same metal alkoxide intermediate.11 Further support for metal alkoxide formation was derived from 2-propanol oxidation with O2 over Ru/Al2O3. Production of H2O and acetone were monitored during the reaction and revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio.10,18 Similarly, the uptake of O2 was monitored during benzyl alcohol oxidation, and a 1:2 molar ratio of O2 to aldehyde was observed.11,14,15,18 These studies demonstrate that simple dehydrogenation of alcohol to aldehyde does not take place.
:1 molar ratio.10,18 Similarly, the uptake of O2 was monitored during benzyl alcohol oxidation, and a 1:2 molar ratio of O2 to aldehyde was observed.11,14,15,18 These studies demonstrate that simple dehydrogenation of alcohol to aldehyde does not take place.
2.2 β-Hydride elimination
A β-hydride elimination is widely accepted as the second step of alcohol oxidation over metal catalysts, producing a carbonyl group and a metal–hydride.10–12,14,15,18,20,21The Hammett methodology has been employed by a few groups to probe the mechanism of benzyl alcohol oxidation and to confirm the β-hydride elimination step.10,13 Comparing the rates of para-substituted benzyl alcohols over Ru catalysts yielded a Hammett ρ value of −0.461, which indicates the formation of a carbocation-type intermediate due to hydride abstraction from the Ru–alkoxide.10 Similarly, a Hammett ρ value of −1.10 was found over a Au catalyst, indicating that a similar mechanism operates despite the higher oxygen coverage on Ru than on Au.13
The hydride abstraction step was also confirmed through the incorporation of deuterium in the α-position of the alcohol. The deuterated alcohol should react significantly slower than the non-deuterated alcohol and the kinetic isotope effect was found to be 1.82. Transfer hydrogenation was noted in a mixture of acetophenone and 2-propanol with a Ru catalyst, further supporting the formation of a Ru–H species.10 Interestingly, the β-hydride elimination step has been proposed as the likely rate-determining step in alcohol oxidation.10,11,14,15
2.3 Oxidation of metal–hydride and regeneration of catalyst surface
The third step in alcohol oxidation to aldehyde is the oxidation of the metal–hydride species generated from the β-hydride elimination step to regenerate either the metal–hydroxide10,14 or metal surface.11,12,15 The oxidation of the metal–hydride likely proceeds through a peroxide intermediate to yield a water molecule and one half of an O2 molecule.112.4 Oxidation of aldehyde to carboxylic acid
It is well known in organic chemistry that aldehydes in H2O undergo reversible hydration to geminal diols and that the rate of hydration is accelerated at elevated pH. The geminal diol will likely adsorb to a metal surface to form a metal alkoxide, which will undergo β-hydride elimination to form a carboxylic acid.222.5 Illustrative example: ethanol and glycerol oxidation
The oxidation of an alcohol to an acid over supported Au and Pt was investigated both experimentally and computationally with density functional theory (DFT).23 Specifically, the oxidation of ethanol to acetic acid and glycerol to glyceric acid over Au and Pt in an aqueous solvent requires dioxygen. In addition, Au requires added base for the reaction to proceed at low temperature whereas the rate of reaction over Pt is significantly increased with the addition of base. The roles of dioxygen and the hydroxide (high pH) in the oxidation mechanism were explored to elucidate the most likely steps in alcohol oxidation. Since ethanol (alcohol) and glycerol (polyol) are oxidized by the same mechanism, it is likely that the mechanism can be extended to most alcohol oxidations.Table 1 (Steps 1 and 2) compares the energetics of the first step of ethanol oxidation to form a metal alkoxide via the initial deprotonation of the alcohol. The deprotonation of an alcohol can occur in solution, where it is controlled by the solution pH and pKa of the alcohol, or it can occur on a metal surface where the deprotonation forms a metal alkoxide. While the intrinsic barrier for deprotonation of the alcohol on a metal surface is high (Step 1), the presence of a surface bound hydroxide decreases the activation energy significantly (Step 2). The activation energy over a Au(111) surface decreases from 204 kJ mol−1 over the bare metal surface to 22 kJ mol−1 over one containing adsorbed hydroxide. Thus, adsorbed hydroxide enhances the deprotonation of an alcohol similar to that in basic solution. It is important to note that Step 1 forms a metal–hydride bond, while Step 2 results in the formation of adsorbed water.
| Step # | Step | Au(111) | Pt(111) | ||
|---|---|---|---|---|---|
| ΔHRXN | EACT | ΔHRXN | EACT | ||
| The * represents a catalytic site on the surface. All values in kJ mol−1.23 | |||||
| 1 | CH3CH2OH* + * → CH3CH2O* + H* | +196 | 204 | +98 | 116 |
| 2 | CH3CH2OH* + OH* → CH3CH2O* + H2O* | +13 | 22 | −5 | 18 |
| 3 | CH3CH2O* + * → CH3CHO* + H* | −40 | 46 | −62 | 15 |
| 4 | CH3CH2O* + OH* → CH3CHO* + H2O* | −222 | 12 | −165 | 24 |
| 5 | CH3CHO* + OH* → CH3CHOOH* + * | −33 | 5 | −5 | 5 |
| 6 | CH3CHOOH* + * → CH3COOH* + H* | −151 | 21 | −154 | 13 |
| 7 | CH3CHOOH* + OH* → CH3COOH* + H2O* | −334 | 29 | −258 | 17 |
After the formation of the metal–alkoxide, a β-hydride elimination can similarly happen on the metal surface alone or be facilitated by surface bound hydroxide. While the energy of activation is again lower over Au(111) with an adsorbed hydroxide (Step 4) compared to just the bare metal (Step 3), the difference in activation energy is not as significant as the initial alcohol deprotonation step. In addition, the β-hydride elimination has similar activation energies over Pt(111) alone and with an adsorbed hydroxide. Once the β-hydride elimination has been completed, an adsorbed aldehyde is formed on the surface.
The aldehyde can undergo reversible hydration to a geminal diol in the solution phase. Under basic conditions, the aldehyde can also react over the metal surface with hydroxide to form an adsorbed diol intermediate (Step 5). A second β-hydride elimination of the geminal diol can form a carboxylic acid over the metal surface (Step 6) or be facilitated by adsorbed hydroxide (Step 7). At this point, O2 does not play a role in the mechanism, yet O2 is required for the oxidation to proceed. Consistent with the above mechanism, when 18O2 was used to oxidize ethanol or glycerol, only unlabeled acetic acid and glyceric acid were produced, regardless of whether the catalyst could easily dissociate 18O2, such as Pt and Pd, or not, such as Au. However, when H218O was used, 18O appeared in the products. The appearance of 18O in the product when using H218O is strong experimental evidence that aqueous-phase alcohol oxidation most likely proceeds through the alkoxy intermediate of the geminal diol which can undergo β-hydride elimination on the metal surface to produce the carboxylic acid.
Because the dissociation of O2 on the Au surface is unlikely (105 kJ mol−1) and most metal surfaces, such as Pt and Pd, are likely inhibited by water and hydroxide adsorbed on the surface, O2 might adsorb associatively on the surface. Experimental evidence of peroxide production formation during the reaction suggests that the associatively adsorbed oxygen is reduced by electrons on the metal catalyst before dissociation into hydroxide.24 The energetics of O2 reduction, summarized in Table 2, show a low barrier of 16 kJ mol−1 over Au(111) (Step 1). The adsorbed peroxide can then react with water or decompose on the surface. The dissociation of peroxide into atomic oxygen and hydroxide on Au(111) (Step 2), however, is less likely energetically compared to the formation of hydrogen peroxide and hydroxide (Step 3), which is consistent with experimental results in which peroxide formation is reported over supported Au.24 The last step closes the catalytic cycle by removing excess electrons from the metal surface via adsorbed hydroxide reacting with any metal hydrides found on the metal surface (Step 5). Thus, the role of associatively adsorbed oxygen is ultimately to remove electrons from the metal surface, oxidize metal–hydride bonds, and regenerate hydroxide ions.
| Step # | Step | Au(111) | Pt(111) |
|---|---|---|---|
| EACT | EACT | ||
| The * represents a catalytic site on the surface.23 | |||
| 1 | O2* + H2O* → OOH* + OH* | 16 | 18 |
| 2 | OOH* + * → O* + OH* | 83 | 52 |
| 3 | OOH* + H2O* → H2O2* + OH* | 48 | 41 |
| 4 | H2O2* + * → OH* + OH* | 71 | 29 |
| 5 | OH* + H* → H2O* + * | 39 | 30 |
It is important to note that hydroxide ions are not conserved during the reaction. Hydroxide ions are reacted during the oxidation of glycerol to glyceric acid and some are used to neutralize the acid product. While the reduction of O2 can regenerate some of the hydroxide ions (Table 2, Step 1), about 0.6 mole of hydroxide was consumed for every mole of glycerol converted, after the hydroxide used to neutralize the acid products was accounted for.25
2.6 Illustrative example. HMF oxidation
The oxidation of 5-hydroxymethylfurfural (HMF) involves both the oxidation of an aldehyde and an alcohol. The oxidation to monoacid hydroxymethylfurancarboxylic acid (HFCA) and diacid 2,5-furandicarboxylic acid (FDCA) at ambient temperatures over Pt and Au catalysts requires both a base (typically NaOH) and gaseous dioxygen and the roles of both base and O2 in the mechanism were elucidated recently.26 Similar to the glycerol study,23 isotopically-labeled H218O and 18O2 were employed to discern the source of oxygen atoms inserted into the products. To ensure that the incorporation of labeled oxygen was due to the mechanism of reaction and not due to oxygen scrambling in solution, control experiments under reaction conditions were conducted. No oxygen scrambling between H218O and the products HFCA and FDCA was detected.The majority product of HMF oxidation was found previously to be affected by the catalyst type and ratio of NaOH:HMF.26 Thus, three different scenarios for HMF oxidation were examined in that study: oxidation to FDCA over Pt, oxidation to HFCA over Au with a relatively low concentration of NaOH (NaOH:HMF 2:1), and oxidation to FDCA over Au with a relatively high concentration of NaOH (NaOH:HMF 20:1). In all of these studies, the incorporation of 18O was found in the product when the reaction was conducted with H218O, but no incorporation of 18O was seen in reactions utilizing 18O2. In the case of a Au catalyst with low NaOH concentration, two 18O atoms were incorporated in the product, HFCA, when the reaction was run in H218O, presumably both in the acid group. In the case of Au with a high concentration of NaOH and in the case of the Pt-catalyzed reaction, four 18O atoms were incorporated in the diacid product, FDCA, when the reaction was run in H218O. Thus, in all cases, oxygen insertion in the acid products occurred through H218O. The proposed mechanism can be seen in Fig. 2.
 | ||
| Fig. 2 Proposed HMF oxidation mechanism, adapted from ref. 26. | ||
In step one, the aldehyde side chain of HMF undergoes reversible hydration in solution to a geminal diol through the nucleophilic addition of a hydroxide ion to the carbonyl and proton transfer from H2O to the alkoxy ion intermediate. The second step is a dehydrogenation of the geminal diol to form a carboxylic acid, likely facilitated by hydroxyl ions on the metal surface, as indicated in the previous section. This step produces two molecules of water and deposits two electrons on the metal surface. In the third step, the hydroxymethyl group is dehydrogenated to an aldehyde. It is believed that base deprotonates the alcohol group, likely in solution, to form an alkoxy intermediate.23 Subsequently, hydroxide ions on the metal surface facilitate the activation of the C–H bond in the hydroxymethyl group to form an aldehyde, producing two molecules of water and depositing two additional electrons on the metal surface. In step four, this aldehyde undergoes the same reversible hydration to a geminal diol as seen in step one. Finally, a dehydrogenation step produces the second carboxylic acid (step five), depositing two more electrons on the metal surface, analogous to step two. In total, six electrons are deposited on the metal catalyst surface.
Though the isotopic labeling studies indicate that oxygen atoms from dioxygen are not directly incorporated in the acid products, it is essential for the oxidation of HMF to FDCA. It was proposed in the ethanol and glycerol oxidation mechanism that O2 is an electron scavenger, removing excess electrons from the metal surface by undergoing the oxygen reduction reaction to peroxide and hydroxide ions.23,26 A test also revealed peroxide in solution during HMF oxidation reactions; therefore, it is believed that the role of O2 in HMF oxidation is to scavenge electrons from the metal catalyst surface, regenerating hydroxide ions and closing the catalytic cycle.26 This mechanism is completely consistent with that described for ethanol and glycerol oxidation.
3. Alcohol oxidation review
3.1 Ethanol
Production of ethanol from biomass has been the investigated thoroughly due to the application of ethanol as a liquid fuel. Much of the literature focuses on electro-oxidation of ethanol in fuel cells, or on oxide materials as catalysts for ethanol oxidation, which is beyond the scope of this review. The oxidation of ethanol can lead to acetic acid, an important industrial chemical in the production of synthetic fibers and fabrics, as well as a chemical reagent in the production of vinyl acetate. Consistent with other alcohol oxidation reactions and the mechanism discussed earlier, the first oxidation product is acetaldehyde and the second oxidation product is acetic acid. The general oxidation scheme for ethanol oxidation is seen in Fig. 3. | ||
| Fig. 3 Reaction scheme for ethanol oxidation. | ||
Zope et al. reported on the oxidation of ethanol over supported Au and Pt catalysts, both in presence and absence of added NaOH.23 The group found the turnover frequencies over Au to be an order of magnitude higher than those over Pt when a 2:1 ratio of NaOH:ethanol was used. In the absence of NaOH, the Au catalysts were inactive for ethanol oxidation at 333 K, whereas Pt/C displayed some activity. The turnover frequencies for ethanol oxidation are summarized in Table 3.
Jorgensen et al.27 investigated the oxidation of aqueous ethanol over Au/TiO2 catalysts at temperatures between 363 and 473 K, and a yield of 95% acetic acid was reached. Preliminary investigations over Au/C did not demonstrate nearly as high a conversion of ethanol or selectivity to acetic acid. Although attempts were made to recycle the catalyst, a significant decrease in activity was noted, likely due to Au particle aggregation (spent catalyst Au size 5–7 nm; unused 3–6 nm). The intermediates and byproducts noted in this reaction were acetaldehyde, CO2, and ethyl acetate. The reaction profile shows that the oxidation of ethanol proceeds through acetaldehyde before reaching acetic acid. Using acetaldehyde as the reactant demonstrated a rapid oxidation to acetic acid (acetic acid yield 98%); thus, it was determined that the dehydrogenation of ethanol was the rate determining step. Because extending the reaction time showed no degradation of acetic acid, formation of CO2 likely occurred via over-oxidation of an intermediate species. One proposed intermediate forms by the adsorption of ethanol to the catalyst surface and can be either oxidized/dehydrogenated to acetaldehyde or cleaved to produce CO2. An attempt was made to increase the yield of ethyl acetate by varying the concentration of ethanol and a maximum selectivity to ethyl acetate (50%) was seen at concentrations between 80–100% ethanol, indicating that water has a significant limiting effect on the production of ethyl acetate from ethanol.
At moderate temperature and pressure (423 K and 0.6 MPa O2), the selective oxidation of aqueous ethanol into acetic acid with 90% yield was demonstrated by Christensen et al.28 Ethanol concentrations corresponding to those obtained during fermentation were utilized. Comparing the yields from Au/MgAl2O4 (conversion 97%, selectivity 86%), Pt/MgAl2O4 (conversion 93%, selectivity 65%), and Pd/MgAl2O4 (conversion 82%, selectivity 20%) at the same reaction conditions, the highest yield was obtained over the Au catalyst. No signs of sintering were seen for the Au catalyst (particle size 3–6 nm) over the course of the reaction (30 h). Under optimized conditions (453 K and 3.5 MPa O2), 92% yield of acetic acid was obtained after 8 h. Increasing the reaction temperature resulted in both increased conversion of ethanol and selectivity to acetic acid.27,28
Gold catalysts are among the most effective of the supported metal nanoparticle catalysts for ethanol oxidation, though much of the literature seems to be focused on metal oxides or electrocatalysts for this transformation. The number of reports of initial TOF is limited, and most reports on ethanol oxidation are at too high conversion to calculate initial rate.
