Metal-catalyzed amide bond forming reactions in an environmentally friendly aqueous medium: nitrile hydrations and beyond
Rocío
García-Álvarez
,
Pascale
Crochet
* and
Victorio
Cadierno
*
Laboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Departamento de Química Orgánica e Inorgánica. Instituto Universitario de Química Organometálica “Enrique Moles”, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: crochetpascale@uniovi.es; vcm@uniovi.es; Fax: +(34) 985103446; Tel: +(34) 985103453
First published on 25th October 2012
Abstract
Amides are versatile building blocks in synthetic organic chemistry, presenting a wide range of pharmacological applications, and are used as raw materials in industry for the large-scale production of engineering plastics, detergents and lubricants. The development of green procedures for the synthesis of this relevant class of compounds from various starting materials, which replace antiquated methods using carboxylic acid derivatives and amines, is therefore of prime interest in modern chemistry. In this review article, a survey of metal-catalyzed synthetic approaches of amides conducted in an environmentally friendly aqueous medium is given.
 Rocío García-Álvarez | Rocío García-Álvarez received her BSc in Chemistry from the University of Oviedo (Spain) in 2007. In 2008 she started her PhD thesis under the supervision of Drs P. Crochet and V. Cadierno at the University of Oviedo, with research on novel ruthenium catalysts for amide syntheses in water. |
 Pascale Crochet | Pascale Crochet studied chemistry at the University of Rennes I (France) and obtained her PhD in 1996 under the supervision of Prof. P. H. Dixneuf and B. Demerseman. After a two-year post-doctoral stay in the group of Prof. M. A. Esteruelas (University of Zaragoza, Spain) and one year as Assistant Professor at the “National High School of Physics and Chemistry” of Bordeaux (France), she moved in 1999 to the University of Oviedo where she is currently Associate Professor of Inorganic Chemistry. Her research interest deals with the design and synthetic applications of organometallic complexes, with a particular focus on hydrosoluble ruthenium catalysts. |
 Victorio Cadierno | Victorio Cadierno received his PhD degree from the University of Oviedo (Spain) in 1996 under the supervision of Prof. J. Gimeno. He then joined the group of Dr J. P. Majoral at the Laboratoire de Chimie de Coordination (LCC-CNRS) in Toulouse (France) for a two-year postdoctoral stay. Thereafter, he returned to the University of Oviedo where he is currently Associate Professor of Inorganic Chemistry. In 2002 he received from the Spanish Royal Society of Chemistry (RSEQ) the Young Investigator Award. His current research involves the use of transition metal complexes for catalytic organic synthesis, with special focus on metal-catalyzed reactions in water. |
Introduction
Amides are one of the most important functional groups in nature (amide linkages are the key chemical connections of proteins), they constitute versatile building blocks in synthetic organic chemistry, and also exhibit a wide range of industrial applications and pharmacological interest.1 Chemical reactions for their formation are among the most executed transformations in organic chemistry. As a representative example, an analysis of drug candidate molecules manufactured by the leading pharmaceutical companies GlaxoSmithKline, AstraZeneca and Pfizer showed that amide bond formation was utilized in the synthesis of 66% of the candidates surveyed.2 Amides are typically prepared from the union of carboxylic acids and derivatives (halides, anhydrides or esters) with amines.1–3 However, although they are remarkably general, these methods present several drawbacks such as the use of toxic, corrosive and/or expensive materials, highly exothermic reactions, low tolerance to sensitive functional groups, complex reaction conditions and wasteful procedures. As a matter of fact, in 2005, the ACS GCIPR (American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable) identified amide formation as one of the most problematic syntheses in the pharmaceutical industry, and labelled it as a high priority research field.4 In the search for improved synthetic methods, metal-catalyzed transformations have emerged in recent years as the most promising alternatives for the atom-economical and cost effective synthesis of amides, opening also previously unavailable routes that start from substrates other than carboxylic acids and their derivatives.5 A couple of reviews covering these innovative approaches have recently appeared in the literature.6On the other hand, over the past two decades, increasing environmental concerns have stimulated the development of new synthetic protocols that minimize the generation of waste to the maximum. Solvents account for 80–90% of mass utilization in a typical pharmaceutical/fine chemical operational process.7 Consequently, they are responsible for most of the waste generated in the chemical industries and laboratories. With the ultimate goal of solving this environmental problem, remarkable research efforts have focused on the replacement of traditional organic solvents by water, since water is the most convenient solvent that one can imagine in terms of cost, availability, safety, and environmental impact.8 Besides its inherent advantages, which fit perfectly with the requirements of the Green Chemistry principles,9 the use of water as a solvent can also provide a notable difference in reactivity, enhancing in some cases the rate or changing the selectivity of a given reaction. Several books, reviews and accounts illustrating the enormous potential of water in developing new organic transformations are currently available, and they are recommended to those readers seeking a broader introduction to the topic.10 In the context of homogeneous catalysis, the use of water as a solvent is also usually associated with an easy catalyst/product separation, thus allowing in some cases the effective recycling of the catalytically active species, which is another crucial factor in realizing a “green” process.11 In addition, it is also worthy of note that the discovery of new techniques in nanofiltration and recovery of metal ions from water are, during water purification and recycling, no longer a barrier for large-scale chemical processes.12
The aim of the present review article is to provide a comprehensive account of metal-catalyzed synthetic approaches of amides in an environmentally friendly aqueous medium. Only those reactions conducted in pure water, without requirement of an organic cosolvent, will be discussed. Aqueous transformations in which the amide functionality already exists in the molecule, and a metal catalyst is employed to promote further structural modifications, are also considered to be out of the scope of this review. Literature published up to September 2012 is covered.
Catalytic synthesis of primary amides in water
In this section the formation of primary amides RC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)NH2 promoted by homogeneous and heterogeneous metal compounds in an aqueous medium is discussed. The field is clearly dominated by the catalytic hydration of nitriles. However, as the reader will see, several synthetic methods that make use of alternative starting materials (aldoximes, aldehydes, amines, azides, alcohols or methylarenes) have also appeared in recent years.
O)NH2 promoted by homogeneous and heterogeneous metal compounds in an aqueous medium is discussed. The field is clearly dominated by the catalytic hydration of nitriles. However, as the reader will see, several synthetic methods that make use of alternative starting materials (aldoximes, aldehydes, amines, azides, alcohols or methylarenes) have also appeared in recent years.
From nitriles
Ideally, the hydration of nitriles is the most atom-economical reaction and a sustainable method for the preparation of primary amides. However, traditional protocols for hydrating nitriles involve the use of strong bases or acids under harsh conditions, methods which are not compatible with many sensitive functional groups. In addition, the base-catalyzed reactions usually cause over-hydrolysis of the amides into the corresponding carboxylic acids, a kinetically favoured reaction compared to the hydration one (Scheme 1).1,3 Although under acidic conditions it is possible to stop the process at the amide stage, in these cases it is necessary to control carefully the temperature and stoichiometry employed in order to avoid the formation of polymeric side products.13 It is also important to note that, from an industrial perspective, the final neutralization step required either in the acid- or base-catalyzed reactions leads to extensive salt formation with inconvenient product contamination and pollution effects.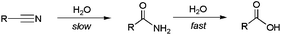
Nitrile hydrations using enzymatic catalysis allow us to circumvent most of these problems, offering cleaner, safer and more selective protocols for the preparation of primary amides.14 In fact, some nitrile hydratases are currently employed for the industrial production of acrylamide, nicotinamide and 5-cyanovaleramide by hydration of the corresponding nitriles.15 However, despite the significant progress made in the field and its commercial success, the isolation costs and the narrow substrate specificity of the current available enzymes severely limit their practical use. Given their greater scope, catalytic methods based on the use of metal compounds represent a more attractive alternative than enzymes. In this context, a great variety of homogeneous and heterogeneous systems have been developed during the last two decades.16 Most of them operate in organic media in the presence of only small amounts of water, but, owing to practical and environmental concerns, current research has focused on the search for catalytic systems able to operate directly in water. The advances reached in the field are summarized in the following lines.
The platinum(II) complex [PtH{(PMe2O)2H}(PMe2OH)] 1, synthesized by Parkins and co-workers in 1995,17 is probably the most versatile catalyst presently available for the hydration of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds. Its remarkable activity under relatively mild conditions (70–100 °C), along with its exquisite functional group tolerance, has allowed the implementation of this catalyst in the synthesis of a huge number of biologically active molecules and natural products.18 As a representative example, the preparation of the steroidal compound 7α-carbamoyl-9(11)Δ-canrenone 3, an advanced intermediate in the production of the orally-active aldosterone antagonist eplerenone 4 used for the treatment of hypertension and congestive heart failure, could be performed in excellent yield by catalytic hydration of 7α-cyano-9(11)Δ-canrenone 2 using this complex (Scheme 2).18e
N bonds. Its remarkable activity under relatively mild conditions (70–100 °C), along with its exquisite functional group tolerance, has allowed the implementation of this catalyst in the synthesis of a huge number of biologically active molecules and natural products.18 As a representative example, the preparation of the steroidal compound 7α-carbamoyl-9(11)Δ-canrenone 3, an advanced intermediate in the production of the orally-active aldosterone antagonist eplerenone 4 used for the treatment of hypertension and congestive heart failure, could be performed in excellent yield by catalytic hydration of 7α-cyano-9(11)Δ-canrenone 2 using this complex (Scheme 2).18e
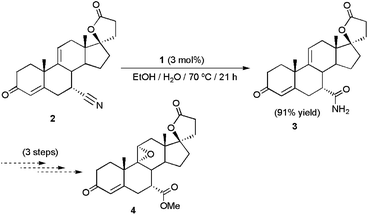 | ||
| Scheme 2 Pt-catalyzed hydration of 7α-cyano-9(11)Δ-canrenone 2. | ||
The presence of a phosphinito ligand in 1, which incorporates a tethered hydroxyl group, eliminates the need for a base in the reactions, the proposed mechanism of action involving the direct participation of this group in the hydration step (Scheme 3).17 Although in the initial work by Parkins’ group the hydration of model acetonitrile and 3-cyanopyridine substrates with 1 could be performed directly in water (yields up to 91%, TOF up to 450 h−1 and TON up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 using 0.004–0.1 mol% of 1 at 100 °C),17 in all the later studies water/organic solvent (ethanol, methanol, tetrahydrofuran or 1,4-dioxane) mixtures were systematically employed as the reaction media (as in the example of Scheme 1).18 Only recently, a pure aqueous medium was used to study the catalytic behaviour of [PtH{(PMe2O)2H}(PMe2OH)] 1 towards cyanohydrins (α-hydroxynitriles).19 Unfortunately, the hydration rates for these substrates were very slow in comparison to those observed with other nitriles, and only a few turnovers could be achieved. The low reactivity was ascribed to the liberation of HCN by partial degradation of the cyanohydrin substrate, the coordination of the cyanide anion leading to the deactivation of the catalyst.
000 using 0.004–0.1 mol% of 1 at 100 °C),17 in all the later studies water/organic solvent (ethanol, methanol, tetrahydrofuran or 1,4-dioxane) mixtures were systematically employed as the reaction media (as in the example of Scheme 1).18 Only recently, a pure aqueous medium was used to study the catalytic behaviour of [PtH{(PMe2O)2H}(PMe2OH)] 1 towards cyanohydrins (α-hydroxynitriles).19 Unfortunately, the hydration rates for these substrates were very slow in comparison to those observed with other nitriles, and only a few turnovers could be achieved. The low reactivity was ascribed to the liberation of HCN by partial degradation of the cyanohydrin substrate, the coordination of the cyanide anion leading to the deactivation of the catalyst.