3.2 Octanol
Octanol is often used as a probe molecule to determine the effectiveness of a catalyst in the oxidation of alkanols. The general reaction scheme for octanol oxidation is seen in Fig. 4. Low reactivity is characteristic of octanol. Accordingly, there is an abundance of low conversion results in the literature, which facilitates the calculation of initial reaction rate. | ||
| Fig. 4 Reaction scheme for octanol oxidation. | ||
Both primary and secondary octanols are investigated frequently, and the rates of 1-octanol oxidation are summarized in Table 4. The highest rate was obtained over bimetallic catalysts containing Au and either Pt or Pd (0.3 s−1 for PdAu/C and 0.2 s−1 for PtAu/C) in H2O solvent with 4 equivalents of NaOH at 323 K.29 Under the same conditions, but in absence of added base, the rates of oxidation were an order of magnitude lower for both bimetallic catalysts. An experiment utilizing a AuPd bimetallic catalyst in water with 1-equivalent NaOH exhibited a TOF of 0.06 s−1, higher than the base-free case but lower than the experiment with higher NaOH concentration.30 Additionally, Au monometallic catalysts were inactive for 1-octanol oxidation without addition of base in both water and toluene solvents.29,30 Thus, the role of base in octanol oxidation over bimetallic PdAu and PtAu catalyst is significant. The role of base is also significant in oxidation over monometallic Au catalysts; in the absence of added base, the Au catalyst was virtually inactive, though some activity was realized with the addition of 4:1 NaOH:substrate. Interestingly, the addition of base to the reaction mixture over Pt and Pd monometallic catalysts had no significant effect on the reaction rate. Aside from the bimetallic catalysts, the rates shown in Table 4 are similar to each other and it would seem that the intrinsic TOF of 1-octanol oxidation in water at temperatures 323 K–373 K over Au, Pt, and Pd metals is on the order of 0.01 s−1.
| Catalyst | Particle size (nm) | T (K) | Pressure (kPa) | Solvent | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a All reactions were performed in batch mode with O2 as oxidant. | ||||||
| Au/CeO2 | Not reported | 373 | 200 | None | 0.007 | 31 |
| Au/C | Not reported | 323 | 310 | H2O + 4 eq. NaOH | 0.05 | 29 |
| Pd/C | Not reported | 323 | 310 | H2O + 4 eq. NaOH | 0.007 | 29 |
| Pd/C | Not reported | 323 | 310 | H2O | 0.007 | 29 |
| Pt/C | Not reported | 323 | 310 | H2O + 4 eq. NaOH | 0.008 | 29 |
| Pt/C | Not reported | 323 | 310 | H2O | 0.008 | 29 |
| PdAu/C | Not reported | 323 | 310 | H2O + 4 eq. NaOH | 0.3 | 29 |
| PtAu/C | Not reported | 323 | 310 | H2O + 4 eq. NaOH | 0.2 | 29 |
| PdAu/C | Not reported | 323 | 310 | H2O | 0.01 | 29 |
| PtAu/C | Not reported | 323 | 310 | H2O | 0.02 | 29 |
| 0.73%Au–0.27%Pd/C | 3.4 | 333 | 155 | H2O | 0.02 | 30 |
| 0.73%Au–0.27%Pd/C | 3.4 | 333 | 155 | H2O + 1 eq. NaOH | 0.06 | 30 |
Dimitratos et al. have speculated that NaOH facilitates the first step of oxidation, H abstraction, which Au alone is unable to catalyze at low temperature.30 Moreover, the addition of NaOH favors formation of Na-carboxylate, which is more stable than metal-carboxylate. Thus, the products are thought to desorb from the metal surface so the catalytic cycle can continue uninhibited.
The addition of NaOH can enhance the reaction rate, but also affects the product selectivity, presumably because of the sequential nature of the oxidation reaction. In the case of a PdAu/C catalyst, 17% conversion of aqueous 1-octanol with 70% selectivity to the aldehyde was realized after 8 h at 323 K without base; adding 4 equivalents of NaOH without changing the other conditions shifted the selectivity toward the acid (98% selectivity) with 93% conversion in just 4 h.29 Under the same conditions but with the monometallic Pd/C catalyst, at 2% conversion of 1-octanol the selectivity to the aldehyde was 70% in absence of NaOH, but with 4 equivalents of NaOH, the reaction was 97% selective to the acid at 2% conversion. The trend continued over Pt and PtAu catalysts.
Prati et al. also noted a negligible effect of O2 pressure on 1-octanol reaction rate. Increasing the reaction temperature enhanced activity but resulted in a decrease in selectivity to aldehyde.29
In addition to 1-octanol, secondary octanols have also been investigated. For example, the rates of oxidation of neat 3-octanol over some supported catalysts are reported in Table 5. The highest rates for 3-octanol oxidation were obtained over Au catalysts supported on nanocrystalline CeO2. Both supported Au catalysts in Table 5 exhibited high selectivity to the ketone product (99 and 97%, respectively). Table 5 also presents a comparative study of Au/CeO2 and Pd/CeO2 demonstrating the enhanced activity of Au (TOFAu = 2.92 s−1 compared to TOFPd = 0.09 s−1 at 333 K), although supporting Pd on apatite improved its activity. The oxidation of 2-octanol was investigated over supported Ru(OH)x and Pd catalysts in toluene and trifluorotoluene, respectively11,14 and both metals were shown to be active for this oxidation.
The literature investigating octanol oxidation highlights the superior activity (rate) of Au catalysts over Pd catalysts for alcohol oxidation, typically exhibiting a TOF an order of magnitude greater. The role of additional homogenous base is significant in reactions utilizing bimetallic Pt or Pd and Au catalysts or Au monometallic catalysts. The role of base is apparently much less important in reactions utilizing monometallic Pt or Pd catalysts.
3.3 Benzyl alcohol oxidation
Benzyl alcohol is often used as a model alcohol to test for catalyst reactivity. While the literature is extensive on this topic, quality rate data obtained at low conversion is lacking. The popularity of benzyl alcohol as a probe molecule results from its extremely high reactivity and the limited number of side products (the intermediate benzaldehyde is a non-enolizable structure).31 Like other alcohols, benzyl alcohol oxidation proceeds through an aldehyde intermediate to the acid final product, as depicted in Fig. 5. Typical metal catalysts for benzyl alcohol oxidation are Pt, Pd, and Au. | ||
| Fig. 5 Reaction scheme for benzyl alcohol oxidation. | ||
It is interesting to note that the TOFs vary by an order of magnitude – from 0.19 s−1 to 2.8 s−1 over a fairly narrow range of experimental conditions.
The role of the support for Pd catalysts was investigated by Mori et al.11 and Chen et al.15 Mori et al. found that benzyl alcohol conversion and the benzaldehyde selectivity depended on support, with the hydroxyapatite (HAP)-supported catalyst having the highest conversion (99%) and selectivity to aldehyde (99%). The other supports examined (Al2O3, SiO2, and C) produced the aldehyde with less than 50% selectivity; the most selective was Pd/SiO2 (47%) though the conversion was only 71%. The highest conversion was achieved over Pd/Al2O3, though the selectivity was the lowest (38%). The Pd/C produced the lowest conversion (46%) and low selectivity (42%). Unfortunately, the reaction rates over these catalysts were not given, so no conclusions can be drawn about the effect of support on reaction rate.
Chen et al. probed the effect of Pd particle size on BA oxidation by varying the ratio of SiO2:Al2O3 in a mixed oxide support to tune the size of Pd particles in the range 2.2–10 nm.15 Decreasing the SiO2:Al2O3 ratio resulted in a decrease in the size of the Pd nanoparticles, due to the relative strength of interactions of the Pd precursor with the support. Though varying the SiO2:Al2O3 ratio changed the relative acidity of the support, the variation in activity correlated only to Pd nanoparticle size. A maximum TOF was found at a mean metal particle size of 3.6–4.3 nm, suggesting that BA oxidation over Pd might be structure sensitive, which was also noted by Mori et al.11 in their work over Pd supported on HAP. The optimized catalysts (Pd particle sizes of 4.3 nm and 3.6 nm) demonstrated high TOF (2.53 s−1 and 2.45 s−1, respectively) as well has high selectivity to the aldehyde (98% selectivity with 73% conversion and 98% selectivity with 99% conversion, respectively, in 10 h) in solvent-free conditions.
Uozumi et al. utilized a novel support, an amphiphilic resin, in the oxidation of aqueous alcohols.35 The resin is highly hydrophobic in its pores, so organic molecules can diffuse from the aqueous media into the matrix where the anchored Pd nanoparticles catalyze the oxidation. A mixture of catalyst and benzyl alcohol was refluxed in water under atmospheric pressure of dioxygen to give 97% yield of the aldehyde product after 1.5 h. The high selectivity to the aldehyde in aqueous medium is interesting particularly because sequential oxidation to the carboxylic acid readily occurs in water. Apparently the hydrophobic nature of the resin prevents the conversion of the aldehyde in water over Pd.
The effect of solvent was investigated by Mori et al.11 and Villa et al.36 Mori found the most effective solvent to be trifluorotoluene (99% conversion in 1 h), although 1,2-diethoxyethane afforded 65% conversion under the same conditions. Aprotic polar solvents were not effective at all, which the authors hypothesized may be due to the coordination of heteroatoms to Pd(II) pre-catalysts, preventing their reduction to Pd0. The oxidation of BA in aqueous NaOAc over Pd/HAP resulted in 90% yield of aldehyde and 10% yield of benzoic acid at 383 K in an O2 atmosphere after 24 h, which is unusual given the typical sequential oxidation of aldehyde to carboxylic acid in high pH solution.
The investigation of role of solvent by Villa et al. used Pd on activated carbon (AC) or on carbon nanotubes (CNT).36 An increased rate of reaction was noted in neat alcohol (TOF 0.83 s−1) as opposed to a mixture of 80% water and 20% alcohol (TOF 0.023 s−1) over Pd/AC catalyst, though the product distribution remained unchanged. The rate of reaction was higher in cyclohexane than in water. It is important to note that the leaching of Pd from the support into the liquid was extensive in this study, 28% for Pd/AC and 25% for Pd/CNTs.
From these results, it appears that the support material for Pd nanoparticles can have a substantial effect on the product selectivity. Interestingly, the appropriate choice of support can suppress the sequential oxidation of aldehyde to carboxylic acid in aqueous solution. Nevertheless, it seems that the intrinsic turnover rate for benzyl alcohol oxidation over Pd catalysts at modest temperatures (<373 K) is on the order of 2 s−1.
| Catalyst | Particle size (nm) | T (K) | Pressure (kPa) | Solvent | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a All reactions were performed in batch mode.b Ar atmosphere. | ||||||
| Au on block co-polymers | 9.5 | 308 | 100 | 1:1 H2O:chloroform with KOH (1:1 KOH:substrate) | 0.04 | 37 |
| Au/CeO2 | ∼4.5 | 373 | 200 | None | 0.042 | 31 |
| Au foil | n/a | 333 | 500 | H2O + 0.6 M NaOH | 2.8 | 38 |
| Au foil | n/a | 363 | 500 | Toluene + equimolar K2CO3 | 2.8 | 38 |
| Au foil | n/a | 363 | 500 | Heptane + equimolar K2CO3 | 4.4 | 38 |
| Au foil | n/a | 333 | 500 | Heptane + equimolar K2CO3 | 2.8 | 38 |
| Au foil | n/a | 383 | 500 | Heptane + equimolar K2CO3 | 5.7 | 38 |
| Au/MgO | 3.3 | 453 | Unknown | Trifluorotoluene | 0.13 | 39 |
| Au/HT | 3.1 | 393 | Flowingb | Toluene | 0.22 | 21 |
Much of the literature on alcohol oxidation over Au catalysts describes results obtained in the presence of a homogeneous base. As Table 7 shows, added base can greatly enhance the reaction rate. Buonerba et al.37 supported Au nanoparticles on block copolymers and used the resulting catalysts in the oxidation of benzyl alcohol in a water and chloroform solvent with added KOH. They attained >99% conversion of benzyl alcohol and 97% selectivity to the aldehyde product after 6 h at mild conditions (308 K, 100 kPa O2). Over longer reaction times (>24 h), further oxidation of benzaldehyde was not observed; the performance of these materials is in stark contrast to other reports which showed partial or complete oxidation to benzoic acid in aqueous media or in the presence of base.22,38,40,41 The authors ascribed this difference to the temperature (407 K) and the hydrophobic nature of the polymer phase in conjunction with the chloroform solvent, similar to what Uozumi and Nakao proposed in their Pd/resin catalysts.35 Thus, a support material may inhibit oxidation of aldehyde to acid in high pH aqueous media. However, the influence of the base might also be limited by the hydrophobic support.
Very recently, Guo et al. reported successful aerobic oxidation of benzyl alcohol over unsupported bulk Au.38 The oxidation of BA was performed in aqueous NaOH, and the distribution of products was a function of the reaction temperature and concentration of NaOH. For benzyl alcohol oxidation at 333 K, the major product was the ester, benzyl benzoate, for lower concentrations of base (≤0.6 M NaOH) and the acid, benzoic acid, at higher concentrations (1.2 M NaOH). Increasing the temperature to 363 K resulted in a majority of the aldehyde product over the range of NaOH concentrations.
Investigation of other solvents revealed that bulk Au was inert in trifluorotoluene but active for BA oxidation in heptane and in toluene, producing the aldehyde as the majority product. The authors hypothesize that the π-electrons in BA enhance interactions with Au, facilitating the oxidation. They found that the π-activation for the –CH2OH group by the phenyl group in BA is critical for oxidation over bulk Au. No oxidation occurred in the presence of inert gas, demonstrating the importance of O2 as oxidant in this case, in contrast to previous work on BA oxidation over supported Au.21,42 Increasing the temperature increased the conversion of BA in heptane solvent, whereas conversion decreased with increasing temperature in water solvent. The authors suggested that the oxidation in aqueous NaOH may be favored by H2O2 produced during the reaction and that the H2O2 may decompose readily at the higher temperatures. At the lower temperature, H2O2 was readily detected, whereas at the higher temperature it was not. No peroxide was detected when heptane was the solvent. Previous work proposed that, in organic solvent, the role of O2 was to remove the hydride from the Au surface, or, in aqueous NaOH, that O2 regenerated catalytic sites by removing electrons from the surface and in the process regenerating hydroxide ions.23
As an alternative to adding homogenous base, some groups investigated solid bases as catalyst supports. The effect of acid and base sites on the support in Au catalysis for BA oxidation has been investigated in the absence of additional homogeneous base.21,31,41 In summary, low conversion of benzyl alcohol was observed after 3 h (maximum conversion 7%, over Au/Fe2O3), which is typical of oxidation reactions in the absence of homogeneous base. Selective oxidation to benzaldehyde was achieved over Au on SiO2, CeO2, and TiO2, with the best results obtained with Au/CeO2 (3.4% conversion, 100% selectivity to aldehyde after 3 h at 373 K and 200 kPa O2). The lowest conversion was seen over Au/TiO2 (0.65% conversion in 3 h; 100% selectivity to aldehyde). The Fe2O3 and C supported catalysts were the only samples to form the ester product (selectivity to benzyl benzoate 12% and 10%, respectively); the authors hypothesized that ester formation is related to the acid sites on the catalyst surface. Indeed, temperature programmed desorption of NH3 revealed the strongest acidic sites on Au/Fe2O3, and addition of a small amount of HCl to a reaction with Au/SiO2 supported this hypothesis.
Villa et al. examined the role of base sites on product selectivity in cyclohexane solvent in the absence of homogeneous base and found a correlation between basicity of the support and catalyst activity.41 Gold catalysts supported on nanosized NiO (3–5 nm) were more active by an order of magnitude than Au on commercial micrometer sized NiO (55% conversion vs. 6% conversion, in 6 h), likely due to both an increase in basicity of the support (basicity per square meter of nNiO: 0.21 mmol g−1; of commercial NiO: 0.12 mmol g−1) and also a cooperative effect of metal and support.
An inverse relationship between the basicity of the support and the selectivity to aldehyde formation was also noted. The more basic nanosized support showed decreased selectivity to aldehyde (66%) compared to the less basic microsized support (selectivity to aldehyde 75%); the increased basicity enhanced sequential oxidation to acid products. The basicity of the support, however, was not the only factor responsible for the activity, as MgO was more basic than nNiO (basic site density 0.42 mmol g−1) but was a much less active support for Au (7% conversion vs. 55%). Investigation via CO adsorption and IR spectroscopy revealed that Au/nNiO did not adsorb CO molecules but Au/TiO2 did, indicating a change in the electronic properties of Au.
Finally, Fang et al. investigated amphoteric materials and the effects of their acidity/basicity on BA oxidation in p-xylene solvent at 393 K in the absence of an oxidant.21 Gold nanoparticles of a similar size were supported on a variety of materials to compare support effects. After 1 h of reaction time, Au/Al2O3, Au/MgO, Au/HAP, and Au/hydrotalcite (HT) catalysts demonstrated BA conversion of about 20–30%; the selectivity to the aldehyde over each catalyst was compared at this conversion. The Au/Al2O3 catalyst showed the highest conversion (32%) but the lowest aldehyde selectivity (18%), whereas Au/MgO showed the highest selectivity (99%) but the lowest conversion (20%). The Au/HT exhibited similar selectivity to the Au/MgO, but at approximately 10% higher conversion. Temperature programmed desorption (TPD) of NH3 and CO2 revealed that Au/HT and Au/MgO possessed higher concentrations of basic sites than the other catalysts. It was also shown that Au/HT had both strong acid and strong base sites, Au/SiO2 had neither acid nor base sites, and Au/MgO had only strong base sites. The catalysts with neither acid nor base sites were almost inactive, whereas the catalyst with the highest acidity and basicity (Au/HT) had the highest conversion of alcohol and selectivity to aldehyde. Moreover, a catalyst with stronger basicity in the absence of acidity (Au/MgO) had higher selectivity to aldehyde, but a catalyst with strong acidity (Au/Al2O3) was more active. This is in direct contrast with Villa et al., who saw a lower selectivity to aldehyde over catalysts with more base sites because of sequential oxidation to carboxylic acid. It should be noted, however, that the work of Fang et al. was in absence of an oxidant so the other products present were toluene and benzene rather than other oxidation products. The role of O2 in conjunction with catalyst support may be significant with respect to product selectivity.
The effect of particle size of Au on BA oxidation was examined by Boronat et al.39 using MgO as a support and by Fang et al. using HT as support.21 In general, the smaller the particle diameter, the higher the TOF. Thus, the groups concluded that alcohol dehydrogenation on Au is a structure-sensitive reaction, which was also supported by DFT calculations.39 Interestingly, a maximum TOF for Au on MgO was observed for a particle size of 3.3 nm (0.09 s−1) while Au/MgO with a smaller particle size (1.9 nm) exhibited lower activity (0.03 s−1).39 The trend is reminiscent of the trend noted by Chen et al.15 who saw an optimum ratio of edge and corner sites to terrace sites in the oxidation of BA over Pd nanoparticles. Others have also noticed structure sensitivity for alcohol oxidation.11,15,43
In summary, the oxidation of benzyl alcohol exhibited the highest rates (5 s−1) over Au catalysts in the presence of homogeneous base. Although the oxidation of alcohol to aldehyde to acid is greatly accelerated by the presence of homogeneous base, the sequential oxidation of aldehyde to acid may be inhibited through careful selection of the solvent and catalyst support. The incorporation of acid sites and/or basic sites on catalyst support can influence the product selectivity of the overall reaction.
| Catalyst | Particle size (nm) | T (K) | Pressure (kPa) | Solvent | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a All reactions performed in batch mode with O2 as oxidant. | ||||||
| Au–Pd/TiO2 | 4 | 353 | 100 | None | 1.6 | 44 |
| Au–Pd/TiO2 | 4 | 353 | 100 He | None | 0.24 | 44 |
| 0.73% Au–0.27% Pd/C | 3.4 | 333 | 155 | Toluene | 0.15 | 30 |
| 0.73% Au–0.27% Pd/C | 3.4 | 333 | 155 | H2O | 0.01 | 30 |
| 0.6% Au–0.4% Pt/C | 3.2 | 333 | 155 | Toluene | 0.004 | 30 |
| 0.6% Au–0.4% Pt/C | 3.2 | 333 | 155 | H2O | 0.05 | 30 |
| 2.5% Au–2.5% Pd/C | Not reported | 433 | 100 | None | 24 | 45 |
Enache et al. noted a very high TOF for BA oxidation (24 s−1) at mild conditions (373 K, 0.1 MPa O2) over a bimetallic Au–Pd/TiO2 catalyst.45 Although the addition of Au to Pd nanoparticles improved the selectivity to aldehyde (92% at 75% conversion for bimetallic, 54% selectivity at 51% conversion for monometallic Pd), the initial rate of the bimetallic catalyst was lower than that of the monometallic Pd. This indicates that the TOF for the monometallic Pd catalyst (not reported) was greater than 24 s−1, which is much higher than any other reports for Pd catalysts for BA oxidation.
Microscopy and XPS showed that the nanocrystals were made up of an Au-rich core with a Pd-rich shell; the enhancement of activity may be due to Au altering the electronic structure of Pd.45 Pure Au and Pd catalysts did not retain high selectivity to the aldehyde at high conversion, whereas the bimetallic catalyst did (selectivity 96% at 100% conversion). Bimetallic catalysts were also synthesized on Al2O3 and Fe2O3, though the selectivity to the aldehyde was lower (87% at 83% conversion on Al2O3, 67% at 63% conversion on Fe2O3) possibly due to acid sites on the support promoting ester formation.
Meenakshisundaram et al. also synthesized bimetallic Au–Pd/TiO2 catalysts for BA oxidation, and found that the initial rate of BA oxidation was an order of magnitude higher over the bimetallic catalyst than over the monometallic Pd catalyst, which showed a rate an order of magnitude higher than the monometallic Au catalyst.44 This contrasts the report above, in which the bimetallic demonstrated a rate between those of the Pd and Au monometallic catalysts.45 Meenakshisundaram et al.'s materials were found to be homogeneous alloys rather than the shell-and-core configuration of Enache et al. This group also looked at the reaction under inert pressure and found that the reaction rates were an order of magnitude lower than in the presence of O2. Moreover, the selectivity to aldehyde was 50% with toluene making up the balance. By comparing results from the monometallic catalysts, it appears that all of the activity of the bimetallic alloys to produce aldehyde under inert pressure was due to the Pd metal and the Au metal did not contribute to the activity.
Dimitratos et al.30 saw an enhancement of the reaction rate over Au, Pd, and Au–Pt and Au–Pd bimetallic catalysts supported on C when water was used as solvent in place of toluene, in all cases. The Au catalysts were inert in H2O and toluene. However, it is important to note that no homogeneous base was added to the reaction so this is not surprising, particularly at such moderate temperatures (333 K). The highest activity was reached over 0.73%Au–0.27%Pd/C catalyst in toluene (96% conversion, 94% selectivity to aldehyde in 3 h).
The reaction rate of BA oxidation over Au–Pd bimetallics was found to be zero order in O2 pressure in the range 100–3000 kPa,44,45 though a direct dependence was noted in pressures up to 100 kPa.44
In summary, the bimetallic catalysts discussed in this review, with the exception of that of Enache et al.,45 do not demonstrate enhanced reaction rates for benzyl alcohol oxidation when compared to their monometallic counterparts. In fact, the highest rate for an Au-containing bimetallic catalyst was 1.6 s−1, which is lower than the highest rate reported for a monometallic Au catalyst (5 s−1). Although there does not seem to be an obvious synergistic effect on rate, alloying two metals for alcohol oxidation catalysts has been shown to inhibit the leaching of metal (Pd) from the support or to prevent catalyst deactivation.30
Beier et al. investigated a number of supported Ag catalysts for the oxidation of alcohols by utilizing a screening method.46 The supports for the Ag particles were SiO2, Al2O3, Celite, CeO2, kaolin, MgO, and activated carbon. Although, Ag/SiO2 and CeO2 were nearly inactive (conversion <10%), a physical mixture of the two was very active for benzyl alcohol oxidation (conversion up to 45%). The catalytic activity and product distribution were found to be a function of the weight loading of Ag on SiO2. Apparently, the selectivity to aldehyde was improved at higher Ag weight loadings, (a 20 wt% Ag/SiO2 catalyst produced >95% selectivity to aldehyde with 20% conversion). The ratio of CeO2:Ag/SiO2 was also investigated, and at higher loadings of CeO2, a higher selectivity to the aldehyde was also achieved. Leaching of Ag and CeO2 were ruled out via ICP analysis of the filtered reaction mixture and hot filtration tests confirming no continued reaction after the catalyst was removed. A soluble cerium source in addition to Ag/SiO2 did not enhance the activity. The work suggests that the CeO2 adsorbs both water and benzaldehyde, thus preventing their adsorption and deactivation of the Ag surface, a phenomenon also noted as a problem by Zotova et al. over Ru catalysts.47
The effect of the calcination temperature of Ag catalysts was also shown to influence activity.46 Calcination of Ag above 773 K reduced both activity and selectivity. Silver-oxygen species in mostly metallic silver particles reportedly played an important role and were greatly affected by the pretreatment temperature. Liotta et al. noted similar results on their Ag/pumice catalyst.34
Although the Ag catalysts required O2 to be catalytically active, the authors surmised that the role of CeO2 may be to act as an oxygen reservoir, reversibly storing oxygen.46 In this scenario, benzyl alcohol adsorbs on the silver surface where it is dehydrogenated to benzaldehyde. The CeO2 activates O2, which can react with hydrogen produced from benzyl alcohol dehydrogenation and thus regenerate the Ag surface.
The work of Liotta et al. also investigated the use of Ag for BA oxidation, both in monometallic form and as a bimetallic with Pd, on a pumice support.34 The Pd monometallic catalyst was the most active, with the bimetallic catalyst having a TOF ten times lower than the Pd, and the Ag monometallic catalyst another order of magnitude lower than the bimetallic. The selectivity to the aldehyde product was ≥95% in all cases. A physical mixture of the two monometallic catalysts resulted in a rate higher than the sum of the rates for the monometallic catalysts used independently, suggesting a synergy between Pd0 and Ag0 in the reactor. The lower activity of the bimetallic catalyst calcined at 773 K may be due to the presence of PdO. Although EXAFS indicated the presence of alloyed PdAg, alloying did not seem to be necessary as the physical mixture of the two metals was adequate for enhanced activity. The authors suggested that the oxidative dehydrogenation mechanism, in which the substrate is dehydrogenated on the metal surface and oxygen removes the hydrogen to regenerate the site, can explain this observation. The Pd can activate the alcohol while the Ag can activate the O2. Close proximity of the two components allows for the activated oxygen to “hop” to the Pd. The importance of the role of O2 was confirmed by testing reactions under inert atmospheres. The reaction rate over both monometallic catalysts was much slower in the absence of O2.
Mitsudome et al.42 used Ag nanoparticles (mean diameter = 3.3 nm) supported on hydrotalcite in the absence of oxidant for high yield of benzaldehyde after 10 h (>99% conversion BA, 90% selectivity to aldehyde) at moderate conditions (403 K, Ar atmosphere). In this way, the over-oxidation to carboxylic acid was avoided, and the only byproduct was H2.
The literature for supported nanoparticle ruthenium catalysts is somewhat sparse, which is perhaps an indication of its ineffectiveness for this particular reaction. Indeed, low ratios of substrate to Ru (10–40 mole substrate per mole metal) are characteristic of many studies. However, the oxidation of benzyl alcohol to aldehyde was possible over Ru on a variety of supports, including Al2O3,10,47 carbon nanotubes (CNT),12 and Fe3O4.14 High selectivity to benzaldehyde (>99%) with high conversion of benzyl alcohol (>99%) was seen at moderate temperatures (356–363 K) over Ru supported on Al2O3 in trifluorotoluene10,16 and toluene,47 as well as over Ru supported on Fe3O4 in toluene.14 Much of the work used toluene12,14,47 and trifluorotoluene10 as solvent, though ethanol, water, and mixtures of toluene and water12 have also been investigated. Unfortunately, none of those studies provided sufficient information to calculate an initial TOF for BA oxidation.
The effect of O2 pressure was also studied over Ru, and a five-fold increase in O2 pressure from 500 kPa to 2500 kPa significantly enhanced the reaction rate. In contrast, Yamaguchi et al.18 found no effect of O2 pressure on reaction rate, though the range of pressures in this case (20–300 kPa) may not have been large enough to note the effect.
A study of the effect of solvent system was conducted by Yang et al.,12 who utilized novel Ru catalysts supported on carbon nanotubes (CNTs). Three different solvents were tested (water, toluene, and ethanol). Although the reaction in water solvent demonstrated the highest conversion, the major product was the acid. The reaction carried out in ethanol resulted in 100% selectivity to the aldehyde, but only 50% conversion of the substrate. The optimized solvent system of 2:1 toluene:water exhibited 98% conversion of benzyl alcohol and 100% selectivity to benzaldehyde after 3 h at mild conditions (358 K and 0.1 MPa O2). The authors suggested that the Ru/CNTs stabilized the emulsion of toluene and water by decreasing the interfacial tension, while also providing a higher interfacial surface area for the oxidation of alcohol to take place. Not only did the oxidation occur at a faster rate in water, but BA also had a higher solubility in water than in toluene. The role of toluene was to selectively extract the aldehyde (which is more soluble in toluene than in water), thus preventing over-oxidation to carboxylic acid.
Alternatively, Zhu et al. investigated the use of various cobalt oxides supported on activated carbon for BA oxidation in the absence of promoters or base.48 Testing of cobalt oxide on a variety of high-surface-area supports verified that high surface area is advantageous to the activity of cobalt oxide, but also demonstrated a synergistic effect on the conversion of benzyl alcohol over cobalt oxide supported on activated carbon. The group proposed that the activated carbon offers the sites for dioxygen adsorption/activation, whereas Co3O4 catalyzes the dehydrogenation reaction.
Although explored to a lesser extent than Au and Pd, Ru, Ag and Co3O4 catalysts offer interesting options for benzyl alcohol oxidation catalysts.
3.4 Cinnamyl alcohol
Cinnamyl alcohol (CA) oxidation is often used together with benzyl alcohol as a model reaction for alcohol oxidation. Like benzyl alcohol, CA is highly reactive; however, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in cinnamyl alcohol can undergo side reactions such as hydrogenation and hydrogenolysis, which add a degree of complexity. The general oxidation scheme for CA oxidation is shown in Fig. 6. Because many studies investigating the oxidation of both BA and CA, the reaction conditions here are similar to those in the previous section. Commonly used catalysts are Au, Pd, and Ru, and the solvents are generally xylene, toluene, and water. The TOF values for cinnamyl alcohol oxidation at temperatures less than 373 K range from 0.17 s−1 to 4.4 s−1, which is of the same order of magnitude as those for benzyl alcohol oxidation. The highest rate was found over Au bulk foil with added homogeneous base. It should be noted that the rate for Pd/hydroxyapatite is not reported on the table and would likely be much higher than that of Pd/SiO2; however, insufficient information was provided to calculate an initial rate for CA oxidation over Pd/HAP (Table 9).11
C bond in cinnamyl alcohol can undergo side reactions such as hydrogenation and hydrogenolysis, which add a degree of complexity. The general oxidation scheme for CA oxidation is shown in Fig. 6. Because many studies investigating the oxidation of both BA and CA, the reaction conditions here are similar to those in the previous section. Commonly used catalysts are Au, Pd, and Ru, and the solvents are generally xylene, toluene, and water. The TOF values for cinnamyl alcohol oxidation at temperatures less than 373 K range from 0.17 s−1 to 4.4 s−1, which is of the same order of magnitude as those for benzyl alcohol oxidation. The highest rate was found over Au bulk foil with added homogeneous base. It should be noted that the rate for Pd/hydroxyapatite is not reported on the table and would likely be much higher than that of Pd/SiO2; however, insufficient information was provided to calculate an initial rate for CA oxidation over Pd/HAP (Table 9).11 | ||
| Fig. 6 Reaction scheme for cinnamyl alcohol oxidation. | ||
There are multiple reports of aldehyde production in the presence of basic aqueous solutions over Au and Pd catalysts.11,37,38 In one instance, Au supported on block co-polymers in a 50% H2O–50% chloroform solution with 1:1 KOH:CA produced 99% conversion of CA with 95% selectivity to aldehyde in 6 h at 273 K.37 Similarly, the Pd/HAP system was effective for 90% conversion of CA with 98% selectivity to aldehyde in aqueous NaOAc in 24 h at 383 K.11 Unsupported bulk Au with 1:1 K2CO3:CA produced similar results, 70% conversion of CA with 100% selectivity to aldehyde, though the solvent was heptane and therefore the high aldehyde selectivity is not surprising. Likewise, Au/CeO2 could convert CA with 73% selectivity to aldehyde (66% conversion) in the absence of solvent and base; using water and Na2CO3 as the solvent system resulted in >99% conversion and 98% selectivity to the expected carboxylic acid.32 The choice of solvent appears to be critical in the bulk Au case, as CA oxidation did not proceed in trifluorotoluene, toluene, or acetophenone. It should be noted that BA oxidation did proceed in heptane and toluene under the same conditions.38
An influence of solvent was also observed over Pd, Pt, Au–Pt, and Au–Pd catalysts supported on C. For example, the CA oxidation rates over all of these catalysts were higher in water rather than in toluene without added base. The Au monometallic catalyst was inactive in both solvents. Bimetallic 0.73 wt% Au–0.27 wt% Pd/C produced the highest conversion and selectivity to aldehyde in both toluene (conversion 72%, selectivity to aldehyde 85%) and water (conversion 95%, selectivity to aldehyde 83%) at 333 K, whereas Pd and Pt monometallic catalysts both exhibited poor conversion (36% and 27%, respectively) but relatively high selectivity to aldehyde (86% and 100%, respectively) in H2O at 333 K.
A Pd/HAP catalyst was likewise effective in toluene solvent in the absence of added base (91% conversion, 87% selectivity to the aldehyde) at a slightly lower temperature (363 K).11 The Pd/C catalyst was nearly as effective (90% conversion, 68% selectivity). Other supported Pd catalysts, under the same conditions, were less effective; Pd/Al2O3 converted 83% CA with only 66% selectivity to aldehyde and Pd/SiO2 produced only 31% conversion with 30% selectivity to the aldehyde.
Similar to what was observed during BA oxidation, a physical mixture of Ag/SiO2 and CeO2 had high activity for CA oxidation (98% conversion in 2 h for BA vs. 83% conversion in 3 h for CA).46 Likewise, a magnetically-separable Ru(OH)x/Fe3O4 catalyst active for BA oxidation was also active for CA oxidation in toluene without addition of base (95% conversion, >99% selectivity to aldehyde in 1.5 h at 378 K).14 Ruthenium supported on carbon was also found to be active for CA oxidation in toluene, with a 79% yield of the aldehyde after 24 h at 343 K.17
The investigation of cinnamyl alcohol oxidation highlights the importance of solvent choice in alcohol oxidation with regards to product selectivity and catalyst activity. As with BA oxidation, the product selectivity is greatly influenced by the solvent as well as the nature of the catalyst support. As in other alcohol oxidations, the Au catalyst features the highest oxidation rate at modest temperature, <373 K (on the same order of magnitude as BA oxidation, ca. 4 s−1), though the solvent and pH of solution must be carefully monitored.