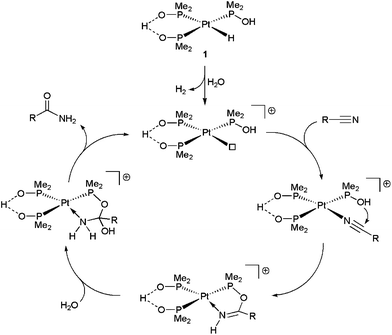 | ||
| Scheme 3 Suggested mechanism for the catalytic hydration of nitriles by complex 1. | ||
Like the platinum–phosphinito complex 1, the molybdocene derivatives [(η5-C5H4R)2Mo(OH)(H2O)][OTs] (R = H 5a, Me 5b) and [{C2Me4(η5-C5H4)2}Mo(OH)(H2O)][OTs] 6 (OTs = p-toluenesulfonate) developed by Tyler and co-workers (Fig. 1) are also slightly soluble in water and effective towards the selective hydration of nitriles, affording the amides without further hydrolysis to the carboxylic acids.20 A wide range of aromatic, aliphatic and α,β-unsaturated nitriles were successfully transformed performing the catalytic reactions in D2O at 80 °C with 0.1–5 mol% of these molybdocene complexes (TOF values up to 4077 h−1). However, we must note that challenging cyanohydrin substrates could not be hydrated despite the rapid conversion observed for β-hydroxynitriles.19 In addition, complexes 5–6 were also intolerant to ether- and ester-containing nitriles (e.g. 2-methylcyanoacetate or 2-methoxyacetonitrile) due to the facile hydrolysis of those groups.20a
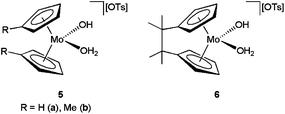 | ||
| Fig. 1 Structure of the water-soluble molybdocene catalysts 5 and 6. | ||
On the basis of kinetic data and H/D exchange experiments a reaction mechanism for the hydration process involving the intramolecular attack of the hydroxo ligand on the metal-coordinated nitrile was proposed for 5b (Scheme 4).20a In addition, it was also observed that increasing the electron-withdrawing ability of the nitrile enhanced the rate of the reaction. This result suggested that the intramolecular attack of OH− is the rate-limiting step of the catalytic cycle. All these facts were later confirmed by Tyler19 and López21 through density functional theory (DFT) calculations on the [Cp2Mo(OH)(H2O)]+ system 5a.
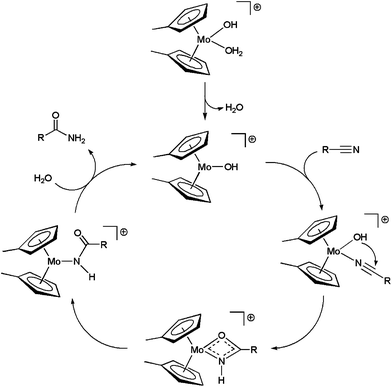 | ||
| Scheme 4 Catalytic cycle for the hydration of nitriles with complex 5b. | ||
The nickel(I) dimer [{Ni(dippe)(μ-H)}2] 7 (dippe = 1,2-bis(diisopropylphosphino)ethane) is another relevant example of a homogeneous metal catalyst active in a pure aqueous medium. For example, hydration of benzonitrile and acetonitrile at 180 °C with this complex led to TON and TOF values of up to 984 and 14 h−1, respectively, using a nickel loading of only 0.04 mol%.22 From a mechanistic point of view (Scheme 5), an initial reduction of Ni(I) to Ni(0) via H2 release was proposed, followed by η2-coordination of the nitrile to the metal. Then, intermolecular nucleophilic attack of water on the η2-nitrile takes place to generate a hydroxy-imine, which decoordinates from the Ni(0) center giving the final amide. In accord with this proposal, isolated complexes [Ni(dippe)(η2-NCR)] proved to be catalytically active.22 In addition to benzonitrile and acetonitrile, hydration of 1,2-, 1,3- and 1,4-dicyanobenzenes,23 as well as a variety of N-heterocyclic nitriles, such as 2- and 3-cyanoquinolines, 2-, 3- and 4-cyanopyridines or 2,6-dicyanopyridine,24 with the aid of complex 7 was also demonstrated. For the latter, selective entries to the mixed cyano/amide and fully hydrated dicarboxamide could be established.24b Moreover, this Ni-based methodology was further optimized for alkyl-nitriles and aliphatic dinitriles of variable chain length making use of p-toluenesulfonic acid (PTSA) as a co-catalyst.25
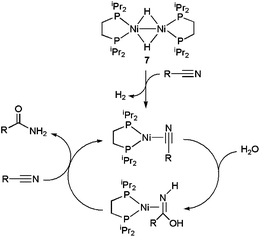 | ||
| Scheme 5 Suggested mechanism for the hydration of nitriles by the Ni dimer 7. | ||
A catalytic system of superior simplicity for the hydration of aryl, benzyl and both sterically hindered and unhindered alkyl nitriles in water, consisting of cheap, commercially available and environmentally friendly compounds (a ZnX2/ketoxime combination), was described by Kukushkin and Pombeiro.26 The nature of the X− anion in the zinc salt (X− = NO3−, Cl−, CF3SO3−) or that of the ketoxime (Me2CNOH, c-C4H8CNOH, c-C5H10CNOH) does not affect strongly the catalytic properties of the system, but the best results were obtained so far with the Zn(NO3)2·6H2O (0.7 mol%)/Me2CNOH (2.8 mol%) combination. TOF and TON values of up to 45 h−1 and 450, respectively, could be reached with this system performing the catalytic reactions in air under refluxing conditions. Interestingly, the presence of both components is imperative as the hydration process does not proceed at all with either the zinc compound or the ketoxime taken alone. Addition of the ketoxime (R2CNOH) to the ligated nitrile (R′CN), to give an imino intermediate [Zn]–NHC(R′)-ONCR2 which by hydrolysis furnishes the amide and regenerates the Zn(II)/ketoxime catalyst, was proposed to explain these experimental observations.
On the other hand, recent work has demonstrated that catalytic nitrile hydration can be facilitated by the presence of functionalized ligands able to activate the nucleophilic water molecule through a hydrogen-bond interaction (Fig. 2).27 Such a cooperative effect of the ligand represents a typical example of the so-called “bifunctional catalysis”; that is, the metal ion acts as a Lewis acid and the ligand as a Lewis base, a concept largely exploited in homogeneous catalysis during the last few years.28
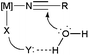
In this context, a series of arene–ruthenium(II) 829 and bis(allyl)–ruthenium(IV) complexes 9–10,30 containing “cage-like” water-soluble phosphines, have been found to operate through this bifunctional catalysis mechanism (Fig. 3). Thus, once the nitrile is coordinated to ruthenium, via dissociation of one of the chloride ligands, its hydration is favoured by H-bonding of the incoming water molecule with the nitrogen atoms of the phosphines. This fact was supported by the remarkably lower effectiveness shown by related complexes bearing the sulphonated phosphine TPPMS ((meta-sulfonatophenyl)-diphenylphosphine sodium salt), in which such an interaction cannot be established.29,30 Complexes 8–10 are all active in pure water, within the temperature regime 100–150 °C (classical or MW heating), without requirement of any acidic or basic co-catalyst. Best results in terms of activity (TOF and TON up to 127 h−1 and 100, respectively) were found with the mononuclear compounds [RuCl2(η6-C6Me6)(PTA-Bn)] and [RuCl2(η3:η3-C10H16)(THPA)], and the dinuclear one [{RuCl2(η3:η3-C10H16)}2(μ-THDP)] (10). Almost quantitative conversions of a wide variety of aromatic, heteroaromatic, α,β-unsaturated and aliphatic nitriles were observed with these systems within 1–15 h, and the reactions tolerated common functional groups such as halides, nitro, hydroxy, ethers, thioethers, amino, ketones, aldehyde, esters or alkynes. After crystallization of the amide, recycling of the aqueous phase containing the active species was also demonstrated for [RuCl2(η6-C6Me6)(PTA-Bn)].29,31
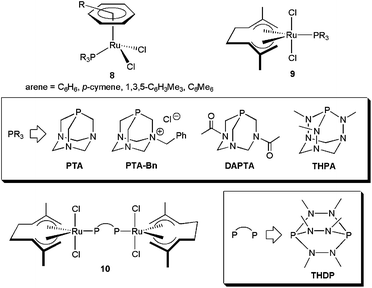
A superior recycling (up to 7 consecutive runs) after amide crystallization was observed with the octahedral ruthenium(II) derivative [RuCl2(PTA)4].32 TOF and TON values of up to 30 h−1 and 22000, respectively, could be reached with this complex performing the catalytic reactions at 100 °C. Other commendable aspects of [RuCl2(PTA)4] are its tolerance to the presence of air (no inert atmosphere required) and its high functional group compatibility.
The Ru(II) complexes [RuCl2(η6-arene){P(NMe2)3}] (arene = C6H6, p-cymene, 1,3,5-C6H3Me3, C6Me6), incorporating the commercially available, inexpensive and H-bond accepting ligand tris(dimethylamino)phosphine, have also shown to be highly effective for nitrile hydration in pure water under neutral conditions.33 Within this family of catalysts, best results were obtained with the hexamethylbenzene derivative [RuCl2(η6-C6Me6){P(NMe2)3}] 11, which selectively provided the desired amides from a wide range of organonitriles in excellent yields and short times (TOF values up to 11400 h−1 could be reached at 150 °C under MW irradiation). Taking advantage of the remarkable activity of this catalyst, an efficient and practical synthesis of the non-steroidal anti-inflammatory drug ibuprofenamide 13 by catalytic hydration of 2-(4-isobutylphenyl)propionitrile 12 could be developed (Scheme 6). In addition, complex 11 proved to be also effective for the one-pot conversion of δ-ketonitrile 14 into the isomeric ene-lactams 15 and 16, via a tandem hydration/condensation sequence.34 The only drawback of these methodologies is the impossibility to recycle 11 due to its progressive decomposition into the less active dimethylamine–ruthenium(II) complex [RuCl2(η6-C6Me6)(NHMe2)] by hydrolysis of the coordinated P(NMe2)3 ligand.33b
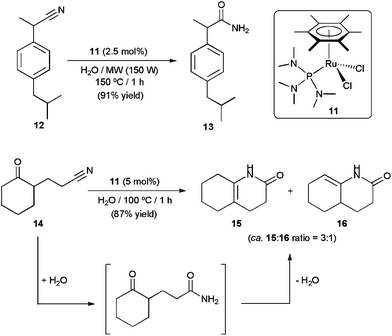 | ||
| Scheme 6 Catalytic synthesis of ibuprofenamide and ene-lactams using complex 11. | ||
Further studies revealed an unprecedented reactivity of [RuCl2(η6-p-cymene){P(NMe2)3}] in the challenging hydration of cyanohydrins to the corresponding α-hydroxyamides.35 Thus, running the catalytic reactions within the pH range 3.5–8.5 to avoid the decomposition of the substrates into the corresponding aldehydes and HCN, complete conversion of glycolonitrile (R = H) and lactonitrile (R = Me) could be achieved at room temperature (Scheme 7). Complex [RuCl2(η6-p-cymene){P(NMe2)3}] was also able to hydrate acetone cyanohydrin, but a lower conversion (15%) to the amide product was in this case observed. Overall, these results represent a benchmark for future studies on cyanohydrins hydration using metallic catalysts.
![Cyanohydrins hydration with complex [RuCl2(η6-p-cymene){P(NMe2)3}].](/image/article/2013/GC/c2gc36534k/c2gc36534k-s7.gif) | ||
| Scheme 7 Cyanohydrins hydration with complex [RuCl2(η6-p-cymene){P(NMe2)3}]. | ||
In the search for cooperative effects of the ligands, other arene–ruthenium(II) complexes with potentially H-bond accepting amino-phosphines36 and pyridyl-phosphines37 were also investigated (Fig. 4). However, in contrast to the previous examples, they showed only modest activities. The results obtained with the amino-phosphine derivatives indicated that the pendant amino group of the ligands acts as a Brønsted base, generating the real nucleophile of the hydration process, i.e. the OH− group. In the case of the pyridyl-phosphine derivatives, bifunctional pathways during the catalytic events were totally ruled out. In fact, the high tendency of PPh2py and PPh2(py-4-NMe2) to adopt a κ2-P,N-chelating coordination results in the formation of very low active species, while in the case of PPh2(py-6-tert-amyl) the presence of the bulky tert-amyl substituent seems to prevent the approach of the substrate to the Lewis acid metal center, leading also to poor results in catalysis.38
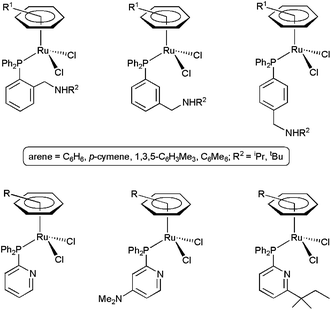 | ||
| Fig. 4 Structure of arene–ruthenium(II) catalysts containing amino-phosphine and pyridyl-phosphine ligands. | ||
Quite recently, the cuprous complex [Cu4(μ3-I)(H2O)4] 17 (2.5 mol%) has been successfully applied in the selective hydration of several benzonitriles, cinnamonitriles and arylacetonitriles in pure water at 100 °C (yields up to 98% after 21 h).39 After completion of the reactions, the mixtures were cooled to ca. 5 °C and crystals of the amides selectively appeared. This fact allowed, once again, the easy separation of the products by simple filtration, and also the recycling of the aqueous solution containing the catalyst (up to 5 consecutive runs). In addition, using a combination of complex 17 and the C–N coupling Cu(II) catalyst 18, 2-oxotetrahydroindole could be prepared in good yields from 2-iodophenylacetonitrile through the one-pot domino protocol depicted in Scheme 8.
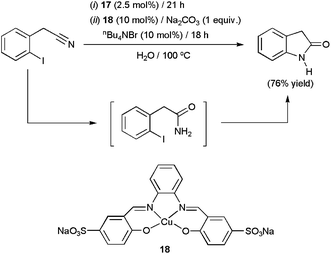 | ||
| Scheme 8 Copper-catalyzed one-pot synthesis of 2-oxotetrahydroindole in water. | ||
In addition to the above-discussed systems, other examples of transition-metal complexes operative in nitrile hydration processes, without the requirement of an organic co-solvent, reported in the literature include: (i) the hydroxo-palladium(II) and -rhodium(I) derivatives [PdCl(OH)(bipy)] (bipy = 2,2′-bipyridine)40 and trans-[Rh(OH)(CO)(PPh3)2],41 (ii) the hydride-iridium(I) and ruthenium(II) complexes [IrH(CO)(TPPTS)3] (TPPTS = P(m-C6H4SO3Na)3·xH2O)42 and [RuH(η5-C9H7)(dppm)] (C9H7 = indenyl; dppm = bis(diphenylphosphino)methane),43 (iii) the recyclable (up to five cycles) Rh(I) system [{Rh(μ-Cl)(cod)}2]/TPPTS (cod = 1,5-cyclooctadiene),44 (iv) the peroxo-iridium(III) derivative [Ir(O2)(PMe3)4][Cl],45 (v) the osmium(II) complex [{OsCl(μ-Cl)(CO)3}2],46 and (vi) the dicationic octahedral ruthenium(II) derivative [Ru(H2O)(NCMe)4(PiPr3)][BF4]2.47 However, they were in general less active and/or selective than the examples discussed above, and their scope was not always demonstrated. In line with this latter point, several Os(II) and Ru(II) complexes have been exclusively studied in the catalytic hydration of chloroacetonitriles (Cl3−nCHnCN) into the corresponding chloroacetamides (Cl3−nCHnC(O)NH2), a particular class of compounds that exhibit a wide range of biological properties and are widely used as building blocks in preparative organic chemistry.48 Among them, the best results were obtained with the osmium(II) derivative cis,fac-[OsCl2(Hbzim)(dmso)3] (Hbzim = benzimidazole) which reached TON values up to 412 for trichloroacetamide (n = 0) and 578 for dichloroacetamide (n = 1) at 75 °C.48a
On the other hand, over the last decade, the use of nanoparticles (NPs) in catalysis has expanded considerably and has led to many interesting applications.49 The nano-sized particles increase the exposed surface area of the active component of the catalyst, thereby enhancing the contact between reactants and catalyst dramatically and mimicking the homogeneous systems. However, their insolubility in reaction solvents renders them easily separable from the reaction mixture like heterogeneous catalysts, which in turn minimizes the effort of the product isolation stage. This bridge between homogeneous and heterogeneous catalysis tended by NPs gives unique opportunities towards the development of efficient reactions under environmentally benign conditions. In this context, colloidal dispersions of Cu/Pd bimetallic nanoclusters proved to be selective and recyclable catalysts for the hydration of acrylonitrile to acrylamide in pure water at 80 °C (up to 95% yield after 8 h), showing much higher activities than those of pure Cu colloids and other Cu-based systems.50,51 PVP-stabilized Pd nanoparticles (PVP = poly(N-vinyl-2-pyrrolidone)) in combination with Cu(acac)2 or CuO acted also as catalysts for the hydration of various nitriles without requiring the preparation of an alloy.52 As an example, more than 99% yield on benzonitrile hydration was achieved with the Pd NP–Cu(acac)2 system (5 mol% of Pd and 10 mol% of Cu) at 180 °C for 16 h.
Studies by Kaneda53a and Park53b have demonstrated the utility of hydroxyapatite-supported and PVP-stabilized silver NPs, respectively, as recyclable catalysts for the selective hydration of nitriles in pure water. Reactions performed with Ag loadings of 0.3–3 mol% delivered the desired amides in high yields (>80%) after 1–6 h of heating at 140–180 °C when starting from aromatic or heteroaromatic nitriles, but proved to be much less effective with aliphatic substrates. The Ag NPs were in both cases readily separated by centrifugation and could be reused 4–5 times without loss of their catalytic activity and selectivity. A mechanism of action involving the initial coordination of water and the nitrile on the silver NP surface was proposed. Subsequently, an OH− anion generated from a proximal silver-coordinated H2O molecule attacks the nitrile carbon atom to form the corresponding amide through an iminol intermediate.53a Further recent studies by Shimizu and co-workers using SiO2-supported Ag NPs pointed out the key role played by the oxygen atoms adsorbed on the silver surface, which seem to act as Brønsted bases facilitating the dissociation of water and the generation of the nucleophilic OH− (Scheme 9).54 Notably, unlike the previous examples, the less reactive aliphatic nitriles could be effectively hydrated with this new Ag-based system (70–90% yield after 24–48 h of heating at 160 °C with a silver loading of 9 mol%).
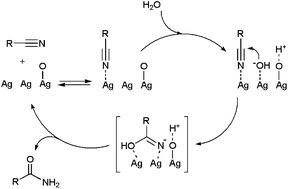 | ||
| Scheme 9 Proposed mechanism for the Ag/SiO2-catalyzed hydration of nitriles. | ||
Gold NPs supported on TiO2 proved to be more efficient and general than the Ag-based systems described above.55 For example, an impressive TOF value of 25000 h−1 was reached in the hydration of pyrazinecarbonitrile to produce pyrazinecarboxamide, an antibacterial agent used for the treatment of tuberculosis (Scheme 10). The reaction cleanly took place in neat water, under air, with a gold loading of only 0.001 mol%. In addition, after recycling the catalyst 10 times by simple filtration, an unprecedented cumulative TON of 1000000 could be realized. The high-yield synthesis of ε-caprolactam by hydration/cyclization of 6-aminocapronitrile is another example of the synthetic usefulness of this heterogeneous system (Scheme 10).55,56
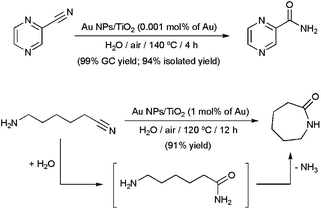 | ||
| Scheme 10 Catalytic synthesis of pyrazinecarboxamide and ε-caprolactam in water using Au NPs supported on TiO2. | ||
Nanoferrites-supported ruthenium hydroxide 19 (0.003 mol% of Ru)57 and the bifunctional arene–ruthenium(II) complex 20 (1.58 mol% of Ru)58 have also been employed to promote nitrile hydration reactions in pure water under neutral conditions, using microwave irradiation as the heating source (Fig. 5).
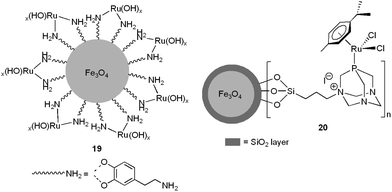 | ||
| Fig. 5 Structure of the nano-catalysts 19 and 20. | ||
Both nano-catalysts showed excellent activities (TOFs up to 30000 h−1) and selectivities for a broad range of aromatic, heteroaromatic, aliphatic and α,β-unsaturated nitriles, leading to the desired primary amides in high yields (70–95%) after 0.5–7 h of MW irradiation at 130–150 °C. Several functional groups, i.e. halide, ether, thioether, amino, nitro, ketone, aldehyde, ester or alkyne, were tolerated and no overhydrolysis to carboxylic acids was observed. Noteworthily, the selective mono and dihydration of dicyano derivatives could also be conveniently achieved using 20 just by controlling the time of MW irradiation (representative examples are given in Scheme 11).58 After completion of the hydration reactions, the magnetic nanocatalysts 19–20 could be easily separated from the reaction mixtures with the help of an external magnet and recycled up to three (19) or six (20) times. In addition, after separation of the nanoparticles, the aqueous solutions were cooled and crystals of the amides with acceptable purity precipitated, thus avoiding the use of organic solvents also in the work-up steps. Overall, the use of relatively low metal concentrations, environmentally friendly water as the reaction medium (with no use of single drop of organic solvent during or after the reactions) and low-energy-consuming microwave heating, along with an easy recycling procedure, makes these protocols “truly” green and sustainable. In a related recent study, the utility of ruthenium hydroxide nanoparticles immobilized on magnetic silica (Fe3O4@SiO2) for aqueous hydration of nitriles under neutral conditions was also demonstrated.59
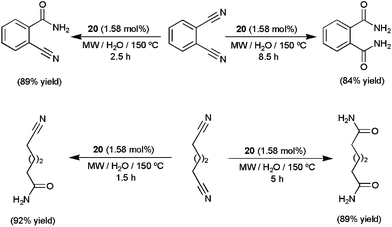 | ||
| Scheme 11 Selective hydration of dinitriles in water using nano-catalyst 20. | ||
In addition to the discussed nanocatalysts, several insoluble metal oxides have been explored as potential heterogeneous catalysts for the hydration of CN bonds in an aqueous medium.60 In general, they exhibited poor activities and, in some cases, low selectivities towards the desired amides. The most relevant results in this field are: (i) the selective generation of nicotinamide from aqueous 3-cyanopyridine over MnO2 (>99% yield after 8 h of heating at 100 °C using 13 mol% of MnO2),60e (ii) the selective hydration of different aromatic and heteroaromatic nitriles by means of the recyclable spinel cobalt oxide Co3O4 (75–99% yield after 7–24 h of heating at 140 °C using 17 mol% of Co3O4),60f and (iii) the remarkable substrate-specificity shown by CeO2.60g This latter oxide proved to be only effective for the hydration of nitriles that contain a heteroatom (N or O) adjacent to the α carbon of the CN group. Such substrates were cleanly converted into the corresponding amides even at 30 °C (as representative examples, the hydrations of 2-cyanopyridine and pyrazinecarbonitrile at this remarkably low temperature are given in Scheme 12). A mechanism involving initial H2O dissociation on CeO2, subsequent adsorption of the nitrile on the solid surface, and final nucleophilic attack of a hydroxyl species on the adsorbed nitrile was proposed, with the latter being the rate-limiting step of the process as determined by kinetic analyses. Favoured adsorption nitriles containing heteroatoms adjacent to the α carbon of the CN group account for the substrate-specificity observed.