3.5 5-Hydroxymethylfurfural oxidation
Oxidation of 5-hydroxymethylfurfural is an interesting and complicated reaction because the substrate HMF is not stable in water at high pH,49–52 subject to conversion via the Cannizzaro reaction and other degradation pathways. However, under most conditions, a high pH is necessary for its oxidation. There has been a push to find a base-free oxidation route, for both economic and environmental benefits, despite the extremely low solubility of the common target product, 2,5-furandicarboxylic acid (FDCA), in neutral water.53 The base-free oxidation of HMF utilizing Au on hydrotalcite (HT) as a solid base support was reported recently;54 however, other work demonstrated the likely leaching of this solid base material into solution as a stoichiometric replacement for homogenous base.25 This is a good demonstration of the complexity and technical barriers to base-free HMF oxidation at present.As described in Section 2.1.5, the oxidation of HMF into FDCA encompasses two steps: aldehyde oxidation and alcohol oxidation. It is generally believed that the alcohol oxidation is the slow step in this oxidation.51,52,55 The main intermediates in HMF oxidation are seen in Fig. 7. The di-aldehyde intermediate, 2,5-diformylfuran, is rarely seen in solution at high pH, probably because of the high reactivity of the aldehyde in aqueous media. The most commonly seen intermediate is the acid–alcohol product, 5-hydroxymethyl-2-furancarboxylic acid (HFCA), which results from oxidation of the aldehyde moiety to acid.
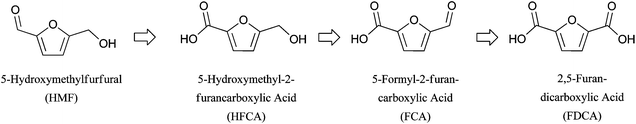 | ||
| Fig. 7 Commonly observed intermediates in HMF oxidation. | ||
| Catalyst | Particle size (nm) | T (K) | Pressure (kPa) | Solvent | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a All reactions were performed in batch mode.b Not given; estimated from data from World Gold Council standard catalyst. | ||||||
| Au/TiO2 | 2.6b | 295 | 100 | Methanol + 8% Sodium Methoxide | 0.14 | 56 |
| Au/CeO2 | 3.5 | 403 | 1000 | Methanol | 0.31 | 58 |
| Au/Fe2O3 | 3.5 | 403 | 1000 | Methanol | 0.12 | 58 |
| Au/TiO2 | 3.5 | 403 | 1000 | Methanol | 0.30 | 58 |
| Au/C | 3.5 | 403 | 1000 | Methanol | 0.47 | 58 |
| Au–Cu/TiO2 | 4.4 | 368 | 1000 | Water + 4:1 NaOH:HMF | 0.18 | 50 |
| Au/TiO2 | 2.6 | 295 | 690 | Water + 2:1 NaOH:HMF | 1.6 | 49 |
| Au/C | 10.5 | 295 | 690 | Water + 2:1 NaOH:HMF | 5.0 | 49 |
| Au/C | 3.0 | 295 | 690 | Water + 2:1 NaOH:HMF | 2.3 | 49 |
| Pt/C | 2.5 | 295 | 690 | Water + 2:1 NaOH:HMF | 0.08 | 49 |
| Pd/C | 3.3 | 295 | 690 | Water + 2:1 NaOH:HMF | 0.15 | 49 |
| Au/TiO2 | 2.6 | 295 | 2000 | Water + 2:1 NaOH:HMF | 1.2 | 49 |
| Au/TiO2 | 2.6 | 295 | 3000 | Water + 2:1 NaOH:HMF | 1.4 | 49 |
Casanova et al.58 explored the reaction over Au/CeO2 at a similar temperature (403 K) but at higher pressure (1000 kPa) and without addition of base, in an attempt to improve the recyclability of catalysts for this reaction. The primary intermediate in that case was the ester of HFCA, and the final product diester was formed in 99% yield after 5 h. They proposed that the rate limiting step in the sequence is the oxidation of the alcohol side chain to an aldehyde, which is analogous to the presumed rate limiting step during HMF oxidation in aqueous solution.
The effect of support on reaction rate and product selectivity was also investigated. The TOFs were calculated from initial rates (at 0.25 h) of both HMF disappearance and of diester formation. However, the TOFs were based on total metal rather than on surface metal, so the values have been recalculated and presented in Table 10. It should be noted that production of diester is the result of subsequent oxidation steps that are presumably slower than the initial oxidation of HMF. According to Table 10, Au/C had the highest rate of HMF oxidation but, relative to the other catalysts in the study, demonstrated low final yield of the diester product. On the other hand, Au/CeO2 had the highest activity of the group for the formation of diester product (99% yield in 5 h). An experiment with Au/CeO2 in the absence of O2 yielded large amounts of the acetal product and 3.8% of the monoester product, which suggests that CeO2 acts as an oxygen donor. When non-nanometric ceria was used as support, the activity of the catalyst was significantly reduced, in agreement with results from CeO2-based catalysts for benzyl alcohol oxidation.32
The effect of temperature was also investigated in the range 353 K–403 K over the Au/CeO2 and yielded an overall activation energy of 34 kJ mol−1. Although other alcohols, such as ethanol and butanol, were also tested for the oxidation-esterification, methanol was the most suitable.58 The effect of water was probed, and a negative effect on the initial reaction rate was noted. In fact, when 20% water was loaded, the reaction did not even reach completion. Interestingly, no trace of carboxylic acid was noted, possibly because of the presence of a Lewis acid site on the catalyst support aiding in the rapid reaction of any formed carboxylic acid to methyl ester.
The work of Davis et al. also showed that the activity of Au catalysts for HMF oxidation was an order of magnitude higher than either Pt or Pd catalysts. The selectivity to the desired diacid, however, was much higher over the Pt and Pd catalysts. At their standard conditions of 295 K and 690 kPa O2 and 2 equivalents NaOH, the major product over Pt and Pd catalysts was FDCA (selectivity of 79% and 71%, respectively) while the major product over the Au catalysts was HFCA (selectivity of 92% over Au/TiO2 catalyst). Gorbanev et al., however, demonstrated high selectivity to FDCA over Au catalysts in water by increasing the amount of catalyst and NaOH (20 equivalents).52
Over a Au/TiO2 catalyst, Gorbanev et al. investigated the effect of NaOH concentration and demonstrated that at 303 K and 2000 kPa O2, the selectivity to diacid did not change in the range 5–20 equivalents NaOH.52 Below 5 equivalents, the selectivity shifted more toward the mono-acid HFCA. In the absence of base at the same conditions, only 13% conversion of HMF was seen, with the majority product HFCA (yield 12%) and little FDCA (selectivity 1%).
Casanova et al.58 and Pasini et al.50 also varied the amount of NaOH to determine the effect of base on product selectivity. Casanova et al. found that using 4 equivalents of NaOH in the presence of Au/CeO2 produced a 96% yield of diacid in 5 h, while 2 equivalents afforded 96% yield in 20 h.51 Using just 1 equivalent of NaOH resulted in only 20% yield in 14 h (76% yield of HFCA) and the oxidation did not proceed further. Pasini et al. found that, in the presence of Au–Cu/TiO2 or Au/TiO2, the product selectivity was influenced by amount of NaOH in the range of 1–4 equivalents.50 Increasing the NaOH concentration in the range 4–10 equivalents did not change product selectivity. The work also showed that using 20 equivalents of NaOH in the absence of catalyst resulted in complete degradation of HMF after 2.5 h.
The role of O2 pressure in HMF oxidation over Au/TiO2 catalysts was investigated by Gorbanev et al.52 and Davis et al.49 Over the range 690 kPa to 3000 kPa, no appreciable difference in the rate of HMF oxidation was noted. In contrast, Vinke et al., reported that reaction rate over Pt/Al2O3 was first order with respect to O2 pressure; however, this assertion is made without showing any data.61
The effect of O2 pressure on product selectivity was investigated by Pasini et al.,50 Davis et al.49 and Gorbanev et al.52 over Au catalysts. A positive correlation between O2 pressure and selectivity to diacid was noted. A control experiment conducted by Gorbanev et al. in the absence of O2 but in the presence of catalyst and 20 equivalents of NaOH found full conversion of HMF after 18 h, with 51% yield of HFCA and the balance comprised of decomposition products. This demonstrates the necessity of dioxygen in this reaction to prevent undesirable side reactions in the highly basic solution.52
Lilga et al., used a flow reactor to investigate the acid/base nature of the medium on HMF oxidation over Pt/Al2O3, Pt/ZrO2, and Pt/SiO2 catalysts.57 Under basic conditions (Na2CO3) at 373 K and 1035 kPa air, complete conversion of HMF to FDCA was observed. However, however, the selectivity shifted to FCA after 2 h of operating at 100% conversion of HMF, which might be attributed to adsorption of products onto the catalyst. Successive experiments in batch reactors report the same phenomenon, yet the original activity could be returned by washing the catalyst with hot water.57 In neutral conditions, the reaction was substantially slower but the downstream separations were easier.57 Product inhibition was apparently much higher than that in the basic conditions, likely because of the low solubility of the reaction products in neutral solution. It was noted that inorganic supports adsorbed less of the products, which also correlated with the surface area of the supports.
Acidic conditions were achieved through addition of acetic acid.57 While the solubility of FDCA is limited in neutral feeds, the solubility in 40% acetic acid and 60% water is twice that in 100% water. At 373 K and with air as the oxidant, the dialdehyde (DFF) is the major product, though at higher temperatures and with O2 as the oxidant, 100% conversion and 85% selectivity to FDCA was realized.
Verdeguer et al. investigated PtPb/C catalysts and found that hydroxyl bases were more effective than carbonate bases at promoting the reaction.55 Vinke et al. found the rate of HMF oxidation over Pt/Al2O3 to be independent of pH between 8–11, but at lower pH values, the rate was lower.61
It is quite clear that the oxidation of HMF in basic aqueous solution is facile over metal catalysts at room temperature.26,49,50,56,57,61 Increasing the temperature, however, can reduce the reaction time or change the product selectivity. For example, Pasini et al. showed that at 333 K over Au–Cu/TiO2 catalysts, the major product was the monoacid HFCA.50 Raising the temperature by just 20 K shifted the selectivity toward the diacid FDCA, and raising the temperature an additional 15 K produced 99% yield of FDCA. This is consistent with a rapid oxidation of HMF to HFCA and a slow oxidation of HFCA to FDCA. Vinke et al. found the observed activation energy to be 37.2 kJ mol−1 over Pt catalysts,61 which is similar to the value found over Au catalysts in MeOH solvent (34 kJ mol−1).58
Casanova et al. varied the temperature between 298 and 403 K and found a positive effect on conversion over their Au/TiO2 catalysts, as expected. However, higher temperatures caused the formation of some undesired byproducts (degradation products).51 Attempted reuse of the catalyst revealed significant deactivation, which was attributed to the buildup of 2.5 wt% C during the reaction. Extensive washing of the spent catalyst did not improve activity. The researchers claimed that the used catalyst was still able to oxidize the alcohol group, but its ability to oxidize the aldehyde group was diminished. This was confirmed by carrying out a reaction with HFCA and a used catalyst; the reaction was completed in 4 h, yielding 93% FDCA. Since the catalyst can be used to oxidize the aldehyde at 298 K without loss of activity, a new reaction protocol was established wherein the first oxidation step of HMF to HFCA was conducted at 298 K, and then the temperature was raised to 403 K to allow for complete and efficient oxidation to the diacid product while limiting catalyst deactivation. With this method, the spent catalyst could be successfully reused 3 times.
A possible promotional role of the support (Fe2O3, C, CeO2 and TiO2) on Au nanoparticles was examined by Casanova et al.51 The Au/CeO2 and Au/TiO2 supported catalysts showed the best activity (over Au/TiO2, 84% selectivity to diacid after 8 h; over Au/CeO2, 96% selectivity to diacid after 5 h), while Au/Fe2O3 and Au/C produced lower yields of FDCA. Highlighting the important collaborative effect of CeO2 and Au, the Au on nanoparticulate CeO2 reached high yields of FDCA in half the time of the Au on non-nanoparticulate CeO2. The reductive pretreatment of the catalyst was shown to increase the activity, likely because it increases amount of Ce3+, which has been shown previously to be important in catalyst activity.32
Monometallic Au and Cu catalysts, as well as bimetallic Au–Cu/TiO2 catalysts, were synthesized by Pasini et al.50 Interestingly, the reaction with Cu/TiO2 produced no FDCA whereas the bimetallic Au–Cu yielded twice the FDCA yield of the monometallic Au catalysts, demonstrating a synergistic effect of alloying the two metals. The optimum metal loading was found to be 1.5 wt% and the preparation method played a role in catalyst activity. Catalysts synthesized from preformed Au–Cu sols supported on TiO2 were more active than those synthesized by post-deposition of a Au sol onto a monometallic Cu/TiO2 catalyst. This may be indicative of the promotional activity of Cu, or because Cu aids the dispersion of Au. The bimetallic catalyst was able to be recycled 5 times without losing activity, whereas the Au monometallic catalyst lost activity after just 1 recycle, highlighting the beneficial effect of alloying Cu with Au.
Pasini et al. also found an effect of pretreatment on catalyst performance.50 The calcined catalyst produced a higher yield of HFCA and a lower yield of FDCA (92% and 8%, respectively) than did the uncalcined catalyst (69% yield HFCA and 31% yield FDCA). In addition, the particle size of the calcined catalyst was larger than the uncalcined (6 nm compared to 4.4 nm, respectively). From this information, the group hypothesizes that the HFCA oxidation may be more sensitive to size of metal nanoparticles than is the HMF oxidation.
3.6 Glycerol oxidation
The oxidation of glycerol by heterogeneous catalysts has both economic and academic appeal. Glycerol is a major byproduct of biodiesel synthesis via transesterification of triglycerides with alcohols. Glycerol oxidation to high value fine chemicals, such as glyceric acid, tartronic acid, or dihydroxyacetone, could help bio-diesel economics become more competitive compared to diesel produced from non-renewable resources. Academic interest in oxidative upgrading of glycerol stems from glycerol being a model polyol with three hydroxyl groups, two primary and one secondary. Fig. 8 shows a reaction scheme of the major glycerol oxidation products. Glycerol oxidation is a sequential reaction that can follow multiple paths. The oxidation of a primary or secondary alcohol of glycerol produces glyceraldehyde or dihydroxyacetone, respectively. These two products are often in equilibrium in aqueous solution, depending on the pH. Glyceraldehyde can be sequentially oxidized to an acid, glyceric acid, and both glyceraldehyde and dihydroxyacetone can be oxidized to an alpha-keto acid product, hydroxypyruvic acid. The sequential oxidation of the primary alcohol of glyceric acid typically produces tartronic acid. Many authors have also reported that glycerol oxidation products can undergo carbon cleavage to form two carbon acids such as glycolic acid and oxalic acid,24 which necessarily means that one carbon products can also be formed.62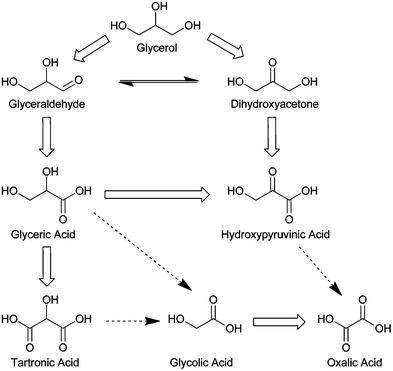 | ||
| Fig. 8 Commonly observed glycerol oxidation products. Dotted lines represent possible carbon cleavage of three carbon products to form two carbon products. | ||
The rate and selectivity of glycerol oxidation depends on the choice of catalyst and the reaction conditions. As the selectivity during glycerol oxidation strongly depends on the pH of the solution, this discussion will focus on the influence of reaction conditions of oxidation on supported Au, Pt, and Pd catalyst performance.
It is known that the rate of glycerol oxidation over supported Au catalysts is enhanced by the addition of base,64 but a true rate of oxidation is difficult to discern from the literature. While the reaction conditions affect the rate of reaction, the initial TOF reported in the literature based on the available Au surface metal can vary by almost three orders of magnitude from 0.2 to 17 s−1. The true rate of reaction, in the absence of external and internal mass transfer limitations, is most likely found at the higher bound of reported TOF's. Thus, selected examples of glycerol oxidation with supported Au catalysts are reported in Table 11.
| Catalyst | Size (nm) | T (K) | pO2 (kPa) | NaOH:Glycerol (mol:mol) | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a TOF calculated or re-calculated based on available surface metal. | ||||||
| 0.5% Au/C | 7.3 | 333 | 1000 | 2 | 17 | 24 |
| 0.8% Au/C | 10.5 | 333 | 1100 | 2 | 6.1 | 23 |
| 1% Au/C | 4.3 | 333 | 100 | pH = 12 | 5.0a | 67 |
| 1.8% Au/TiO2 (WGC) | 3.5 | 333 | 1100 | 2 | 4.9 | 23 |
| 1% Au/C | 3.7 | 333 | 1000 | 2 | 4.8a | 68 |
| 1.47% Au/TiO2 | 3.7 | 333 | 1000 | 2 | 4.1 | 64 |
| 0.75% Au/C | 3.7 | 333 | 1000 | 2 | 3.9a | 69 |
| 0.59% Au/C | 6.8 | 333 | 1000 | 2 | 3.4a | 65 |
| 0.6% Au/C | 7.7 | 333 | 500 | 2 | 2.6a | 71 |
| Au powder | — | 333 | 1000 | 2 | 2.5 | 66 |
| 0.4% Au/MWCNT | 5.5 | 333 | 300 | 2 | 1.7a | 72 |
| 1% Au/C | 2.5 | 323 | 300 | 4 | 0.8a | 73 |
| 1% Au/TiO2 | 3.8 | 363 | 100 | 4 | 0.7a | 74 |
| 1% Au/C | 3.5 | 323 | 300 | 4 | 0.7a | 75 |
| 1.1% Au/C | 13.3 | 333 | 500 | 2 | 0.5a | 71 |
| 1.5% Au/TiO2 (WGC) | 4 | 323 | 300 | 4 | 0.2a | 76 |
| 3% Pd/C | 3 | 333 | 1100 | 2 | 2.2 | 23 |
| 3% Pt/C | 2.3 | 333 | 1100 | 2 | 1.6 | 23 |
| 0.5% Pt/CS | 5 | 343 | 1000 | 0.8 | 1.2a | 77 |
| 2.9% Pd/C | 2.9 | 333 | 1000 | 2 | 1 | 24 |
| 1.28% Pd/C | 5.6 | 323 | 300 | 4 | 0.4a | 78 |
| 1% Pt/TiO2 | 2.5 | 363 | 100 | 4 | 0.3a | 74 |
| 0.5% Pt/C | 3.8 | 333 | 300 | 2 | 0.3a | 79 |
| 1% Pt/C | 2.5 | 323 | 300 | 4 | 0.3a | 80 |
| 0.65% Pd/C | 2 | 333 | 300 | 2 | 0.09a | 65, 79 |
| 5% Pt/C | 2.3 | 333 | 300 | 2 | 0.06a | 81 |
| 1% Pd/TiO2 | 3 | 363 | 100 | 4 | 0.008a | 74 |
| 5% Pt/MWNTs | 6.7 | 333 | Flow | 0 | 0.6a | 82 |
| 5% Pt/MWNTs | 2.1 | 333 | Flow | 0 | 0.5a | 83 |
| 5% Pt/C | 3.6 | 398 | Flow | pH = 4 | 0.2a | 84 |
| 5% Pt/C | 3.2 | 333 | Flow | 0 | 0.2a | 85 |
| 3% Pt/C | 3.2 | 333 | Flow | 0 | 0.1a | 83 |
| 3% Pt/C | 2.3 | 333 | 1100 | 0 | 0.06 | 23 |
| 5% Pt/C | 5 | 343 | 500 | pH = 7 | 0.05 | 86 |
| 3% Pd/C | 3 | 333 | 1100 | 0 | 0.004 | 23 |
The initial TOF for glycerol oxidation over well-dispersed supported Au catalysts at 333 K, presumably in the absence of mass transfer limitations, appears most frequently between the values of 2–6 s−1. Four different research labs have produced initial rates within this range and the supported Au catalysts vary in synthesis procedure, support type, and Au weight percent. In addition, a wide range of Au loadings in the reactor were utilized, with glycerol to Au ratios ranging from 200065 to over 50000.24 It is noteworthy that several of the authors of the higher glycerol oxidation rates have addressed the external mass transfer limitations of O223,65–69 and one addressed internal mass transfer limitations as well.67
The two highest rates for glycerol oxidation were 17 s−1 by a 0.5% Au/C catalyst with a glycerol to Au ratio of 5000024 and 6.1 s−1 by a 0.8% Au/C catalyst with a glycerol to Au ratio of 8000, both at 333 K.23 Both authors calculated the maximum O2 transfer rate for their semi-batch reactor configuration and purposely chose catalyst loadings in the reactor to ensure glycerol oxidation rates fell below the maximum rate of dioxygen transfer. The oxidation of sodium sulfite to sodium sulfate by O2 was used to calculate the maximum oxygen diffusion rate into the liquid phase at 298 K. In addition, the authors used two different titania-supported catalysts and reported that for a 1.47% Au/TiO2 the TOF was 4.1 s−164 and over 1.8% Au/TiO2 it was 4.9 s−1.23 Interestingly, the 0.8% Au/C catalyst investigated had a particle size approximately three times larger than both Au/TiO2 catalysts at 10.5 nm, yet still had an initial TOF of 6.1 s−1.
An accurate initial rate can be determined regardless of reaction conditions as long as they are not mass transfer limited. Demirel et al. investigated a semi-batch process in which the pH was controlled at 12 by the addition of NaOH and the dioxygen pressure was only 100 kPa.67 Instead of increasing the dioxygen pressure, the loading of Au in the reactor was limited to ensure there were no external mass transfer limitations. An initial TOF of 5 s−1 was reported with a 1% Au/C catalyst. The external and internal mass transfer limitations of glycerol oxidation were subsequently modeled by Demirel et al. and shown to fall within the gas–liquid and liquid–solid criterion established by Chaudhari et al. for liquid phase reactions of fine chemicals.68,70 The kinetic modeling of glycerol oxidation was fitted with the Langmuir–Hinshelwood mechanism and suggested that only the adsorption of glycerol, glyceric acid, and tartronic acid is relevant and that oxalic acid is most likely the product of glycolic oxidation. In addition, an initial rate was calculated for the 1% Au/C catalyst to be 4.8 s−1.
A good example of an initial rate that is limited by external mass transfer of O2 was presented by Rodrigues et al. for glycerol oxidation over a 0.59% Au/C catalyst.65 An increase in the dioxygen pressure from 300 to 1000 kPa resulted in an increase in the initial rate from 1.3 to 3.4 s−1. In addition, they noticed that modification of the carbon support to increase the number of oxygenated acid groups six-fold severely decreased the initial rate of reaction, while carbon supports with a low content of oxygenated surface groups all had a similarly higher initial rate.
Finally, Ketchie et al. illustrated that bulk Au powder can catalyze glycerol oxidation at a rate of 2.5 s−1 in the presence of base at 333 K.66 Even very large 20 nm and 40 nm sized particles of Au on carbon supports had oxidation rates of 2.3 and 2.2 s−1, respectively. All evidence in the literature shows that a good guideline for the initial TOF of a well dispersed supported Au catalyst is on the order of magnitude of ∼1 s−1 at 333 K and O2 pressure that does not introduce mass transfer limitations.
If the initial rate of supported Au catalysts is on the order of magnitude of ∼1 s−1, the question of why there are many examples of authors reporting lower initial rates is raised. We suspect that, in the case of supported Au catalysts, low rates can be explained by diffusion limitations of O2 and rapid deactivation of the catalyst.
Demirel-Gulen et al. determined that the initial rate of reaction over a 0.75 wt% Au/C catalyst increased from 2 to 6 s−1 as the ratio of NaOH to glycerol increased from 1 to 4.69 No conversion was realized in the absence of NaOH. Thus, the initial rate was still the same order of magnitude as long as there was a high level of base. The effect of temperature on the initial rate of reaction is summarized in Table 12. The temperature was found to influence the initial rate of reaction by an order of magnitude over the range 298–373 K. The rate followed a typical Arrhenius relationship with temperature with an apparent activation energy of 50 ± 5 kJ mol−1. At 298 K the initial TOF for glycerol oxidation was calculated to be about 0.5 s−1, but even at a temperature of 313 K the initial TOF was already 1.5 s−1, which is comparable to other initial rates reported.
The selectivity of the glycerol oxidation reaction over supported Au catalysts in the presence of base is primarily to glyceric acid and tartronic acid. Cleavage of the three carbon acids can result in two carbon acids, glycolic acid and oxalic acid. The two carbon products are typically produced by cleavage of three carbon glycerol oxidation products, presumably with hydrogen peroxide formed as a byproduct.24 Oxidation of tartronic acid can also produce mesoxalic acid. Selectivity is difficult to compare between papers because it depends not only on level of conversion but also whether the reaction is mass transfer limited.
Gil et al. determined that the selectivity can change significantly when the reaction is not run in the kinetic regime as seen in Table 13.71 The TOF of 2.6 s−1 at a glycerol:Au ratio of 3500 was not mass transfer limited and had a selectivity to glyceric acid of 20.2% with a 1.1% Au/C catalyst with 13.3 nm Au particles at 30% conversion. However, a clearly mass transfer limited TOF of 0.5 s−1 at a glycerol:Au ratio of 1000 has a selectivity to glyceric acid of 8.5%. In addition, one can see a higher selectivity to the diacid, tartronic acid, at higher catalyst loadings. Thus, selectivity comparisons require not only equivalent levels of conversion, but also results free from transport limitations.
| Selectivity | |||||||
|---|---|---|---|---|---|---|---|
| Glycerol:Au (mol:mol) | TOFb (s−1) | Glyceric acid | Glycolic acid | Tartronic acid | Oxalic acid | Mesoxalic acid | Hydroxypyruvic acid |
| a Reaction conditions: 0.3 M glycerol, NaOH:Glycerol = 2 (mol:mol), 500 kPa O2, 333 K. Catalyst: 1.1 wt% Au/C (particle size 13.3 nm). Selectivities reported at 30% conversion. Ref. 71.b TOF re-calculated based on available surface metal. | |||||||
| 3500 | 2.6 | 20.2 | 39.4 | 9.1 | 8.6 | 17.8 | 4.9 |
| 2000 | 0.95 | 18.6 | 40.6 | 15.1 | 5.0 | 13.3 | 7.4 |
| 1000 | 0.5 | 8.5 | 46.9 | 19.9 | 4.8 | 12.7 | 7.1 |
In most cases, the selectivity of supported Au catalysts in the presence of base is typically toward monoacids, with a minority of diacid products. Although 100% selectivity to monoacids has been reported,63 the diacid product is the subsequent oxidation of the monoacid. Thus, the conversion and the amount of Au loaded in the reactor can play a major role in determining the concentration of diacid. Still, the highest reported selectivity to tartronic acid is only 44% over a 1% Au/TiO2 catalyst after 4 h at 393 K.87 Gold catalysts have difficulty producing the diacid in high yield because of inhibition by strongly adsorbed ketone intermediates and condensation products in addition to cleavage and over-oxidation reactions, as noted earlier.88 Significant amounts of one carbon products, such as carbon dioxide (in the solution as carbonate) and formic acid can also be observed depending on the temperature of the reaction.89 Even selectivity to dihydroxyacetone in basic solutions was reported over MWCNT-supported Au catalysts at very low conversions of glycerol.65
The initial rate of glycerol oxidation over supported Pt and Pd catalysts has also been investigated, although less thoroughly than supported Au catalysts. Table 11 provides the initial TOF of a few examples of Pt and Pd catalyzed glycerol oxidation reactions. Glycerol oxidation over Pt and Pd is slower than over Au, even in the presence of base. However, as discussed earlier, the glycerol oxidation mechanism is likely to be similar over the different metals. Carrettin et al. investigated the role of the type of basic promoter on glycerol oxidation with a 5% Pt/C catalyst.81 Sodium hydroxide (NaOH) was determined to promote the highest conversion when compared to other alkali metal hydroxides. One major advantage of using supported Pt and Pd catalysts is that oxidation is possible even without base. Also, the selectivity during glycerol oxidation can be significantly different at acidic and basic conditions. However, a major disadvantage of Pt and Pd catalysts is that they can deactivate quickly under certain reaction conditions. The noble nature of Au prevents substrates, including oxygen, from adsorbing strongly to the surface. Deactivation of the Pt and Pd catalysts can be caused by over-oxidation of the metal to form oxides on the surface and by poisoning of the surface with acid products, byproducts, or cleavage products.90
The initial rate of reaction for supported Pt catalyst in the presence of base was observed both by Zope et al.23 at 333 K and Huang et al.77 at 343 K to be on the order of ∼1 s−1. Huang et al. noted that the TOF was sensitive to the dioxygen pressure, decreasing to about 0.4 s−1 at a pressure of 300 kPa. Zope et al. and Ketchie et al. reported TOFs for Pd/C catalysts of 1 s−1 and 2.2 s−1, respectively, in the presence of base at 333 K. Interestingly, while Pt and Pd exhibit a slower oxidation rate of glycerol than Au in the presence of base, the order of magnitude is similar. The inhibition of the Pt and Pd catalysts by more strongly adsorbed reactants and products compared to the more inert Au, might explain the slightly lower rate.
The majority of examples cited in Table 11, however, have initial rates significantly below ∼1 s−1. A TOF of 0.3 s−1 was calculated over a 1% Pt/TiO2 catalyst, a 0.5% Pt/C catalyst, and a 1% Pt/C catalyst.65,74,80 Dimitratos et al. reported an initial rate of 0.4 s−1 over a 1.28% Pd/C catalyst78 and Rodrigues et al. found an initial rate of approximately 0.09 s−1 with a 0.65% Pd/C catalyst.65 It is possible that rapid deactivation could play a role in the low initial rate, but a second reason might be mass transfer limitations of O2 at low pressure and high catalyst loading used in the study. For example, Shen et al. was able to form lactic acid (presumably a solution-mediated reaction) by limiting the amount of available dioxygen at increased temperature, but also had a low TOF of 0.008 s−1 over a 1% Pd/TiO2 catalyst.74
In the absence of base, the TOF for glycerol oxidation is about ∼0.5 s−1 as observed by Liang et al. over 5% Pt/MWNT.83 Again, this value could be influenced by rapid deactivation. It has been suggested that over-oxidation can form metal oxides on the surface that are not active for oxidation and that byproducts, intermediates, or acids can strongly adsorb on the metal surface. Gao et al. observed quick deactivation with the rate of reaction dropping in half from 0.6 s−1 at 20 min to approximately 0.3 s−1 at 60 min in the absence of base over 5% Pt/MWNT.82 The TOF of 0.06 s−1 reported by Zope at 5 h over 1% Pt/C, even at the low conversion of 5%, is probably measured after too long to accurately gauge the initial rate of reaction without interference from deactivation.23
Villa et al. tested both 1% Pt/C and 1% Pt/H-mordenite, but did not report the reaction profile or an estimation of particle size, thus no TOF could be calculated.62 Villa did note that the Pt/H-mordenite catalyst had a lower selectivity to one-carbon products. However, the conversion was significantly higher for the Pt/C catalyst, 78% compared to 20% for the Pt/H-mordenite; thus the difference in selectivity could just be a result of over-oxidation of the glyceric and tartronic acid at higher conversions.
Villa et al. reported very low activity for a 1% Pd/C catalyst in base-free glycerol oxidation at 373 K with a conversion similar to Au at about 5%.62 It is unknown what the blank conversion is at 373 K, but it is noted that no conversion with Au and Pd was seen at 323 K. This is consistent with the low TOF reported by Zope et al. of 0.004 s−1 for 3% Pd/C in the absence of base.23 Either Pd deactivates very quickly without base or it is relatively inactive for glycerol oxidation in water.