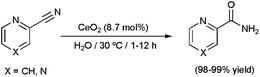 | ||
| Scheme 12 Hydration of 2-cyanopyridine and pyrazinecarboxamide with CeO2. | ||
Ruthenium hydroxide supported on alumina (Ru(OH)x/Al2O3),61 ruthenium-substituted hydroxyapatite ((RuCl)2Ca8(PO4)6(OH)2)62 and a Nafion-Ru solid resin63 proved to be also useful heterogeneous and recyclable catalysts for the hydration of various kinds of nitriles in water at 140–175 °C. Worthy of note, using Ru(OH)x/Al2O3, a totally organic solvent free process was developed since the solid catalyst can be easily separated from the reaction mixture by hot filtration at 90 °C, and the amides crystallize in a pure form from the filtrate upon cooling at 0 °C.
From aldoximes
The metal-catalyzed rearrangement of aldoximes represents an alternative atom efficient method for forming primary amides (Scheme 13(a)). Despite the similitude of this process with the classical Beckmann rearrangement in which N-substituted amides are generated by the acid-catalyzed rearrangement of ketoximes (Scheme 13(b)),64 it is important to note that the reaction of aldoximes in the presence of acid catalysts usually gives the corresponding nitriles due to the propensity of the H atom to act as a leaving group. In fact, the synthesis of primary amides from aldoximes is a specially exigent reaction that traditionally requires harsh conditions.65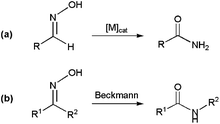
In recent years, several Ni-,66 Pd-,66a,b,67 Cu-,68 Au-,69 Ru-,70 Rh-,71 Ir-,72 Zn-73 and In-based73b systems able to promote efficiently this atom-economical transformation in organic media have been developed, thus expanding its synthetic potential. From a mechanistic point of view, two different reaction pathways have been proposed. The first one involves the initial dehydration of the aldoxime into a nitrile intermediate,74 which is subsequently hydrated by the water released in the previous step to generate the final amide (path (a) in Scheme 14).
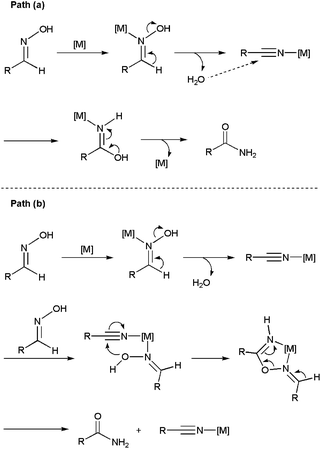
In the second one, metal-promoted dehydration of the aldoxime to form a nitrile also takes place initially, but the nitrile now evolves into the final amide with the aid of a second molecule of aldoxime which acts as a water surrogate (path (b) in Scheme 14).75 Thus, intramolecular attack on the nitrile by a coordinate aldoxime leads to a five-membered cyclic intermediate, which decomposes into the final amide product and another coordinated nitrile which continues with the catalytic cycle. Remarkably, despite the dehydration/rehydration pathway involving water (a) has been the generally accepted mechanism for the rearrangement of aldoximes to amides, a recent study by Williams and co-workers using 18O labelled substrates has pointed out that most of the homogeneous catalysts described so far in the literature for this reaction really operate through the pathway (b).76
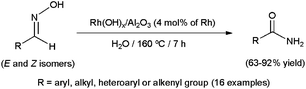
To date, little attention has been paid to this rearrangement in aqueous media. In fact, the first approach was made by Mizuno and co-workers only in 2007 employing supported rhodium hydroxide (Rh(OH)x/Al2O3) as catalyst (Scheme 15).77 The reactions, performed in pure water with a rhodium loading of 4 mol%, delivered the desired amides in high yields after 7 h of heating at 160 °C. Both aromatic, heteroaromatic, aliphatic and α,β-unsaturated aldoximes were tolerated, and the heterogeneous catalyst could be recovered by filtration and reused with retention of its activity. Remarkably, in contrast to the transformations in water, nitriles were formed as the major products when the same reactions were performed in common organic solvents. This fact, along with the ability of Rh(OH)x/Al2O3 to catalyze the hydration of nitriles under the same experimental conditions, seems to indicate that this heterogeneous system operates through pathway (a) in which the nitrile intermediates are hydrated by water (Scheme 14).
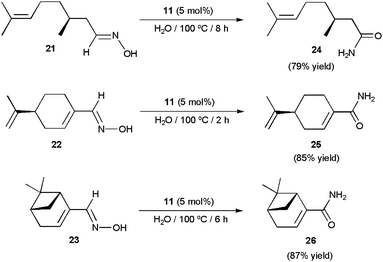
More recently, the arene–ruthenium(II) complex [RuCl2(η6-C6Me6){P(NMe2)3}] 11 (5 mol%) proved to be also effective in water.78 The reactions proceeded cleanly at 100 °C without the assistance of any co-catalyst, affording the desired amides in high yields (70–90%) after short reaction periods (1–7 h). Again, the process was operative with both aromatic, heteroaromatic, α,β-unsaturated and aliphatic aldoximes, and tolerated several functional groups. The synthetic utility of this homogeneous system was fully demonstrated in the preparation of the chiral amides 24–26 (Scheme 16), which were isolated in high yields by rearrangement of the optically pure aldoximes 21–23 derived from the naturally occurring aldehydes (S)-(−)-citronellal, (S)-(−)-perillaldehyde and (1R)-(−)-myrtenal, respectively. Reaction profiles, kinetic analyses, and experiments using 18O-labelled water supported in this case the involvement of both dehydration/rehydration pathways depicted in Scheme 14, with that involving the hydration of the corresponding nitrile intermediates by a second molecule of aldoxime (b) being predominant.
From aldehydes
Aldoximes are generally synthesized by condensation of aldehydes with hydroxylamine. Consequently, significant efforts have been devoted in recent years to the development of one-pot processes enabling the direct formation of primary amides from aldehydes and hydroxylamine derivatives, via rearrangement of the in situ formed aldoximes (Scheme 17). This protocol is an attractive alternative to the more classical oxidative amidations of aldehydes (see below).79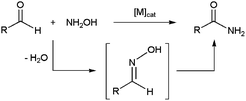 | ||
| Scheme 17 Catalytic synthesis of primary amides from aldehydes and hydroxylamine. | ||
Several catalytic systems based on copper,68c,80 scandium,81 zinc,73b iron,82 ruthenium,70b,c,83 rhodium,77 iridium,72 palladium,84 and indium73b compounds able to effect efficiently this sequential transformation have already been discovered, some of them operating in aqueous media. In particular, using catalytic amounts of Rh(OH)x/Al2O3 (4 mol% of Rh)77 and FeCl3 (5 mol%),82 a large variety of aromatic, heteroaromatic, α,β-unsaturated and aliphatic aldehydes were converted into the corresponding primary amides in high yields performing the reactions in pure water at 100–160 °C for 6–32 h with the hydroxylammonium salts (NH2OH)2·H2SO4 or NH2OH·HCl. Remarkably, while heterogeneous Rh(OH)x/Al2O3 could be reused without a significant loss of its activity, the homogeneous one FeCl3 proved to be useful for the preparation of biologically relevant compounds, such as the β-D-ribofuranose derivatives 27 and 28 (Scheme 18).
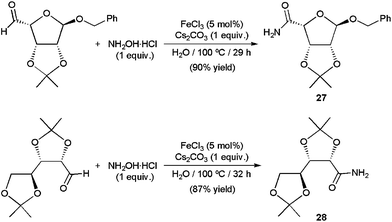 | ||
| Scheme 18 FeCl3-catalyzed synthesis of amides 27 and 28. | ||
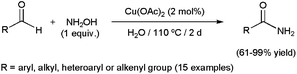
Faster transformations of any type of aldehydes were described by Singh and co-workers employing scandium(III) triflate (10 mol%) under controlled MW irradiation (300 W).81 The reactions, performed in water at 135 °C with stoichiometric amounts of NH2OH·HCl and Na2CO3, selectively delivered the desired amides in excellent yields (82–95%) after very short irradiation periods (15–35 min). More recently, a protocol involving recyclable (up to ten times) copper(II) acetate and hydroxylamine has been described by Ramón and co-workers (Scheme 19).68c The direct use of hydroxylamine instead of hydroxylammonium salts avoids in this case the introduction of a base in the reaction medium to catch the acid released during the generation of the aldoxime, thus minimizing the generation of waste. However, the extremely long reaction times required (2 days) may be a drawback for further applications of this catalytic system.