Bianchi et al. reports a TOF based on the total metal loading of 0.32 s−1 at 323 K, 304 kPa O2, NaOH:glycerol = 4, and glycerol:Pd = 500, but does not report an estimation of the particle size for Pd.73 Similarly, Dimitratos et al. calculated a TOF based on total metal loading of 0.09 s−1, but does not report an estimation of particle size.78 However, these conditions are identical to those reported by Bianchi et al. Without knowing the dispersion of the catalyst it is difficult to assess the catalysts and determine whether the difference in TOF is due to particle size variations or external mass transfer limitations.
| Catalyst | Size (nm) | T (K) | pO2 (kPa) | NaOH:glycerol (mol:mol) | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a TOF calculated or re-calculated based on available surface metal. | ||||||
| 2.8% Pd–2.2Au%/C | 4.4 | 333 | 1000 | 2 | 7 | 24 |
| 0.6% Au–0.35% Pd/C | 2.6 | 323 | 300 | 4 | 1.3a | 73 |
| 1% (1:1 Au–Pd)/C | 3.7 | 323 | 300 | 4 | 1.9a | 78, 80, 91 |
| 1% (1:1 Au–Pt)/C | 2.5 | 323 | 300 | 4 | 0.6a | 80 |
| 0.96% Au–1.71% Pd/C | 6.1 | 323 | 300 | 4 | 1.6 | 78 |
| 1% (6Au:4Pt)/H-Mordenite | 4 | 373 | 300 | 0 | 0.2 | 62 |
| 1% AuPd(1:3)/MgO | 4.1 | 333 | 300 | 0 | 0.04 | 92 |
| 1% AuPt(1:3)/MgO | 4 | 333 | 300 | 0 | 0.12 | 92 |
Ketchie et al. reported that a bimetallic Au–Pd/C catalyst had a TOF of 7 s−1 which is fairly typical of Au catalysts, but with increased selectivity to glyceric acid.24 Palladium apparently accelerated the decomposition of H2O2, which prevented cleavage of glyceric acid and tartronic acid to two and one carbon products. Other bimetallic Au–Pd catalysts synthesized with slightly less and more Au were of about the same rate while maintaining the same selectivity at 50% conversion. Bianchi et al. saw similar results with Au–Pd/C catalysts, reporting that glyceric acid was not “over-oxidized” to tartronic acid. However, the opposite trend was noticed for rate as the Au–Pd alloy had a faster reaction rate with a TOF of 1.3 s−1 than just the monometallic Au which had a TOF of 0.8 s−1. The low initial rate of the monometallic Au catalyst, however, hints at possible artifacts in the results. In addition, a Au–Pt/C catalyst was synthesized that did see oxidation to tartronic acid and glycolic acid. Since the particle size was not reported, however, a direct comparison of TOF is not is possible.73
A bimetallic Au–Pd catalyst was also synthesized by Dimitratos et al. and showed no significant effect on activity of glycerol oxidation compared to a monometallic catalyst (TOF = 1.6 s−1).78 As the particle size of the bimetallic catalyst increased the TOF decreased and the selectivity to glyceric acid increased. Most likely the activity and selectivity were linked since less over-oxidation products were produced (glycolic and tartronic acid) on the larger particles. In a separate paper, a rate increase of a factor of five is reported for a 1% (Au–Pd)/C compared to a 1% Au/C catalyst, but an estimation of the particle size was not reported.91
Prati et al. reported that a 1% Au/C catalyst had a TOF of approximately 0.7 s−1, based on the available surface metal.75 A bimetallic catalyst synthesized by deposition of Ru on the Au/C catalyst (Ru@(Au/C)) using the sol method showed enhanced conversion of glycerol of 99% compared to 73% over the monometallic Au/C catalyst after 1 h, but it is difficult to perform a quantitative rate comparison of such high levels of conversion. The authors noted a progressive segregation of the metals on the catalyst surface, which would make an initial rate of greater importance.
Villa et al. reported a TOF of 0.28 s−1 over a 1% Pd/C catalyst and of 0.22 s−1 over a 1% Au/C catalyst at 323 K and 300 kPa O2 with NaOH:glycerol of 4.93 The monometallic Pd and Au catalysts were compared to bimetallic Pd–Au catalysts of different molar ratios it was determined that the highest TOF (1.2 s−1) was found at a bimetallic ratio of 90%Au–10%Pd/C, but an estimation of particle size was not reported. This is a significant increase in the rate of reaction for the bimetallic catalyst even though the amount of available surface metal for the reaction is uncertain. However, if a particle size between two to four nanometers is assumed, the TOF value would appear to be similar to that seen for monometallic Au catalysts in other work.
A Pd–Au/C catalyst was explored in the absence of base for glycerol oxidation by Prati et al.89 A higher conversion of 11.9% for the 1% 1:1 Pd–Au/C catalyst, compared to 3% conversion for the monometallic 5% Pd/C catalyst, was reported after 1 h at 353 K and 300 kPa O2. If a TOF is calculated a the total metal basis after 1 h, the 1% Pd/C catalyst has a TOF of 0.004 s−1 and the bimetallic Au–Pd/C catalyst has a rate of 0.017 s−1. The authors speculated that the alloy prevented irreversible adsorption of byproducts and thus the deactivation of the Pd, which is the active metal in the absence of base. The same bimetallic phenomena was not observed for a 1% (1:1) Pt–Au/C that had a TOF of 0.08 s−1, while the 5% Pt/C catalyst had a TOF of 0.07 s−1 calculated on a total metal basis.
In the absence of base at the relatively higher temperature of 373 K, Villa et al. observed only 5% conversion of glycerol and 70% selectivity to glyceric acid over a 1% Au/H-mordenite catalyst.62 While it is interesting that the supported Au catalyst had any activity at all without base, a 1% (Au : Pt 6:4)/H-mordenite catalyst had a 70% conversion and 83% selectivity to glyceric acid. Although comparing the selectivities is not appropriate because of the different levels of conversion, the significant increase in activity at the same reaction conditions is noteworthy. A TOF calculated at the 70% conversion, which is admittedly high for an initial rate, is approximately 0.2 s−1 without base for the bimetallic catalyst. A monometallic Pt/H-mordenite catalyst only had 20% conversion and 79% selectivity to glyceric acid.
Brett et al. also investigated the effect of bimetallic AuPt and AuPd on an MgO support at 333 K and 300 kPa O2 in the absence of added base.92 No monometallic catalysts were tested. While the particle size was similar between the two catalysts, the Pt–Au bimetallic was significantly more active with a TOF of 0.12 s−1. This is similar to the initial activity Villa et al. observed for the bimetallic alloy on the H-mordenite support. The low rate of reaction is consistent with the idea that Mg was not likely acting as a sacrificial base, which was confirmed by elemental analysis.
An interesting application of bimetallic catalysts was reported by Hirasawa et al., who alloyed Pd and Ag on carbon by co-impregnation.94 An increase in conversion from 2.8% for the monometallic 1% Pd/C to 6.7% for the 1% 1:1 Pd–Ag/C catalyst was reported at 353 K and 300 kPa O2 in the absence of base. In addition, the amount of CO chemisorbed did not vary significantly (<20%) for the monometallic and bimetallic catalysts which led the authors to suggest that the two catalysts have similar metal particle sizes. The researchers speculate that the increase in conversion is due to Ag preventing deactivation of the active Pd catalyst. A physical mixture of Pd/C and Ag/C did not affect the overall conversion. In addition, the selectivity to dihydroxyacetone increased from 66.1% to 74.6%, although these selectivities were reported at different conversion levels. Liang et al. also attempted to alloy Cu into Pt to prevent deactivation of the Pt during glycerol oxidation at 333 K and 300 kPa O2 in the absence of base. Although the catalyst demonstrated a small increase in rate (<15%), it delayed deactivation significantly.
Diacid production is difficult to achieve over monometallic Pt/C catalysts, with the highest selectivity to tartronic acid being reported by Carrettin et al. at 23%.63 The production of diacid can be improved by using a promoter, such as Bi. For example, a Pt–Bi/C catalyst was able to achieve an 80% selectivity to tartronic acid from glyceric acid (not glycerol) oxidation at a pH = 10–11 at 323 K under 0.75 cm3 min−1 of flowing air.95 At a lower pH (3–4), a high selectivity to hydroxypyruvic acid was reported confirming the major role that pH plays in glycerol oxidation over Pt catalysts. Kimura utilized a 5% Pt–1% Bi/C catalyst to lessen the influence deactivation during glycerol oxidation in acidic solutions.96 He was able to achieve almost 80% selectivity to dihydroxyacetone (DHA) at 323 K and a pH between 2 and 3.
Bradner et al. and Hu et al. both observed that a monometallic Pt catalyst had an initial TOF of about 0.25 s−1 and a low selectivity to dihydroxyacetone, whereas the addition of Bi increased the TOF of the catalyst significantly and increased the selectivity to dihydroxyacetone (see Table 15). Loss of DHA selectivity after consecutive reactions led Bradner et al. to speculate that Bi leached from the catalyst during the reaction. Garcia et al. showed a significant increase in selectivity to dihydroxyacetone with a Pt–Bi/C catalyst at 333 K in flowing air in which they claimed Bi sterically-hindered strong adsorption of acid products and increased selectivity to dihydroxyacetone by sterically hindering Pt nanoparticles thus preventing further oxidation.97
3.7 Glucose and other sugars
The catalytic oxidation of glucose and other sugars or sugar alcohols has been thoroughly investigated over the past several decades because of their low cost and simple production from biomass. Both monosaccharides and disaccharides can be oxidized to sugar acids, which are useful chemicals for the food and cosmetics industries. Sugars are also interesting because of their unique long-chain polyol structure and the equilibrium between their cyclic tautomers in aqueous solution. Sugars are primarily in the closed ring form in neutral water, but both basic and acidic conditions catalyze ring opening. Similar to glycerol, the rate of sugar oxidation depends primarily on the pH and catalytic metal, whereas the product distribution depends mostly on the solution pH and temperature. As discussed earlier, Pt and Pd catalysts suffer deactivation in both acidic and basic environments whereas Au is more resistant to deactivation, presumably because of its noble nature. In this section, the oxidation of several different sugars over supported metal catalysts will be discussed, with a focus on glucose oxidation.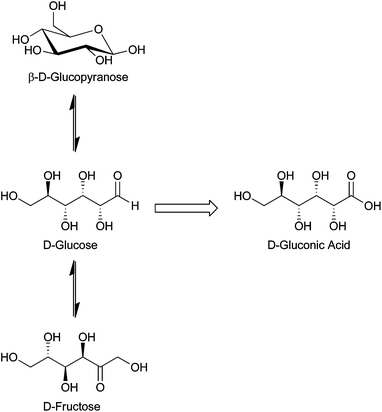 | ||
| Fig. 9 Major glucose oxidation products reported in the literature. Fructose is an isomerization product of glucose at high pH and temperature. Only β-D-glucopyranose is shown to simplify the scheme. Other possible cyclic isomers include α-D-glucopyranose, α-D-glucofuranose, and β-D-glucofuranose. | ||
Table 16 reports the TOF from selected examples of glucose oxidation over supported Au catalysts. In basic conditions, the initial TOF of glucose oxidation was in the range of about 1 to 10 s−1. The high rate of glucose oxidation at relatively mild basic conditions is simply because the ring-open form of the sugar is an aldehyde instead of an alcohol. As discussed earlier with HMF, the oxidation of the aldehyde side chain is much more rapid than the alcohol side chain. The highest TOFs are reported by Ishida et al. and are 45 s−1 for Au/ZrO2 and 42 s−1 over 0.86% Au/Al2O3 at 323 K.100 The same authors report a TOF over a 1% Au/C catalyst of 10 s−1, which suggests a potential support effect for glucose oxidation over Au. In addition, Ishida et al. showed that the synthesis procedure for preparing Au catalysts can greatly affect the final activity. Solid grinding of the support with a volatile organo gold complex works best for Al2O3- and ZrO2-supported Au, but deposition–precipitation works well for TiO2- and CeO2-supported Au. In similar work, Benko et al. tested glucose oxidation over SiO2-, TiO2-, CeO2-, and C-supported Au catalysts,101 but the highest TOF of 4.1 s−1 was found on a Au/C catalyst.
| Catalyst | Size (nm) | T (K) | pO2 (kPa) | pHc | TOF (s−1) | Reference |
|---|---|---|---|---|---|---|
| a TOF calculated or re-calculated based on available surface metal.b No pressure reported.c The pH was controlled by addition of NaOH solution. | ||||||
| 0.86% Au/Al2O3 | 2.6 | 323 | —b | 9 | 42 | 100 |
| Au Nanoparticles | 3.6 | 303 | —b | 9.5 | 18a | 102 |
| 1% Au/C | 3.4 | 323 | —b | 9.5 | 16.2a | 103 |
| 1% Au/C | 3.8 | 323 | —b | 9.5 | 15.2a | 104 |
| 1% Au/C | 5.4 | 323 | —b | 9 | 10 | 100 |
| 0.3% Au/Al2O3 | 1.5 | 313 | —b | 9 | 6.2a | 105 |
| 1.06% Au/NPC | 2.6 | 323 | —b | 9.5 | 6.0 | 106 |
| 0.1% Au/Al2O3 | 2.25 | 313 | —b | 9 | 4.5a | 107 |
| 0.8% Au/C (WGC) | 10.5 | 318 | —b | 9.5 | 4.1a | 101 |
| 0.89% Au/C | 4.42 | 323 | —b | 9.5 | 3.6a | 98 |
| 0.45% Au/TiO2 | 1.5 | 333 | —b | 11 | 2.7a | 108 |
| 0.3% Au/Al2O3 | 6 | 313 | 900 | 9 | 1.7a | 109 |
| 0.45% Au/TiO2 | 1.5 | 313 | —b | 11 | 1.1a | 108 |
| 0.89% Au/C | 4.42 | 373 | 300 | Not controlled | 0.6a | 98 |
| 1% Au/C | 3.4 | 343 | 300 | Not controlled | 0.05a | 103 |
High TOFs for glucose oxidation were also reported by Comotti et al. in several publications. For example, a 1% Au/C catalyst with 3.8 nm and 2.4 nm particles had TOFs of 15.2 and 16.2 s−1, respectively.103,104 It should be noted that glucose concentration in solution may affect the oxidation rate. Prusse et al. determined the initial rate of oxidation increased with glucose concentration up to 20 wt% and then decreased at higher concentrations of glucose. A volcano-type relationship with a peak at 20 wt% glucose concentration was found and had a maximum TOF of 1.7 s−1 at 313 K and 900 kPa O2.109
Comotti et al. also tested the catalytic activity of unsupported Au nanoparticles in solution for glucose oxidation.102 The soluble Au nanoparticles had a TOF of 18 s−1, which is consistent with many of the other reports for supported Au catalysts (see Table 16). The authors also noticed that Au particle size of the homogeneous nanoparticles increased during glucose oxidation from 3.6 nm to 10 nm, which could affect the measured rates. Interestingly, Comotti et al.103 reported a lower TOF for Au/C in the absence of added base than Biella.98 The pH dropped throughout the run from 6.0 to 2.6. Evidently, Au was not active at low pH.110,111
Baatz et al. studied the long term stability of Au catalysts for glucose oxidation.107 In particular, a 0.3% Au/Al2O3 catalyst with a Au particle size ranging from 1.2 to 3 nm was recycled 20 times at 313 K and maintained a fairly consistent TOF of 6.2 s−1. Mirescu et al. also demonstrated high selectivity and activity of 0.45% Au/TiO2 at 313 K, with no deactivation after 17 recycle experiments with a TOF of 2.7 s−1.108
Mirescu et al. explored the initial oxidation activity of a 0.45% Au/TiO2 catalyst for a variety of sugar substrates as summarized in Table 17 and Fig. 10.112 Although the selectivity to the sugar acid was very high in all cases, the substrate affected the TOF of the reaction. It should be mentioned that the TOF of glucose oxidation reported in Table 17 is about two orders of magnitude lower than many of those reported in Table 16. However, the result reported in Table 17 corresponds to an experiment utilizing a very low concentration of glucose, which may account for the much lower TOF. The oxidation of the disaccharide maltose, which is formed from the condensation of two molecules of glucose, had a TOF of 0.27 s−1, which is almost identical to the TOF of glucose (0.28 s−1) at identical conditions. It appears that the ring opening of the disaccharide (maltose) to an open chain on one side is identical to the ring opening of the monosaccharide (glucose). When the maltose concentration was increased from 0.01 M to 0.1 M the TOF increased to 1.3 s−1. The oxidation of lactose, which is formed by the condensation of two different sugars (galactose and glucose), had a lower TOF (0.09 s−1) than either of its monosaccharides. Galactose had a lower TOF (0.17 s−1) than its stereoisomer glucose.
 | ||
| Fig. 10 Sugars used as substrates in Table 17 and their ring opening products that are in equilibrium in aqueous solution. Only the beta and dextro ring and linear forms were shown to simplify the scheme. Maltose and lactose are disaccharides in which one ring opens. | ||
| Substrate | TOF (s−1) | Selectivity to product acid (%) |
|---|---|---|
| a Reaction conditions: 0.01 M substrate, 450 substrate:Au (mol:mol), 313 K, 500 cm3 min−1 O2, pH = 9. Catalyst: 0.45 wt% Au/TiO2 (particle size: 1.5 nm). Ref. 112.b 0.1 M Maltose, 4500 maltose:Au (mol:mol). | ||
| Glucose (hexose) | 0.28 | >99.5 |
| Galactose (hexose) | 0.17 | >99.5 |
| Arabinose (pentose) | 0.12 | >99.5 |
| Xylose (pentose) | 0.10 | >99.5 |
| Maltose (disaccharide) | 0.27 | >99.5 |
| Maltose (disaccharide)b | 1.3 | >99.5 |
| Lactose (disaccharide) | 0.09 | >99.5 |
The rates reported by Mirescu et al. (Table 17) are similar to other reports on galactose and arabinose oxidation over supported Au catalysts. Kusema et al. performed galactose oxidation with a 2% Au/Al2O3 catalyst with 2.6 nm particle size and found a TOF of 0.18 s−1 at a pH = 10 and 333 K.113 An acidic medium had a significantly lower TOF of 0.012 s−1, and shifted the product distribution toward galactonolactone (45%). The oxidation of arabinose with supported Au catalysts has been investigated by Kusema et al. as well.114 A 1% Au/Al2O3 catalyst at a pH = 8 and 333 K had a TOF of 0.320 s−1 for conversion of 0.1 M arabinose to arabinonic acid (99%). Simakova et al., however, reported a TOF of 0.29 s−1 for arabinose oxidation with 2% Au/Al2O3 at 333 K (presumably the same catalyst).115 The authors proposed that the reaction is structure sensitive since when rate was plotted versus particle size, 2.3 nm size Au particles showed the greatest rate of reaction. Smolentseva et al. also reported the rate of arabinose oxidation of 0.101 s−1 over a 4% Au/CeO2 catalyst with Au particles about 2.3 nm in size at 333 K and a pH = 8.116 It should be noted that the TOF for arabinose oxidation over Au is significantly lower than that for glucose oxidation under similar conditions. Smolentseva et al. also reported that bimetallic Pd–Au/CeO2 catalyst had a TOF of 0.275 s−1, higher than both the rate of monometallic Pd, 0.029 s−1, and Au, 0.090 s−1 under similar conditions. The bimetallic alloy was suggested to have a higher activity than the monometallic catalysts because of a bifunctional mechanism in which Pd dissociated dioxygen while Au oxidized arabinose.