From amines
Aerobic oxidation of amines is an emerging research field that offers great opportunities for the sustainable production of a large variety of key nitrogen-containing compounds.85 With regard to the synthesis of primary amides, the direct oxygenation of α-methylene groups in primary RCH2NH2 amines is a highly desirable reaction since the latter are readily available and inexpensive starting materials. However, it is very difficult to oxygenate primary amines, and highly reactive stoichiometric reagents are usually required.86 In addition, the significantly higher reactivity of the amine functionality over the methylene α-carbon necessitates in some cases of functional-group protection, with the consequent decrease in atom efficiency.86c Recent research in this direction has brought to light a promising alternative route for the direct catalytic conversion of primary amines to primary amides through the oxidative dehydrogenation/hydration sequence depicted in Scheme 20, in which hydration of an in situ formed nitrile takes place.62,87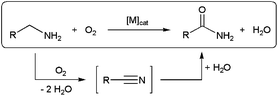 | ||
| Scheme 20 Oxidative hydration of primary amines to amides via nitrile intermediates. | ||
The number of catalytic systems capable of carrying out this reaction is still very limited and organic co-solvents or solvent-free conditions are required in most cases.87 However, given the unique ability of Ru(OH)x/Al2O3 to selectively oxidize amines to nitriles,88 various kinds of primary amines could be effectively converted into the corresponding amides performing the catalytic reactions in pure water at 140 °C, and using air (5 atm) as the sole oxidant (77–92% yield after 10–24 h using a ruthenium loading of 5 mol%).87a
From azides
Although they are generally considered hazardous, azides are attractive functional groups in synthesis because of their enhanced reactivity (dipolar character) and easy introduction into organic molecules by many convenient methods, such as the simple SN2 displacement of alkyl halides with NaN3. The Staudinger and Schmidt reactions, the Curtius rearrangement and the “click” synthesis of triazoles are the most popular applications of these relevant molecules.89 More contemporary developments revealed that azides that contain α-hydrogens are useful precursors for oxidation to the corresponding nitriles.90 In this context, Mizuno and co-workers have described the aerobic oxidation of a wide range of azides by means of the supported ruthenium hydroxide catalyst Ru(OH)x/Al2O3 (Scheme 21).91 In addition, they also demonstrated that primary amides can be generated in high yields by performing the catalytic transformations in water, via Ru-promoted hydration of the initially formed nitriles (Scheme 21). Although only a couple of examples were shown, this unprecedented sequential transformation represents a new synthetic tool for amide-bond formation which makes use of readily available starting materials.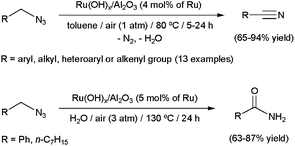 | ||
| Scheme 21 Aerobic oxidative transformations of primary azides. | ||
From alcohols
Since the sound work of Milstein and co-workers in 2007 using PNN pincer-type ruthenium complexes,92 transition-metal catalyzed direct amide synthesis from primary alcohols and amines has become a major field within current research.93 Although most examples described to date are focused on the preparation of secondary amides (see below), some catalytic systems have also proven effective for primary amide formation using ammonia.94 In this context, employing MnO2 nanorods supported on graphene oxide (GO), Hou and co-workers were able to convert several primary alcohols to the corresponding primary amides employing aqueous ammonia (Scheme 22).94f Contrary to other catalytic systems, the reactions proceeded cleanly in the absence of any organic co-solvent. In addition, the heterogeneous catalyst showed good recyclability (up to five cycles). A reaction pathway involving the initial oxidation of the alcohol to the corresponding aldehyde, via Mn(IV)/Mn(II) reduction (O2 is needed to regenerate the active Mn(IV)), followed by its reaction with NH3 to form a hemiaminal that is subsequently dehydrogenated to the amide, was proposed. In complete accord with this, MnO2/GO proved to be also effective for primary amide formation starting from aldehydes and aqueous ammonia.94f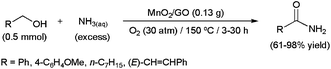 | ||
| Scheme 22 MnO2-catalyzed synthesis of primary amides from primary alcohols and aqueous ammonia. | ||
Organic solvent-free amidation of benzyl alcohol with aqueous ammonia by virtue of water-soluble DNA-stabilized gold nanoparticles has also been described.94c
From methylarenes
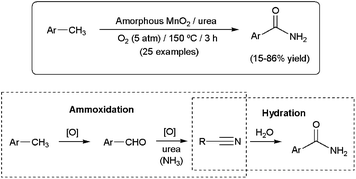
Quite recently, an unprecedented aerobic oxidative amidation reaction of methylarenes to primary amides in the presence of amorphous MnO2 has been described (Scheme 23).95 Although the best results were obtained using urea as the nitrogen source, aqueous ammonia proved to be also effective for the present MnO2-catalyzed amidation. The process is believed to proceed through the initial ammoxidation of the methylarenes to generate the corresponding nitriles (via aldehydes), followed by hydration of the CN bond.
 | ||
| Scheme 23 Aerobic oxidative amidation of methylarenes promoted by amorphous MnO2. | ||
Catalytic synthesis of secondary and tertiary amides in water
Catalytic reactions leading to the formation of N-substituted amides in water have been comparatively much less studied. However, some synthetic approaches starting from nitriles, aryl halides, aldehydes, alcohols or alkynes are currently known, and they will be presented in this section.From nitriles
In addition to the well-known hydration reactions, which provide an easy access to primary amides (see above), different methodologies leading to secondary and tertiary amides have also been developed,6 although they still remain little exploited. In this context, the Ritter reaction, based on the coupling of a nitrile with a carbocation (generated in situ from alcohols, halides or olefins) and further hydrolysis, offers an attractive entry to secondary amides (Scheme 24).96 Since the stability of carbocations increases with the number of substituents, this procedure is especially useful for the preparation of bulky amides.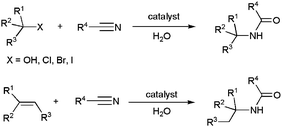 | ||
| Scheme 24 Formation of secondary amides through Ritter-type reactions. | ||
Traditionally promoted by stoichiometric amounts of a strong Brønsted acid (typically H2SO4), the process can also be catalyzed by Lewis acids under more environmentally friendly conditions.96,97 In particular, some metallic salts showed to be suitable for performing the reaction in water. The first example reported was based on the coupling of alcohols with nitriles using 20 mol% of Bi(OTf)3 (Scheme 25).98 A wide range of aliphatic, aromatic and heteroaromatic nitriles with different substitution patterns was tolerated, giving rise in general to high yields of the corresponding amides. However, the process seems to be restricted to tertiary alcohols, such as tert-butanol, 2-methylbutan-2-ol, adamantol, 2-methyl-1-phenylpropan-2-ol and 1-methylcyclopentanol, in line with their propensity to generate carbocations. Interestingly, tert-butyl methyl ether could also be used as a carbocation precursor in this reaction, via ether bond cleavage. Although most of the reactions were carried out in pure water, in the case of poorly soluble solid nitriles, the addition of nitrobenzene as a co-solvent was required to achieve high conversions. It is also worth noting that the use of 20 mol% TfOH instead of Bi(OTf)3 resulted in similar yields of the amides. This fact suggests that Bi(OTf)3 generates TfOH under the reaction conditions employed, and that this is probably the effective catalyst involved in these transformations.
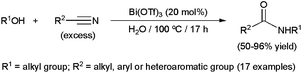 | ||
| Scheme 25 Bi(OTf)3-promoted Ritter reactions in water. | ||
Related Ritter reactions between different benzonitriles and alcohols were successfully performed in water by Firouzabadi and Iranpoor using 5 mol% of the tungsten-oxoacid H3PW12O40.99 As in the precedent case, the transformation could be extended to protected alcohols (methyl-, trimethylsilyl- or tetrahydropyranyl-ethers) containing a tertiary alkyl or benzyl substituent subject to easy generation of a carbocation. This tungsten compound, and its molybdenum analogue, were also applied in the synthesis of N-bornyl amides through Ritter-type reactions of camphene with nitriles. However, in these cases, high catalyst loadings (50 mol%) were necessary to achieve only modest conversions.100
A more general copper-catalyzed procedure has been recently developed by Qu and co-workers employing halides and nitriles (Scheme 26).101 In this case, not only tertiary alkyl substituents, but also primary and secondary ones, as well as allyl and benzyl groups, could be introduced on the nitrogen atom of the resulting amides. Among the different copper precursors evaluated, Cu(OTf)2 showed the best catalytic performances, leading to the desired amides in high yields using a metal loading of only 5 mol%.
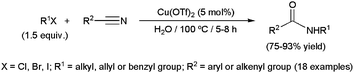
Efficient couplings between 1-bromoadamantane and various alkyl-, alkenyl- or aryl-nitriles in water have also been described using the acetylacetonate derivative [Mn(acac)3] (3 mol%).102 High selectivities in the corresponding N-(adamantan-1-yl)amides were reached at 130 °C in short reaction times (75–100% yield after 1–5 h). In contrast, the formation of adamantan-1-ol, by hydrolysis of the initial 1-bromoadamantane, became predominant at a lower temperature (70 °C). This alcohol is not an intermediate in the coupling process, since it was recovered unchanged after heating, even under harsh conditions (175 °C for 45 h), in the presence of the manganese(III) precursor and nitriles. Only when [Mn(acac)3] was associated with one equivalent of HBr, Ritter reactions between adamantan-1-ol and nitriles took place.
Metal-catalyzed hydrolytic amidation with amines represents an alternative synthetic method to access N-substituted amides starting from nitriles (Scheme 27). The process, first developed by Murahashi and co-workers,34c,103 consists of the reaction between a nitrile, an amine and one equivalent of water to generate the corresponding amide along with ammonia. Although still little studied, this methodology has already shown to be applicable to a wide range of aliphatic and aromatic substrates, including both primary and secondary amines, and was also used in the synthesis of different alkaloids,103 biologically active molecules104 and polyamides.103
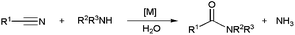
Most of these catalytic processes, promoted by ruthenium-,34c,103 platinum-105 or iron-based104 homogeneous systems, have been carried out in wet organic solvents. However a few examples, recently reported by Shimizu and co-workers, were performed in pure water using heterogeneous catalysts.106 In particular, they explored the activity and selectivity of different metal oxides in the coupling of heteroaromatic or aliphatic nitriles with primary amines, CeO2 showing the best performances (Scheme 28). The reaction also proceeded efficiently with a secondary amine, such as morpholine. Interestingly, the insoluble catalyst could be separated by centrifugation and, after calcination at 300 °C, reused in at least three further runs.
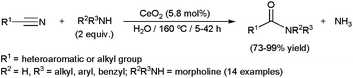
Monitoring the course of the reactions, the authors evidenced that these hydrolytic amidations take place through the rapid hydration of the nitrile and further amination of the resulting primary amide, which furnishes the desired N-substituted amide with concomitant release of NH3. In complete accord with these observations, Duchateau and co-workers also demonstrated that the hydrolytic amidation of pentanenitrile with n-hexylamine promoted by the ruthenium complex [RuH2(PPh3)4] or the heterogeneous catalyst ZrO2 in water proceeds through the initial formation of the primary amide which either directly, or after hydrolysis to the corresponding carboxylic acid, reacts with the amine to give the N-hexylpentamide product.107,108 These reaction pathways contrast with those generally proposed for processes realized in wet organic solvents where an amidine intermediate, which evolves to the final product by hydrolysis, is initially formed.104,105
On the other hand, zirconia oxide was found to be a promising catalyst for the selective formation of the polyamide nylon-6 from 6-aminocapronitrile (H2N–(CH2)5–CN), via intermolecular hydrolytic amidation.108 However, although important advances have been realized in the field, the results obtained are still unsatisfactory since the presence of water required for the process prevented the build-up of the molecular weight. Formation of ε-caprolactam using Au NPs supported on TiO2, through the intramolecular hydrolytic amidation of 6-aminocapronitrile (Scheme 10), has also been described.55
From aryl halides
Palladium-catalyzed aminocarbonylation of aryl halides has emerged in recent years as a powerful synthetic tool to generate secondary and tertiary amides. This three-component process, which involves the coupling between an aryl halide, a primary or secondary amine and CO, has been extensively studied in organic media with excellent results in terms of catalyst efficiency, selectivity and substrate scope.109 In contrast, the development of such reactions in aqueous media has received much less attention. This is most likely due to the occurrence of competing hydroxycarbonylation of the aryl halides, in which water instead of the amine acts as a nucleophile, giving rise to the formation of undesirable carboxylic acids instead of the amides.110 The first report on the amidation of aryl halides in water appeared in 2005.111 Under optimal conditions (i.e. 5 mol% of the palladacycle complex 29, microwave irradiation (170 °C), 1 equiv. of amine, 2 equiv. of aryl bromide, 3 equiv. of Na2CO3 and 0.5 equiv. of Mo(CO)6 as the CO source), the hydroxycarbonylation could be minimized and the expected amides were generated in moderate to good yields, with only small amounts of the carboxylic acid by-products (Scheme 29). This protocol proved to be effective with a wide range of aryl bromides, bearing both electron-donating or -withdrawing functionalities, and primary or secondary aliphatic amines. However, we must note that highly sterically demanding amines, such as di-n-butylamine, dramatically affected the conversion.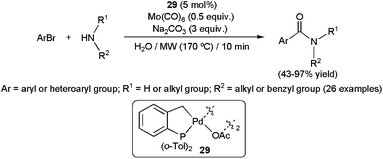 | ||
| Scheme 29 The first examples of aminocarbonylations of aryl halides in water. | ||
Remarkably, the palladacycle 29 was also operative in the aminocarbonylation of aryl chlorides,112 usually poorly reactive.109,113 Nevertheless, longer reaction times (30 min of MW irradiation at 170 °C) and the addition of [tBu3PH][BF4] to the medium were in these cases mandatory to reach acceptable conversions. Under the basic conditions employed, the phosphonium salt liberates tributylphosphine which presumably coordinates onto the metallic center, leading to a more catalytically active Pd-species.114 Remarkably, this methodology was applied to the synthesis of compound 30, a HIV-1 protease inhibitor, albeit only a modest yield could be achieved (Scheme 30).112
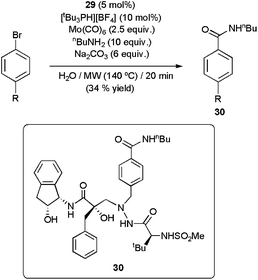 | ||
| Scheme 30 Synthesis of the HIV-1 protease inhibitor 30 by aminocarbonylation. | ||
In line with the relative C–X bond energies, amidation of aryl iodides results by far easier than that of bromide or chloride derivatives. Thus, even phosphine-free catalytic systems, known to be less effective,109 are suitable for promoting the transformation of ArI substrates. Accordingly, aromatic or heteroaromatic iodides were smoothly converted into the corresponding secondary amides, in water under a CO atmosphere (100 psi), using the commercially available palladium catalyst Pd(OAc)2 (Scheme 31).115 A low metal loading (0.5 mol%) and relatively smooth conditions (100 °C) were enough to attain high conversions. In addition, not only aliphatic amines, but also less nucleophilic anilines, with different electron-donating or -withdrawing substituents, including the highly hindered ortho-substituted ones, were tolerated. Worthy of note, the catalytic performances attained in an aqueous medium exceeded those observed in polar or apolar organic solvents, as well as those under solvent-free conditions. Similar transformations of aryl iodides into the corresponding secondary or tertiary amides promoted by Pd(OAc)2 have also been described using [Mo(CO)6] as the CO source under MW heating.112
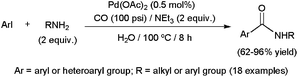 | ||
| Scheme 31 Pd(OAc)2-promoted aminocarbonylation of aryl iodides in water. | ||
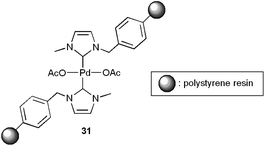
Very recently, Bhanage and co-workers have reported that the heterogeneous system 31, based on a polystyrene-supported NHC–palladium complex (Fig. 6), associated with Na2CO3 features a similar reactivity to homogeneous Pd(OAc)2, leading also to efficient couplings between aryl iodides, primary and secondary aliphatic or aromatic amines and gaseous CO.116 High selectivities in the desired amides were attained, albeit small amounts of dehalogenation products of the aryl iodides were in all cases detected. This heterogeneous catalyst could be easily recycled by simple filtration without loss of activity and selectivity (up to four consecutive runs).
From aldehydes
In 1966, Nagakawa and co-workers discovered that aromatic aldehydes can be directly converted into primary amides in the presence of ammonia and stoichiometric amounts of nickel peroxide.117 Since this pioneering work, this oxidative amidation of aldehydes has been successfully extended to primary and secondary amines to furnish the corresponding N-substituted amides,79 and the process could be performed under catalytic conditions in the presence of only small amounts of ruthenium,118 palladium,119 rhodium120 or lanthanide121 derivatives. In most of the cases, the transformation was assumed to take place through a sequential two-step mechanism involving the initial coupling between the aldehyde and the amine, followed by the subsequent oxidation of the resulting hemiaminal via formal H2 release (Scheme 32). Accordingly, stoichiometric or excess amounts of an oxidant, such as aryl bromides, hydrogen peroxide, amine N-oxides, hypervalent iodine compounds, etc., are necessary to achieve good conversions. In some cases, the own aldehyde can act as the hydrogen acceptor and, therefore, three-fold excess with respect to the amine must be employed.121 Formation of the desired amides is in these cases accompanied by the generation of alcohol and ester byproducts.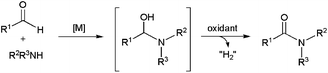
Although amidation reactions of aldehydes often require strict moisture-free conditions,120a,121 with some catalytic systems the addition of a small quantity of water resulted crucial to reach high selectivities towards the amides.118a,122,123 Despite this, examples of this reaction in a pure aqueous medium are extremely limited. In this context, Wong and co-workers reported recently the coupling of several cyclic diamines with the oligosaccharide derivative D-raffinose aldehyde 32 in water (Scheme 33).122 The process, promoted by KAuCl4 (50 mol%), delivered the corresponding tertiary amides 33 in good to excellent yields under smooth conditions (40 °C), demonstrating a high functional group tolerance. Using a 1:1 mixture of acetonitrile and water as the reaction medium, this catalytic system was also operative with only 1–10 mol% of KAuCl4 for a wide range of aromatic and aliphatic aldehydes, including formaldehyde.122
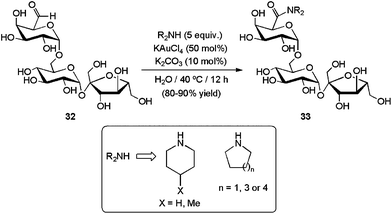 | ||
| Scheme 33 Gold-catalyzed oxidative amidation of D-raffinose aldehyde 32. | ||
Copper(I) iodide, combined with the additive AgIO3 and a stoichiometric amount of the oxidant tert-butyl hydroperoxide (TBHP), was also found to be suitable to promote the amidation of benzaldehyde in water (Scheme 34).123 The primary amines involved in the process were in this case generated in situ from the appropriate hydrochloride salt in the presence of a base. Under these conditions, low to moderate yields of the desired secondary amides were reached. However, we must note that better results, in terms of both conversion and substrate scope, were attained performing the catalytic transformations in an organic solvent (MeCN) with only a small quantity of water (T-HYDRO®, i.e. aqueous TBHP, was in this case used as an oxidant).
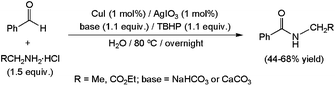 | ||
| Scheme 34 Copper-promoted oxidative amidation of benzaldehyde. | ||
From alcohols
Closely related oxidative-amidation protocols have been developed starting from primary alcohols, via in situ generation of the corresponding aldehydes (Scheme 35). These chemical transformations, known for a long, were first applied to the synthesis of five-, six- and seven-membered ring lactams, through intramolecular couplings in aminoalcohols, with the aid of ruthenium or rhodium catalysts.118a,124 At least, two-fold excess of a hydrogen acceptor, such as benzalacetone or acetone, was needed to observe full conversions.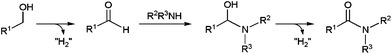
Remarkably, this methodology regained popularity after Milstein's report on a highly atom-economical protocol that does not need a hydrogen acceptor.92,93a In this case, the two oxidation steps take place with formation of molecular hydrogen as the only by-product. Another important advantage of the Milstein's catalytic system, based on a PNN-pincer ruthenium complex, is the possibility to extend the reaction to intermolecular couplings of alcohols and amines. From then on, several ruthenium93c,118c,125 and rhodium93c,94a complexes, associated or not with a hydrogen acceptor, as well as silver and gold nanoparticles,126 proved to be effective for this reaction in organic media. The advances achieved in the field have rendered the process feasible at room temperature,94a allowing high functionality tolerance and the use of a wide range of aldehydes. Both primary and secondary amines, as well as NH3, could be employed, although highly hindered non-cyclic N,N-disubstituted amines usually led to unsatisfactory conversions. Worthy of note, polyamines with both primary and secondary groups could react with complete chemoselectivity at the –NH2 positions keeping untouched the NH functions, without the need of a protection/deprotection strategy.125i
Very recently, the first examples of this transformation in an aqueous medium have been reported by Wang and co-workers.94c They developed a new water-soluble catalytic system immobilizing gold nanoparticles on inexpensive natural fish sperm DNA (Au/DNA) (Scheme 36). In contrast with other systems which required cumbersome hydrogen acceptors, this nanohybrid catalyst cleanly proceeded under an oxygen atmosphere, water being then the sole by-product of the oxidation steps. Moreover, the Au–DNA system smoothly promoted the reaction of challenging anilines with different aliphatic or aromatic aldehydes, in spite of their low nucleophilicity. Both primary and secondary amines were also tolerated, although steric hindrance strongly affected the efficiency. For example, high yields were observed for dimethylamine or piperidine, while the reaction completely failed starting from N-methylaniline. Although water-soluble, this nano-catalyst could only be recycled (up to five consecutive runs) by precipitation with EtOH and EtOAc and centrifugation, without significant loss in activity.
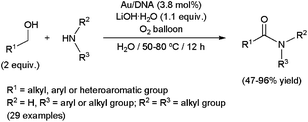
Polymer-supported gold nanoparticles were also successfully employed to convert alcohols into amides, albeit water–organic solvent mixtures were used in these cases as the reaction media.94b,127
From alkynes128
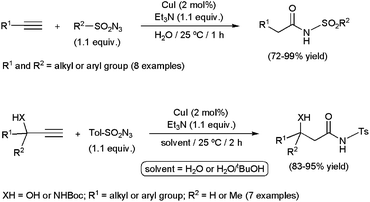
Several metal-catalyzed synthetic routes to N-substituted amides have been established starting from alkynes, such as aminocarbonylation reactions,129 oxidative couplings with amines130 or hydrative amidations with azides.131 However, as far as we know, only the latter have found application in aqueous media. In fact, a great rate acceleration has been evidenced when performing the reactions in pure water, the catalytic activity and selectivity being significantly lower in 1:2 mixtures of water with tBuOH, DMF, MeCN or CHCl3, or using CHCl3 with only the stoichiometric quantity of water.132 This three-component coupling, which involves a terminal alkyne, a sulfonyl azide and one equivalent of water, could be efficiently promoted by different copper(I) catalytic systems (Scheme 37). In particular, CuI combined with Et3N delivered the corresponding secondary amides in good to excellent yields under smooth conditions (25 °C) and short reaction times (1 h). Aromatic and aliphatic substrates were readily transformed, with conversions being not significantly affected by electronic and/or steric variations. Not only 1-alkynes but also acetylene gas was allowed to react with the azides and water. However, a mixture of tBuOH and water was required in this case to enhance the solubility of HCCH in the medium. The synthetic protocol was also applicable to the preparation of synthetically useful β-hydroxy- or β-amino-amides starting from propargylic alcohols or N-Boc protected propargylamines, respectively (Scheme 37). But, once again, these reactions needed the use of tBuOH–water mixtures, transformations in pure water being so exothermic that thermal degradations took place lowering significantly the selectivity. Interestingly, enantiomerically enriched propargyl alcohols and amines were converted without racemization. Taking advantage of this property, a synthetic approach of chiral polyhydroxy amides, hardly accessible by other methods, was designed.
Copper(I) cyanide also proved to be active in the hydrative coupling of alkynes with the 4-acetamidobenzenesulfonyl azide, and was employed in the gram-scale production of sulfonyl amides in an aqueous medium (Scheme 38).133 Aromatic, heteroaromatic and aliphatic 1-alkynes, as well as 1,7-octadiyne, were tolerated in the process.