Since the selectivity of glucose oxidation is nearly 100% to the acid at low temperatures and moderately basic conditions, bimetallic particles of Pt or Pd have been investigated as a possible way to reduce the rapid deactivation. Abbadi et al. showed that the pH had a significant effect on rate of reaction and rate of deactivation.118 At acidic conditions (i.e. no control of pH) a 5% Pt/C quickly deactivated during oxidation of 0.05 M glucose at 333 K. While attempts to reactivate the catalyst by dihydrogen flow for 30 min were not successful, addition of KOH did reactivate the Pt catalyst. Addition of Bi as a promoter helped delay deactivation of the catalyst. Besson et al. also showed the same effect of reduced deactivation during glucose oxidation after promoting Pd with Bi. More specifically, a 4.7% Pd/C catalyst demonstrated 82.6% conversion of glucose after 24 h whereas a 4.7% Pd–0.47% Bi/C catalyst reached 99.6% conversion after only 2.5 h. The Bi-promoted catalyst was also recycled successfully, although leaching was not investigated.119 The addition of Bi to a bimetallic Pt–Pd catalyst was also shown by Biella et al. to increase the TOF of glucose oxidation based on total surface metal versus just Pd, where the Bi promoted Pt–Pd tri-metallic catalyst was reported to have a TOF of about 0.2 s−1 and demonstrated limited deactivation.98
Witonska et al. has shown that Te can be used instead of Bi as a promoter for Pd-catalyzed oxidation of glucose and that a significant increase in TOF was realized.120 A 5% Pd/SiO2 catalyst had a TOF of 0.79 s−1, while a 5% Pd–5% Te/SiO2 catalyst had a TOF of 48.1 s−1 at 333 K. The 5% Pd–2% Te/Al2O3, however, only had a TOF of 1.64 s−1. The authors believed that both Pd and Te had an increased dispersion on Al2O3, which may have reduced the effectiveness of Te as a promoter of the Pd. Whereas Bi has been shown to leach into solution, especially at high weight loadings, the leaching of Te was about five times less. Karsi et al. extended the use of oxophilic promoters by studying the effect of Bi, Tl, Sn, and Co promoters on Pd catalysts. The Bi-promoted catalysts exhibited the highest TOF of 34.2 s−1, followed by Tl, Sn, and then Co. The authors suggest that Bi does not just act as a steric promoter of Pd particles by preventing the adsorption of acid byproducts, but also prevents over-oxidation of Pd metal to PdO. Bismuth, however, leached at about the same rate as Tl and Co. The Sn promoter did not leach extensively into solution, but it did not prevent deactivation of the Pd either.121,122 A high TOF of glucose oxidation over promoted Pd catalysts, which is of the same magnitude as that on Au, might suggest that the mechanism for the aldehyde to acid oxidation of glucose is similar over both Au and Pd. The deactivation profile of the two catalysts, however, appears to be very different, most likely because Au is so noble.
4. Factors affecting the evaluation of reaction rates
4.1 Deactivation of the catalyst
The deactivation of metal catalysts is a serious issue that must be addressed prior to commercialization of oxidation processes. Not only are transition metals expensive, but purification of bio-renewable feedstocks from biomass remnants can also be expensive. Deactivation of catalysts is often reported in papers without attributing it to a specific cause. Sintering, leaching, and over-oxidation of the metal as well as strong adsorption of products or byproducts on the metal surface are among the possibilities. While the addition of a promoter, such as Bi on Pt and Pd prevents some deactivation, specific modes of deactivation need to be identified so that the catalyst stability can be improved.Although Au is a noble metal that is resistant to over-oxidation and leaching, recent work has shown that inhibition of Au (and Pt) by trace ketone byproducts formed during oxidation can inhibit oxidation reactions.88 At high pH, the presence of compounds with secondary alcohol groups, such as sugar acids or other polyols, can produce ketones and enones that strongly adsorb to the active catalytic sites. Indeed, the addition of simple ketones at the beginning of a reaction can decrease oxidation rate by over 90%. Acetone and its aldol condensation product, mesityl oxide, both significantly poisoned Au and Pt surfaces used in glycerol oxidation. These results suggest that Au-catalyzed alcohol oxidation reactions may have difficulty dealing with carbohydrate impurities present in biomass feedstocks since basic conditions are needed for high activity.
As discussed earlier, Pt and Pd catalysts are thought to be deactivated by the adsorption of gluconic acid and other byproducts from glucose oxidation.123–125 Moreover, the deactivation of Pt during glucose oxidation was enhanced at neutral and acidic pH. For example, Abbadi et al. showed a rapid deactivation of 5% Pt/C at any pH below 7 whereas little deactivation was observed above a pH of 7.118 Attempts to reactivate the catalyst by reduction with H2 after poisoning did not restore catalytic activity, indicating that over-oxidation of the metal catalyst was not the sole mechanism of deactivation. Deactivation was attributed to the strong adsorption of CO formed by decarbonylation or the strong adsorption of free gluconic acid. Recently, the decarbonylation of benzaldehyde, a product of benzyl alcohol oxidation, produced adsorbed CO on the surface of Pd/Al2O3 as observed by ATR-IR spectroscopy. The authors reported that decarbonylation occurs preferentially on Pd(111) sites.126
Although many authors claim that over-oxidation of Pt and Pd metal catalysts is the cause of deactivation, very few provide experimental evidence. A brief discussion of the oxidation state of Pt and Pd during alcohol oxidation was provided by Mallet and Baiker.127 The active site for dissociative adsorption of the alcohol and β-hydride elimination is considered to be the reduced metal, Pt(0) and Pd(0).117 Deactivation by over-oxidation of the metal has been deduced when the catalyst is regenerated by reduction with dihydrogen or by treatment with alcohol in an inert atmosphere. Gogova et al. showed that deactivation by over-oxidation was reversible for supported Pd in a CSTR by replacing dioxygen with dinitrogen for a short amount of time at reaction temperature.128 The metal oxide surface was thought to be re-reduced in situ by the alcohol groups of glucose. Markusse et al. studied the formation of surface PtO by measuring the Pt–O distance using EXAFS during cyclohexanol oxidation.129 The use of cyclohexanol as a substrate was thought to prevent decarbonylation of the product aldehyde and therefore minimize formation of CO or CxHy species that could strongly adsorb on the surface. Markusse et al. also found that treating the catalyst with dihydrogen or with cyclohexanol in dinitrogen re-activated the catalyst and reduced the Pt–O contribution measured in EXAFS. The same result was found using the quick EXAFS technique for Pd/Al2O3 during cinnamyl alcohol oxidation in organic solvent at 333 K.130 Keresszegi et al. used EXAFS to reveal that both Pd and Bi were oxidized on a Bi-promoted Pd/Al2O3 catalyst used for 1-phenylethanol oxidation at 333 K.131 As expected, when both metals were oxidized, their rate of leaching into solution was accelerated.132 It appears that over-oxidation of Pt and Pd catalysts can contribute to deactivation under certain oxidizing conditions.
4.2 Utilizing basic supports to produce organic acids
The previous sections have discussed extensively the positive role of added base on the rate of metal-catalyzed alcohol oxidation reaction. The addition of homogeneous base, however, presents negative environmental and economic impacts since the high pH of the medium is corrosive and the product salts need to be neutralized to release free acid. Thus, there has been a recent push to find alternatives to the use of homogeneous bases.A logical area of exploration is the use of solid bases as supports for metal catalysts. However, the acid products of the oxidation will likely adsorb strongly on the solid bases, or worse, react with the support, thereby leaching it into solution. If solid bases are to be used for catalyst supports, the interaction between them and the organic acid products would need to be quite weak, which likely precludes their use in aqueous media where acids are formed.
There are a number of reports in the literature investigating the use of hydrotalcite materials as catalyst supports in alcohol oxidation reactions.21,42,43,133,134 Typically, these investigations were conducted in non-polar solvents (i.e. toluene or xylene), which presents no problem when aldehydes were the final product. Leaching of hydrotalcite supports in aqueous solutions can be significant.135 Even if the supported transition metal does not leach, it is possible that the hydrotalcite support is dissolved by the product acid. Therefore, analysis of leaching of the basic support is imperative when aqueous solutions are involved.25
4.3 Mass transfer limitations
The initial TOF of glycerol oxidation has been shown to be about 1 to 6 s−1 at approximately 333 K, however there are a number of reports with much lower initial rates. One possible reason for a low observed rate of reaction is the slow rate of transfer of O2 from the gas phase to the liquid, or diffusivity of O2 in the liquid and catalyst pores. If the O2 in the aqueous solvent is consumed faster than the rate of diffusion of O2 from the gas phase to the liquid phase, then the reactor system is considered to be mass transfer limited. The solubility of dioxygen in water is quite low compared to other non-polar solvents; at 333 K and 1000 kPa, Henry's law predicts an O2 solubility of approximately 0.007 M. Thus, the amount of catalyst loaded into the reactor must be kept relatively low so that there is an adequate flux of O2 from gas phase to the liquid phase.4.4 Structure sensitivity and TOF calculation
Mori et al. gave careful thought to the analysis of the oxidation rate of phenylethanol over Pd catalysts with various Pd particle sizes.11 First, the initial rates were determined at 10% conversion of alcohol at 363 K without base. Based on the size of Pd nanoparticles, assuming an FCC cuboctahedral shape, the number of face atoms and the number of edge and corner atoms were estimated and used to normalize the rate of oxidation. The turnover frequencies were therefore normalized in three different ways: by the number of surface atoms, by the number of face atoms (those with a high coordination number), and by the number of edge and corner atoms (those with a low coordination number). If all of the surface atoms of Pd were active species, then the TOF based on total surface atoms would be independent of the Pd particle size. However, the results showed a clear dependence of TOF on the metal particle size. Since only the TOF values normalized by the number of low coordination sites were independent of the cluster size, Mori et al. concluded that phenylethanol alcohol oxidation is structure-sensitive and the low coordination Pd atoms are the active species.In contrast, glycerol oxidation does not appear to depend on the particle size of supported Au catalysts in basic aqueous solution. The structure insensitive oxidation of glycerol over supported Au catalysts has a TOF of about 5 s−1 at about 333 K. The structure insensitivity of supported Au for alcohol oxidation in basic aqueous solution is consistent with the work of Abad et al.136 and Shang et al.,137 which suggests that the rate of alcohol oxidation is directly proportional to the amount of exposed Au. The support also did not appear to play an important role in glycerol oxidation over supported Au catalysts. While the majority of the catalysts contained Au particles smaller than 5 nm, Ketchie et al. showed that even very large particles of Au (20 to 40 nm) on a carbon support had oxidation rates of about 2 s−1 at 333 K with a 2 to 1 ratio of substrate to base. However, glycerol oxidation over a 0.5% Au/C catalyst with 7.3 nm surface average diameter particles had a TOF of 17 s−1, which is the highest rate reported in the literature by three times and does not fit the idea of structure insensitivity for the reaction.24 Gold is a difficult metal to accurately quantify the number of active sites available, which is needed to calculate a TOF. The relative inability of Au particles to adsorb dihydrogen, dioxygen, and carbon monoxide at room temperature rules out the use of chemisorption as a method to count sites. The lack of a good evaluation of the amount of available surface metal by the chemisorption of a gas makes the TEM a valuable resource to estimate the amount of surface metal. The images obtained from the TEM and the nanoparticle size measurements are typically a small sample size of the overall catalyst and do not always accurately reflect the actual average diameter of the supported metal. In addition, larger particles (10 to 30 nm) from the agglomeration or sintering of Au during catalyst synthesis can increase the average particle size, even if a significant portion of the particles are small. While a histogram of Au particle size was not included for the 0.5% Au/C catalyst, the micrograph clearly shows a bimodal distribution with many particles 2–3 nm in size and some particles 10–30 nm in size. A bimodal distribution of particles can artificially inflate the TOF because the smaller particles have a greater amount of surface atoms in comparison to the larger particles. If it were assumed that the average particle size was 2.5 nm (estimated as 40% dispersion), the previous TOF of 17 s−1 would be about 6 s−1 which is close to the TOF for glycerol oxidation found in the literature. Thus, the oxidation of glycerol is most likely structure insensitive and a TOF significantly greater than 10 s−1 can be explained by inaccurate quantification of available surface metal.
5. Conclusions
The oxidation of alcohols and aldehydes is an important reaction in chemicals production and is especially relevant in the production of chemicals from biomass sources. While oxidations have long been performed using highly toxic oxidants, the environmental and economic benefits of supported metal catalysts are significant. The oxidation of alcohols over supported transition metals is rapid at mild temperatures (295–333 K) and facilitated by the addition of base.Mechanistically, the oxidation of alcohols proceeds through a first oxidation step to aldehydes and through a second oxidation step to carboxylic acids. The first oxidation, alcohol to aldehyde, is difficult, whereas the second oxidation of aldehyde to acid is rapid in water. The second oxidation step is so fast that the transport limits must be carefully considered and avoided if a kinetic rate is to be determined. Kinetic results should be obtained at low levels of substrate conversion since deactivation of the metal surface can be rapid. This deactivation can be due to over-oxidation of the metals (Pt and Pd) but also can be the result of product or byproduct adsorption, especially by ketones, acids, and carbon monoxide. Adjusting reaction conditions, alloying materials to create bimetallic catalysts, and/or utilizing oxophilic promoters can be used to increase the stability and thus longevity of heterogeneous catalysts.
Tuning the product selectivity to acid or aldehyde may be most easily achieved by selection of the solvent, since water solvent favors the formation of acid. As with any sequential reaction, comparison of catalyst activity with respect to product selectivity should be made at equal levels of conversion of the substrate. The rapid rate of alcohol oxidation, as well as the potential to tune product selectivity, makes transition metal catalysts attractive materials for use in the emerging biomass-derived chemicals market.
Acknowledgements
Support from the National Science Foundation (Grant Nos. OISE 0730277 and EEC-0813570) is gratefully acknowledged.References
- T. L. Donaldson and O. L. Culberson, Energy, 1984, 9, 693–707 CrossRef.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554, 10.1039/b922014c.
- T. Werpy and G. R. Petersen, Top Value Added Chemicals from Biomass, No. DOE/GO-102004-1992, Department of Energy, Office of Scientific and Technical Information, Washington, DC, 2004.
- D. Romano, R. Villa and F. Molinari, ChemCatChem, 2012, 4, 739–749, DOI:10.1002/cctc.201200042.
- L. Palmisano, V. Augugliaro, M. Bellardita, A. Di Paola, E. García López, V. Loddo, G. Marcì, G. Palmisano and S. Yurdakal, ChemSusChem, 2011, 4, 1431–1438, DOI:10.1002/cssc.201100196.
- C. Della Pinna, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369, 10.1039/c1cs15089h.
- B. Katryniok, H. Kimura, E. Skrzynska, J. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979, 10.1039/c1gc15320j.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 3, 520–540, 10.1039/c0gc00639d.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4541 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666, DOI:10.1021/ja04088683.
- X. Yang, X. Wang and J. Qui, Appl. Catal., A, 2010, 382, 131–137 CrossRef CAS.
- P. Fristrup, L. B. Johansen and C. H. Christensen, Catal. Lett., 2008, 120, 184–190, DOI:10.1007/s10562-007-9301-8.
- M. Kotani, T. Koike, K. Yamaguchi and N. Mizuno, Green Chem., 2006, 8, 735–741, 10.1039/b603204d.
- J. Chen, Q. Zhang, Y. Wang and H. Wan, Adv. Synth. Catal., 2008, 350, 453–464, DOI:10.1002/adsc.200700350.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145, DOI:10.1021/ja001325i.
- S. Mori, M. Takubo, K. Makida, T. Yanase, S. Aoyagi, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. Commun, 2009, 34, 5159–5161, 10.1039/b908451g.
- K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4353–4361, DOI:10.1002/chem.200304916.
- K. B. Sharpless, K. Akashi and K. Oshima, Tetrahedron Lett., 1976, 17, 2503–2506 CrossRef.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- W. Fang, J. Chen, Q. Zhang, W. Deng and Y. Wang, Chem.–Eur. J., 2011, 17, 1247–1256, DOI:10.1002/chem.201002469.
- G. An, H. Ahn, K. A. de Castro and H. Rhee, Synthesis, 2009, 3, 477–485, DOI:10.1055/s-0029-1217115.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS.
- W. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 264–273, DOI:10.1016/j.jcat.2007.06.011.