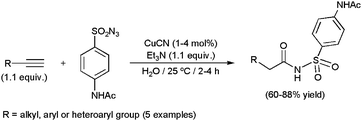 | ||
| Scheme 38 CuCN-promoted gram-scale synthesis of sulfonyl amides. | ||
Conclusions
Catalytic methods are nowadays the most powerful tools for the atom-economical and cost effective synthesis of amides. A large number of previously unavailable routes, starting from substrates other than the classical carboxylic acid derivatives, have been disclosed in recent years with the aid of transition metals.6 In line with the increasing environmental concerns, remarkable efforts have also been devoted to the development of these catalytic processes in the aqueous medium, since water is the most reliable alternative to the harmful petroleum-based solvents.8 Advances reached in the field have been comprehensively outlined in this review article, from which the following conclusions can be drawn. Formation of primary amides has been comparatively much more studied than that of N-substituted amides, with most of the published work being devoted to the catalytic hydration of nitriles. For this particular transformation, a large number of homogeneous and heterogeneous catalysts, as well as nanocatalysts, able to operate efficiently in water and showing a wide substrate scope are now available. Examples of recyclable systems, and of catalysts active under mild temperature regimes, are also increasingly common for this reaction. Several synthetic methods of primary amides in water that make use of alternative starting materials (aldoximes, aldehydes, amines, azides, alcohols or even methylarenes) have also appeared in recent years. However, in most of the cases, the processes have been comparatively much less studied than in organic media, and the number of catalysts currently active in water is very limited. The same can be said for the synthesis of secondary and tertiary amides, where the methods described in an aqueous medium, regardless of the starting materials employed, present a low generality. Chemists should take note of the opportunities existing in the field, and devise new catalytic systems for these poorly studied transformations. The authors hope that this comprehensive account will stimulate further work in this direction.Acknowledgements
Financial support by the Ministerio de Economía y Competitividad of Spain (Projects CTQ2010-14796/BQU and CSD2007-00006) is gratefully acknowledged.Notes and references
- See, for example: (a) The Chemistry of Amides, ed. J. Zabicky, Wiley-Interscience, New York, 1970 Search PubMed; (b) The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, John Wiley & Sons, New York, 2000 Search PubMed; (c) Polyesters and Polyamides, ed. B. L. Deopura, B. Gupta, M. Joshi and R. Alagirusami, CRC Press, Boca Raton, 2008 Search PubMed; (d) I. Johansson, in Kirk-Othmer Encyclopedia of Chemical Technology, JohnWiley & Sons, New York, 2004, vol. 2, pp. 442–463 Search PubMed.
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (b) See also: R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253 CrossRef CAS.
- See, for example: (a) Methoden Org. Chem. (Houben Weyl), ed. D. Dopp and H. Dopp, Thieme Verlag, Stuttgart, 1985, vol. E5 (2), pp. 1024–1031 Search PubMed; (b) P. D. Bailey, T. J. Mills, R. Pettecrew and R. A. Price, in Comprehensive Organic Functional Group Transformations II, ed. A. R. Katritzky and R. J. K. Taylor, Elsevier, Oxford, 2005, vol. 5, pp. 201–294 Search PubMed; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- A drastic waste reduction in classical amide syntheses from carboxylic acids and amines has been recently achieved by using a continuous method based on the recycling of the reaction mixture: K. Thalluri, K. C. Nadimpally, A. Paul and B. Mandal, RSC Adv., 2012, 2, 6838 RSC.
- (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- See, for example: (a) W. M. Nelson, in Green Solvents for Chemistry: Perspectives and Practice, Oxford University Press, New York, 2003 Search PubMed; (b) J. H. Clark and S. J. Taverner, Org. Process Res. Dev., 2007, 11, 149 CrossRef CAS; (c) F. M. Kerton, in Alternative Solvents for Green Chemistry, RSC Publishing, Cambridge, 2009 Search PubMed.
- (a) P. T. Anastas and J. C. Warner, in Green Chemistry Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed; (b) A. S. Matlack, in Introduction to Green Chemistry, Marcel Dekker, New York, 2001 Search PubMed; (c) Handbook of Green Chemistry and Technology, ed. J. H. Clark and D. J. Macquarrie, Blackwell Publishing, Abingdon, 2002 Search PubMed; (d) M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS; (e) M. Lancaster, in Green Chemistry: An Introductory Text, RSC Publishing, Cambridge, 2010 Search PubMed.
- See, for example: (a) C.-J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS; (b) A. Lubineau, J. Auge and Y. Queneau, Synthesis, 1994, 741 CrossRef CAS; (c) Organic Synthesis in Water, ed. P. A. Grieco, Blackie Academic & Professional, London, 1998 Search PubMed; (d) U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef; (e) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (f) C. K. Z. Andrade and L. M. Alves, Curr. Org. Chem., 2005, 9, 195 CrossRef CAS; (g) C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC; (h) L. Chen and C.-J. Li, Adv. Synth. Catal., 2006, 348, 1459 CrossRef CAS; (i) C. I. Herrerías, X. Yao, Z. Li and C.-J. Li, Chem. Rev., 2007, 107, 2546 CrossRef; (j) C.-J. Li and T. H. Chan, in Comprehensive Organic Reactions in Aqueous Media, John Wiley & Sons, New York, 2007 Search PubMed; (k) Organic Reactions in Water: Principles, Strategies and Applications, ed. U. M. Lindström, Blackwell Publishing, Oxford, 2007 Search PubMed; (l) Handbook of Green Chemistry (vol. 5), ed. P. T. Anastas and C.-J. Li, Wiley-VCH, Weinheim, 2010 Search PubMed; (m) M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC; (n) Water in Organic Synthesis, ed. S. Kobayashi, Thieme, Stuttgart, 2012 Search PubMed.
- See, for example: (a) Aqueous-Phase Organometallic Catalysis: Concepts and Applications, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1998 Search PubMed; (b) Aqueous Organometallic Catalysis, ed. I. T. Horváth and F. Joó, Kluwer, Dordrecht, 2001 Search PubMed; (c) Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley & Sons, Chichester, 2009 Search PubMed; (d) Metal-Catalyzed Reactions in Water, ed. P. Dixneuf and V. Cadierno, Wiley-VCH, Weinheim, 2013 Search PubMed.
- See, for example: (a) J. Yeo, S. Y. Kim, S. Kim, D. Y. Ryu, T.-H. Kim and M. J. Park, Nanotechnology, 2012, 23, 245703/1 CAS; (b) B. Al-Rashdi, C. Somerfield and N. Hilal, Sep. Purif. Rev., 2011, 40, 209 CrossRef CAS; (c) A. K. Goyal, E. S. Johal and G. Rath, Curr. Nanosci., 2011, 7, 460 Search PubMed; (d) Nanotechnology in Water Treatment Applications, ed. T. E. Cloete, M. de Kwaadsteniet, M. Botes and J. M. López-Romero, Caister Academic Press, Norfolk, 2010 Search PubMed.
- See, for example: (a) J. T. Edward and S. C. R. Meacock, J. Chem. Soc., 1957, 2000 RSC; (b) P. J. Steynberg, Z. Denga, R. Steyn, B. C. Bezuidenhout and N. L. Stark, PCT Int. Appl., WO 0026178, 2000 Search PubMed.
- For selected reviews covering the use of enzymes in catalytic nitrile hydrations, see: (a) M. Kobayashi and S. Shimizu, Curr. Opin. Chem. Biol., 2000, 4, 95 CrossRef CAS; (b) I. Endo, M. Nojori, M. Tsujimura, M. Nakasako, S. Nagashima, M. Yohda and M. J. Odaka, J. Inorg. Biochem., 2001, 83, 247 CrossRef CAS; (c) J. A. Kovacs, Chem. Rev., 2004, 104, 825 CrossRef CAS; (d) G. De Santis and R. Di Cosimo, in Biocatalysis for the Pharmaceutical Industry: Discovery, Development and Manufacturing, ed. J. Tao, G.-Q. Lin and A. Liese, Wiley-VCH, Weinheim, 2009, pp. 153–181 Search PubMed; (e) S. Prasad and T. C. Bhalla, Biotechnol. Adv., 2010, 28, 725 CrossRef CAS.
- See, for example: (a) H. Yamada and M. Kobayashi, Biosci., Biotechnol., Biochem., 1996, 60, 1391 CrossRef CAS; (b) S. van Pelt, F. van Rantwijk and R. A. Sheldon, in Focus in Catalysis Applications (supplement to Chimica Oggi/Chemistry Today), Teknoscienze Srl, Milano, 2008, vol. 26, pp. 2–4 Search PubMed; (c) S. Sanchez and A. L. Demain, Org. Process Res. Dev., 2011, 15, 224 CrossRef CAS; (d) B. Li, J. Su and J. Tao, Org. Process Res. Dev., 2011, 15, 291 CrossRef CAS.
- For reviews on metal-catalyzed nitrile hydration reactions, see: (a) A. W. Parkins, Platinum Metals Rev., 1996, 40, 169 CAS; (b) V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771 CrossRef CAS; (c) N. A. Bokach and V. Y. Kukushkin, Russ. Chem. Rev., 2005, 74, 153 CrossRef CAS; (d) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1 CrossRef CAS; (e) T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949 CrossRef CAS.
- (a) T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657 CrossRef CAS; (b) A. W. Parkins and T. Ghaffar, PCT Int. Appl., WO 9630379, 1996 Search PubMed; (c) T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249 CrossRef CAS.
- Examples of the synthetic applications of this catalyst can be found in: (a) J. Akisanya, A. W. Parkins and J. W. Steed, Org. Process Res. Dev., 1998, 2, 274 CrossRef CAS; (b) A. Papakyprianou, A. W. Parkins, P. D. Prince and J. W. Steed, Org. Prep. Proced. Int., 2002, 34, 436 CrossRef CAS; (c) M. North, A. W. Parkins and A. N. Shariff, Tetrahedron Lett., 2004, 45, 7625 CrossRef CAS; (d) X.-B. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327 CrossRef CAS; (e) R. G. Gieling and M. B. Groen, PCT Int. Appl., WO 2006097342, 2006 Search PubMed; (f) T. J. Greshock and R. L. Funk, Org. Lett., 2006, 8, 2643 CrossRef CAS; (g) A. Myers, S. B. Herzon, J. E. Wulff, R. Siegrist, J. Svenda and M. A. Zajac, PCT Int. Appl., WO 2006102097, 2006 Search PubMed; (h) X. Jiang, N. Williams and J. K. De Brabander, Org. Lett., 2007, 9, 227 CrossRef CAS; (i) B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS; (j) T. Kan, Y. Kawamoto, T. Asakawa, T. Furuta and T. Fukuyama, Org. Lett., 2008, 10, 169 CrossRef CAS; (k) L. Tafesse, PCT Int. Appl., WO 2008133973, 2008 Search PubMed; (l) R. A. Jones and M. J. Krische, Org. Lett., 2009, 11, 1849 CrossRef CAS; (m) D. R. Saito, D. D. Long, P. van Dyke, T. J. Church, L. Jiang and B. Frieman, PCT Int. Appl., WO 2009029256, 2009 Search PubMed; (n) L. E. Brown, Y. R. Landaverry, J. R. Davies, K. A. Milinkevich, S. Ast, J. S. Carlson, A. G. Oliver and J. P. Konopelski, J. Org. Chem., 2009, 74, 5405 CrossRef CAS; (o) F. D. J. Cortez and R. Sarpong, Org. Lett., 2010, 12, 1428 CrossRef; (p) C.-K. Mai, M. F. Sammons and T. Sammakia, Angew. Chem., Int. Ed., 2010, 49, 2397 CrossRef CAS; (q) T. A. Brugel, R. W. Smith, M. Balestra, C. Becker, T. Daniels, G. M. Koether, S. R. Throner, L. M. Panko, D. G. Brown, R. Liu, J. Gordon and M. F. Peters, Bioorg. Med. Chem. Lett., 2010, 20, 5405 CrossRef CAS; (r) M. K. M. Tun, D.-J. Wüstmann and S. B. Herzon, Chem. Sci., 2011, 2, 2251 RSC; (s) L. Yao, B. Pitta, P. C. Ravikumar, M. Purzycki and F. F. Fleming, J. Org. Chem., 2012, 77, 3651 CrossRef CAS; (t) R. S. Andrews, J. L. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 4140 CrossRef CAS.