- B. N. Zope, S. E. Davis and R. J. Davis, Top. Catal., 2012, 55, 24–32, DOI:10.1007/s11244-012-9777-3.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147, 10.1039/c1gc16074e.
- B. Jorgensen, S. E. Christiansen, M. L. D. Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–337 CrossRef CAS.
- C. H. Christensen, B. Jorgensen, J. Rass-Hansen, K. Egeblad, R. Madsen, S. K. Klitgaard, S. M. Hansen, M. R. Hansen, H. C. Andersen and A. Riisager, Angew. Chem., Int. Ed., 2006, 45, 4648–4651, DOI:10.1002/anie.200601180.
- L. Prati, A. Villa, C. Campione and P. Spontoni, Top. Catal., 2007, 44, 319–324, DOI:10.1007/s11244-007-0305-9.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121, DOI:10.1016/j.jcat.2006.08.019.
- D. I. Enache, D. W. Knight and G. J. Hutchings, Catal. Lett., 2005, 103, 43–52, DOI:10.1007/s10562-005-6501-y.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069, DOI:10.1002/anie.200500382.
- A. Abad, C. Almala, A. Corma and H. Garcia, Tetrahedron, 2006, 62, 6666–6672, DOI:10.1016/j.tet.2006.01.118.
- L. F. Liotta, A. M. Venezia, G. Deganello, A. Longo, A. Martorana, Z. Schay and L. Guczi, Catal. Today, 2001, 66, 271–276 CrossRef CAS.
- Y. Uozumi and R. Nakao, Angew. Chem., Int. Ed., 2003, 42, 194–197 CrossRef CAS.
- A. Villa, D. Wang, N. Dimitratos, D. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8–15, DOI:10.1016/jcattod.2009.06.009.
- A. Buonerba, C. Cuomo, S. O. Sanchez, P. Canton and A. Grassi, Chem.–Eur. J., 2012, 18, 709–715, DOI:10.1002/chem.201101034.
- H. Guo, A. Al-Hunaiti, M. Kemell, S. Rautiainen, M. Leskela and T. Repo, ChemCatChem, 2011, 3, 1872–1875, DOI:10.1002/cctc.201100286.
- M. Boronat, A. Corma, F. Illas, J. Radilla, T. Rodenas and M. J. Sabater, J. Catal., 2011, 278, 50–58, DOI:10.1016/j.jcat.2010.11.013.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375, DOI:10.1021/ja052161e.
- A. Villa, C. E. Chan-Thaw, G. M. Veith, K. L. More, D. Ferri and L. Prati, ChemCatChem, 2011, 3, 1612–1618, DOI:10.1002/cctc.201100161.
- T. Mitsudome, Y. Mikami, H. Funai, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 138–141, DOI:10.1002/anie.200703161.
- T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawa and K. Kaneda, Adv. Synth. Catal., 2009, 351, 1890–1896, DOI:10.1002/adsc.200900239.
- S. Meenakshisundaram, E. Nowicka, P. J. Miedziak, G. L. Brett, R. L. Jenkins, N. Dimitratos, S. H. Taylor, D. W. Knight, D. Bethell and G. J. Hutchings, Faraday Discuss., 2010, 145, 341–356 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- M. J. Beier, T. W. Hansen and J. D. Grunwaldt, J. Catal., 2009, 266, 320–330, DOI:10.1016/j.jcat.2009.06.022.
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157–2163, 10.1039/c0gc00493f.
- J. Zhu, J. L. Faria, J. L. Figueiredo and A. Thomas, Chem.–Eur. J., 2011, 17, 7112–7117, DOI:10.1002/chem.201003025.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef CAS.
- T. Pasini, M. Piccinini, M. Blosi, R. Bonelli, S. Albonetti, N. Dimitratos, J. A. Lopez-Sanchez, M. Sankar, Q. He, C. J. Kiely, G. J. Hutchings and F. Cavani, Green Chem., 2011, 13, 2091–2099, 10.1039/c1gc15355b.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144, DOI:10.1002/cssc.200900137.
- Y. Y. Gorbanev, S. K. Klitgaard, J. M. Woodley, C. H. Christensen and A. Riisager, ChemSusChem, 2009, 2, 672–675, DOI:10.1002/cssc.200900059.
- S. M. Payne and F. M. Kerton, Green Chem., 2010, 12, 1648–1653, DOI:10.1030/c0gc00205d.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- P. Verdeguer, N. Merat and A. Gaset, J. Mol. Catal., 1993, 85, 327–344 CrossRef CAS.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78, DOI:10.1002/cssc.200700033.
- M. A. Lilga, R. T. Hallen and M. Gray, Top. Catal., 2010, 53, 1264–1269, DOI:10.1007/s11244-010-9579-4.
- O. Casanova, S. Iborra and A. Corma, J. Catal., 2009, 265, 109–116, DOI:10.1016/j.cat.2009.04.019.
- Y. Y. Gorbanev, S. Kegnaes and A. Riisager, Catal. Lett., 2011, 141, 1752–1760, DOI:10.1007/s10562-011-0707-y.
- B. Saha, S. Dutta and M. M. Abu-Omar, Catal. Sci. Technol., 2012, 2, 79–81, 10.1039/c1cy00321f.
- P. Vinke, H. E. van Dam and H. van Bekkum, New Dev. Select. Oxidat., 1990, 55, 147–157 Search PubMed.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., 2010, 49, 4499–4502, DOI:10.1002/ange.201000762.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 7, 696–697, 10.1039/b201112n.
- W. C. Ketchie, M. Murayama and R. J. Davis, Top. Catal., 2007, 44, 307–317, DOI:10.1007/s11244-007-0304-x.
- E. G. Rodrigues, M. F. R. Pereira, X. Chen, J. J. Delgado and J. J. M. Órfão, J. Catal., 2011, 281, 119–127, DOI:10.1016/j.jcat.2011.04.008.
- W. Ketchie, Y. Fang, M. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94–101, DOI:10.1016/j.jcat.2007.06.001.
- S. Demirel, K. Lehnert, M. Lucas and P. Claus, Appl. Catal., B, 2007, 70, 637–643, DOI:10.1016/j.apcatb.2005.11.036.
- S. Demirel, M. Lucas, J. Wärnå, T. Salmi, D. Murzin and P. Claus, Top. Catal., 2007, 44, 299–305, DOI:10.1007/s11244-007-0303-y.
- S. Demirel-Gülen, M. Lucas and P. Claus, Catal. Today, 2005, 102–103, 166–172, DOI:10.1016/j.cattod.2005.02.033.
- P. L. Mills and R. V. Chaudhari, Catal. Today, 1997, 37, 367–404, DOI:10.1016/s0920-5861(97)00028-x.
- S. Gil, M. Marchena, L. Sánchez-Silva, A. Romero, P. Sánchez and J. L. Valverde, Chem. Eng. J., 2011, 178, 423–435, DOI:10.1016/j.cej.2011.10.048.
- E. G. Rodrigues, S. A. C. Carabineiro, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, J. Catal., 2012, 285, 83–91, DOI:10.1016/j.jcat.2011.09.016.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102–103, 203–212, DOI:10.1016/j.cattod.2005.02.003.
- Y. Shen, S. Zhang, H. Li, Y. Ren and H. Liu, Chemistry, 2010, 16, 7368–7371, DOI:10.1002/chem.201000740.
- L. Prati, F. Porta, D. Wang and A. Villa, Catal. Sci. Technol., 2011, 1, 1624–1629, 10.1039/c1cy00218j.
- N. Dimitratos, A. Villa, C. L. Bianchi, L. Prati and M. Makkee, Appl. Catal., A, 2006, 311, 185–192, DOI:10.1016/j.apcata.2006.06.026.
- Z. Huang, F. Li, B. Chen, F. Xue, Y. Yuan, G. Chen and G. Yuan, Green Chem., 2011, 13, 3414–3422, 10.1039/c1gc15811b.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Lennon, F. Porta, L. Prati and A. Villa, Catal. Lett., 2006, 108, 147–153, DOI:10.1007/s10562-006-0036-8.
- E. G. Rodrigues, S. A. C. Carabineiro, X. Chen, J. J. Delgado, J. L. Figueiredo, M. F. R. Pereira and J. J. M. Órfão, Catal. Lett., 2010, 141, 420–431, DOI:10.1007/s10562-010-0515-9.
- N. Dimitratos, C. Messi, F. Porta, L. Prati and A. Villa, J. Mol. Catal., A, 2006, 256, 21–28, DOI:10.1016/j.molcata.2006.04.019.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329–1336, 10.1039/b212047j.
- J. Gao, D. Liang, P. Chen, Z. Hou and X. Zheng, Catal. Lett., 2009, 130, 185–191, DOI:10.1007/s10562-009-9849-6.
- D. Liang, J. Gao, H. Sun, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 106, 423–432, DOI:10.1016/j.apcatb.2011.05.050.
- A. Brandner, K. Lehnert, A. Bienholz, M. Lucas and P. Claus, Top. Catal., 2009, 52, 278–287, DOI:10.1007/s11244-008-9164-2.
- D. Liang, J. Gao, J. Wang, P. Chen, Z. Hou and X. Zheng, Catal. Commun., 2009, 10, 1586–1590, DOI:10.1016/j.catcom.2009.04.023.
- W. Hu, D. Knight, B. Lowry and A. Varma, Ind. Eng. Chem. Res., 2010, 49, 10876–10882, DOI:10.1021/ie1005096.
- M. Sankar, N. Dimitratos, D. W. Knight, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Thomas and G. J. Hutchings, ChemSusChem, 2009, 2, 1145–1151, DOI:10.1002/cssc.200900133.
- B. N. Zope and R. J. Davis, Green Chem., 2011, 13, 3484–3491, 10.1039/c1gc15953d.
- L. Prati, P. Spontoni and A. Gaiassi, Top. Catal., 2009, 52, 288–296, DOI:10.1007/s11244-008-9165-1.
- A. P. Markusse, B. F. M. Kuster and J. C. Schouten, Catal. Today, 2001, 66, 191–197, DOI:10.1016/s0920-5861(00)00648-9.
- N. Dimitratos, F. Porta and L. Prati, Appl. Catal., A, 2005, 291, 210–214, DOI:10.1016/j.apcata.2005.01.044.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139, DOI:10.1002/anie.201101772.
- A. Villa, C. Campione and L. Prati, Catal. Lett., 2007, 115, 133–136, DOI:10.1007/s10562-007-9077-x.
- S. Hirasawa, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2012, 2, 1150–1152, 10.1039/c2cy20062g.
- P. Fordham, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 133, L179–L184, DOI:10.1016/0926-860x(95)00254-5.
- H. Kimura, Appl. Catal., A, 1993, 105, 147–158, DOI:10.1016/0926-860x(93)80245-l.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176, DOI:10.1016/0926-860x(95)00048-8.
- S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 206, 242–247, DOI:10.1006/jcat.2001.3497.
- Y. Önal, S. Schimpf and P. Claus, J. Catal., 2004, 223, 122–133, DOI:10.1016/j.jcat.2004.01.010.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268, DOI:10.1002/anie.200802845.
- T. Benkó, A. Beck, O. Geszti, R. Katona, A. Tungler, K. Frey, L. Guczi and Z. Schay, Appl. Catal., A, 2010, 388, 31–36, DOI:10.1016/j.apcata.2010.08.008.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815, DOI:10.1002/anie.200460446.
- M. Comotti, C. D. Pina and M. Rossi, J. Mol. Catal., A, 2006, 251, 89–92, DOI:10.1016/j.molcata.2006.02.014.
- M. Comotti, C. D. Pina, R. Matarrese, M. Rossi and A. Siani, Appl. Catal., A, 2005, 291, 204–209, DOI:10.1016/j.apcata.2004.11.051.
- C. Baatz, N. Decker and U. Pruse, J. Catal., 2008, 258, 165–169, DOI:10.1016/j.jcat.2008.06.008.
- H. Okatsua, N. Kinoshitaa, T. Akitab, T. Ishidaa and M. Haruta, Appl. Catal., A, 2009, 369, 8–14 CrossRef CAS.
- C. Baatz, N. Thielecke and U. Prüße, Appl. Catal., B, 2007, 70, 653–660, DOI:10.1016/j.apcatb.2006.01.020.
- A. Mirescu, H. Berndt, A. Martin and U. Prüße, Appl. Catal., A, 2007, 317, 204–209, DOI:10.1016/j.apcata.2006.10.016.
- U. Prüße, M. Herrmann, C. Baatz and N. Decker, Appl. Catal., A, 2011, 406, 89–93, DOI:10.1016/j.apcata.2011.08.013.
- S. Hermans, A. Deffernez and M. Devillers, Appl. Catal., A, 2011, 395, 19–27, DOI:10.1016/j.apcata.2011.01.019.
- S. Hermans, A. Deffernez and M. Devillers, Catal. Today, 2010, 157, 77–82, DOI:10.1016/j.cattod.2010.04.010.
- A. Mirescu and U. Prüße, Appl. Catal., B, 2007, 70, 644–652, DOI:10.1016/j.apcatb.2006.01.017.
- B. T. Kusema, B. C. Campo, O. A. Simakova, A. Leino, K. Kordás, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, ChemCatChem, 2011, 3, 1789–1798, DOI:10.1002/cctc.201100183.
- B. T. Kusema, B. C. Campo, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2010, 386, 101–108, DOI:10.1016/j.apcata.2010.07.037.
- O. A. Simakova, B. T. Kusema, B. C. Campo, A. Leino, K. Kordás, V. Pitchon, P. Mäki-Arvela and D. Y. Murzin, J. Phys. Chem. C, 2011, 115, 1036–1043, DOI:10.1021/jp105509k.
- E. Smolentseva, B. T. Kusema, S. Beloshapkin, M. Estrada, E. Vargas, D. Y. Murzin, F. Castillon, S. Fuentes and A. Simakov, Appl. Catal., A, 2011, 392, 69–79, DOI:10.1016/j.apcata.2010.10.021.
- I. V. Delidovich, O. P. Taran, L. G. Matvienko, A. N. Simonov, I. L. Simakova, A. N. Bobrovskaya and V. N. Parmon, Catal. Lett., 2010, 140, 14–21 CrossRef CAS.
- A. Abbadi and H. van Bekkum, J. Mol. Catal., A, 1995, 97, 111–118, DOI:10.1016/1381-1169(94)00078-6.
- M. Besson, F. Lahmer, P. Gallezot, P. Fuertes and G. Fleche, J. Catal., 1995, 152, 116–121, DOI:10.1006/jcat.1995.1065.
- I. Witonska, M. Frajtak and S. Karski, Appl. Catal., A, 2011, 401, 73–82, DOI:10.1016/j.apcata.2011.04.046.
- S. Karski, T. Paryjczak and I. Witonñska, Kinet. Catal., 2003, 44, 618–622 CrossRef CAS.
- S. Karski and I. Witońska, J. Mol. Catal., A, 2003, 191, 87–92 CrossRef CAS.
- A. Abbadi and H. van Bekkum, Appl. Catal., A, 1995, 124, 409–417, DOI:10.1016/0926-860x(94)00285-1.
- P. C. C. Smits, B. F. M. Kuster, K. van der Wiele and S. van der Baan, Appl. Catal., 1987, 33, 83–96, DOI:10.1016/s0166-9834(00)80585-0.
- M. Wenkin, P. Ruiz, B. Delmon and M. Devillers, J. Mol. Catal., A, 2002, 180, 141–159, DOI:10.1016/s1381-1169(01)00421-6.
- D. Ferri and A. Baiker, Top. Catal., 2009, 52, 1323–1333, DOI:10.1007/s11244-009-9310-5.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–283, DOI:10.1016/0920-5861(94)80187-8.
- Z. Gogová and J. Hanika, Chem. Eng. J., 2009, 150, 223–230 Search PubMed.
- A. P. Markusse, B. F. M. Kuster, D. C. Koningsberger and G. B. Marin, Catal. Lett., 1998, 55, 141–145, DOI:10.1023/a:1019007601442.
- C. Keresszegi, T. Mallat and A. Baiker, J. Catal., 2003, 213, 291–295 CrossRef CAS.
- C. Keresszegi, J. Grunwaldt, T. Mallat and A. Baiker, J. Catal., 2004, 222, 268–280, DOI:10.1016/j.jcat.2003.10.013.
- J. H. Vleeming, B. F. M. Kuster and G. B. Marin, Catal. Lett., 1997, 46, 187–194, DOI:10.1023/a:1019066401732.
- K. Ebitani, K. Motokura, T. Mizugaki and K. Kaneda, Angew. Chem., Int. Ed., 2005, 44, 3423–3426, DOI:10.1002/anie.200462600.
- T. Matsushita, K. Ebitani and K. Kaneda, Chem. Commun., 1999, 3, 265–266, 10.1039/a809082c.
- M. Jobbagy and A. Regazzoni, Appl. Clay Sci., 2011, 51, 366–369, DOI:10.1016/j.cly.2010.11.027.
- A. Abad, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 212–222, DOI:10.1002/chem.200701263.
- C. Shang and Z. P. Liu, J. Am. Chem. Soc., 2011, 133, 9938–9947, DOI:10.1021/ja203468v.
| This journal is © The Royal Society of Chemistry 2013 |