- T. J. Ahmed, B. R. Fox, S. M. M. Knapp, R. B. Yelle, J. J. Juliette and D. R. Tyler, Inorg. Chem., 2009, 48, 7828 CrossRef CAS.
- (a) K. L. Breno, M. D. Pluth and D. R. Tyler, Organometallics, 2003, 22, 1203 CrossRef CAS; (b) K. L. Breno, M. D. Pluth, C. W. Landorf and D. R. Tyler, Organometallics, 2004, 23, 1738 CrossRef CAS; (c) K. L. Breno, T. J. Ahmed, M. D. Pluth, C. Balzarek and D. R. Tyler, Coord. Chem. Rev., 2006, 250, 1141 CrossRef CAS; (d) T. J. Ahmed, L. V. Zakharov and D. R. Tyler, Organometallics, 2007, 26, 5179 CrossRef CAS; (e) T. J. Ahmed and D. R. Tyler, Organometallics, 2008, 27, 2608 CrossRef CAS.
- E. Tílvez, M. I. Menéndez and R. López, Organometallics, 2012, 31, 1618 CrossRef.
- (a) M. G. Crestani, A. Arévalo and J. J. García, Adv. Synth. Catal., 2006, 348, 732 CrossRef CAS; (b) A. Arévalo and J. J. García, Eur. J. Inorg. Chem., 2010, 4063 CrossRef.
- C. Crisóstomo, M. G. Crestani and J. J. García, J. Mol. Catal. A: Chem., 2007, 266, 139 CrossRef.
- (a) C. Crisóstomo, M. G. Crestani and J. J. García, Chim. Oggi, 2009, 27, 36 Search PubMed; (b) C. Crisóstomo, M. G. Crestani and J. J. García, Inorg. Chim. Acta, 2010, 363, 1092 CrossRef.
- M. G. Crestani and J. J. García, J. Mol. Catal. A: Chem., 2009, 299, 26 CrossRef CAS.
- M. N. Kopylovich, V. Y. Kukushkin, M. Haukka, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Inorg. Chem., 2002, 41, 4798 CrossRef CAS.
- See, for example: (a) T. Oshiki, H. Yamashita, K. Sawada, M. Utsunomiya, K. Takahashi and K. Takai, Organometallics, 2005, 24, 6287 CrossRef CAS; (b) T. Šmejkal and B. Breit, Organometallics, 2007, 26, 2461 CrossRef; (c) T. Oshiki, I. Hyodo and A. Ishizuka, J. Synth. Org. Chem. Jpn., 2010, 68, 41 CrossRef CAS; (d) M. Muranaka, I. Hyodo, W. Okumura and T. Oshiki, Catal. Today, 2011, 164, 552 CrossRef CAS; (e) P. Daw, A. Sinha, S. M. W. Rahaman, S. Dinda and J. K. Bera, Organometallics, 2012, 31, 3790 CrossRef CAS.
- For leading accounts and reviews on bifunctional catalysts, see: (a) D. B. Grotjahn, Chem.–Eur. J., 2005, 11, 7146 CrossRef CAS; (b) A. S. Borovik, Acc. Chem. Res., 2005, 38, 54 CrossRef CAS; (c) T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393 RSC; (d) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef CAS; (e) D. B. Grotjahn, Dalton Trans., 2008, 6497 RSC; (f) D. B. Grotjahn, Chem. Lett., 2010, 39, 908 CrossRef CAS; (g) D. B. Grotjahn, Pure Appl. Chem., 2010, 82, 635 CrossRef CAS; (h) T. Ikariya and I. D. Gridnev, Top. Catal., 2010, 53, 894 CrossRef CAS; (i) D. B. Grotjahn, Top. Catal., 2010, 53, 1009 CrossRef CAS.
- V. Cadierno, J. Francos and J. Gimeno, Chem.–Eur. J., 2008, 14, 6601 CrossRef CAS.
- V. Cadierno, J. Díez, J. Francos and J. Gimeno, Chem.–Eur. J., 2010, 16, 9808 CrossRef CAS.
- Performing the catalytic reactions in a water–glycerol mixture (1:1 v/v), this catalyst could also be recycled by selective extraction of the reaction products with ethyl acetate.
A. E. Díaz-Álvarez, R. García-Álvarez, P. Crochet and V. Cadierno, in Glycerol: Production, Structure and Applications, ed. M. D. S. Silva and P. C. Ferreira, Nova Science Publishers, New York, 2012, pp. 249–262 Search PubMed.
- W.-C. Lee and B. J. Frost, Green Chem., 2012, 14, 62 RSC.
- (a) R. García-Álvarez, J. Francos, P. Crochet and V. Cadierno, Tetrahedron Lett., 2011, 52, 4218 CrossRef; (b) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2011, 30, 5442 CrossRef.
- Such a tandem process promoted by [RuH2(PPh3)4] in an organic medium was earlier described by Murahashi and co-workers: (a) S.-I. Murahashi, S. Sasao, E. Saito and T. Naota, J. Org. Chem., 1992, 57, 2521 CrossRef CAS; (b) S.-I. Murahashi, S. Sasao, E. Saito and T. Naota, Tetrahedron, 1993, 49, 8805 CrossRef CAS; (c) S.-I. Murahashi and H. Takaya, Acc. Chem. Res., 2000, 33, 225 CrossRef CAS.
- S. M. M. Knapp, T. J. Sherbow, J. J. Juliette and D. R. Tyler, Organometallics, 2012, 31, 2941 CrossRef CAS.
- R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2010, 29, 3955 CrossRef.
- R. García-Álvarez, S. E. García-Garrido, J. Díez, P. Crochet and V. Cadierno, Eur. J. Inorg. Chem., 2012, 4218 CrossRef.
- Modest catalytic activities were also achieved with the related complexes [RuCl2(η6-arene)(PR3)] (arene = C6H6, p-cymene; PR3 = PPh3, PiPr3, P(OEt)3, PPh(OEt)2, PPh2(OEt)). In addition, surfactants were needed to ensure the solubilization of these catalysts in water: A. Cavarzan, A. Scarso and G. Strukul, Green Chem., 2010, 12, 790 RSC.
- Z. Li, L. Wang and X. Zhou, Adv. Synth. Catal., 2012, 354, 584 CrossRef CAS.
- (a) G. Villain, P. Kalck and A. Gaset, Tetrahedron Lett., 1980, 21, 2901 CrossRef CAS; (b) G. Villain, G. Constant, A. Gaset and P. Kalck, J. Mol. Catal., 1980, 7, 355 CrossRef CAS; (c) G. Villain, A. Gaset and P. Kalck, J. Mol. Catal., 1981, 12, 103 CrossRef CAS.
- M. A. Bennett and T. Yoshida, J. Am. Chem. Soc., 1973, 95, 3030 CrossRef CAS.
- C. S. Chin, S. Y. Kim, K.-S. Joo, G. Won and D. Chong, Bull. Korean Chem. Soc., 1999, 20, 535 CAS.
- W. K. Fung, X. Huang, M. L. Man, S. M. Ng, M. Y. Hung, Z. Lin and C. P. Lau, J. Am. Chem. Soc., 2003, 125, 11539 CrossRef CAS.
- M. C. K.-B. Djoman and A. N. Ajjou, Tetrahedron Lett., 2000, 4845 CrossRef CAS.
- M. G. Crestani, A. Steffen, A. M. Kenwright, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Organometallics, 2009, 28, 2904 CrossRef CAS.
- E. Cariati, C. Dragonetti, L. Manassero, D. Roberto, F. Tessore and E. Lucenti, J. Mol. Catal. A: Chem., 2003, 204–205, 279 CrossRef CAS.
- M. Martín, H. Horváth, E. Sola, Á. Kathó and F. Joó, Organometallics, 2009, 28, 561 CrossRef.
- (a) I. N. Stepanenko, B. Cebrián-Losantos, V. B. Arion, A. A. Krokhin, A. A. Nazarov and B. K. Keppler, Eur. J. Inorg. Chem., 2007, 400 CrossRef CAS; (b) S. M. Ashraf, I. Berger, A. A. Nazarov, C. G. Hartinger, M. P. Koroteev, E. E. Nifant'ev and B. K. Keppler, Chem. Biodiversity, 2008, 5, 1640 CrossRef CAS; (c) S. M. Ashraf, W. Kandioller, M. G. Mendoza-Ferri, A. A. Nazarov, C. G. Hartinger and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2060 CrossRef CAS.
- See, for example: (a) Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743 RSC; (c) G. A. Somorjai and Y. Li, Top. Catal., 2010, 53, 832 CrossRef CAS; (d) H. Cong and J. A. Porco, ACS Catal., 2012, 2, 65 CrossRef CAS; (e) S. B. Kalidindi and B. R. Jagirdar, ChemSusChem, 2012, 5, 65 CrossRef CAS.
- (a) N. Toshima and Y. Wang, Chem. Lett., 1993, 22, 1611 CrossRef; (b) N. Toshima and Y. Wang, Langmuir, 1994, 10, 4574 CrossRef CAS; (c) Y. Wang, H. Liu and N. Toshima, J. Phys. Chem., 1996, 100, 19533 CrossRef CAS; (d) N. Toshima, P. Lu and Y. Wang, Stud. Surf. Sci. Catal., 2011, 132, 243 CrossRef.
- For related works using Cu/Ni alloys, Cu NPs, and copper suspended on PVP or silica-magnesia see: (a) H. Hayashi, H. Nishi, Y. Watanabe and T. Okazaki, J. Catal., 1981, 69, 44 CrossRef CAS; (b) N. Ravindranathan, N. Kalyanam and S. Sivaram, J. Org. Chem., 1982, 47, 4812 CrossRef; (c) K. Sugiyama, H. Miura, Y. Watanabe, Y. Ukai and T. Matsuda, Chem. Lett., 1986, 15, 47 CrossRef; (d) K. Sugiyama, H. Miura, Y. Watanabe, Y. Ukai and T. Matsuda, Bull. Chem. Soc. Jpn., 1987, 60, 1579 CrossRef CAS.
- (a) A. Ishizuka, Y. Nakazaki and T. Oshiki, Chem. Lett., 2009, 38, 360 CrossRef CAS; (b) T. Oshiki and A. Ishizuka, Jpn. Kokai Tokkyo Koho, JP 2009214099, 2009 Search PubMed.
- (a) T. Mitsudome, Y. Mikami, H. Mori, S. Arita, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 3258 RSC; (b) A. Y. Kim, H. S. Bae, S. Park, S. Park and K. H. Park, Catal. Lett., 2011, 141, 685 CrossRef CAS.
- K. Shimizu, N. Imaiida, K. Sawabe and A. Satsuma, Appl. Catal., A, 2012, 421–422, 114 CrossRef CAS.
- Y.-M. Liu, L. He, M.-M. Wang, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2012, 5, 1392 CrossRef CAS.
- (a) The use of gold NPs supported on different metal-oxides has been covered by a recent patent: Y. Cao, Y. Liu and L. He, Faming Zhuanli Shenqing, CN 102285850, 2011 Search PubMed; (b) A catalytic system consisting of hydrotalcite-clay supported nickel nanoparticles, active in an aqueous medium, has also very recently appeared: T. Subramanian and K. Pitchumani, Catal. Commun., 2012, 29, 109 CrossRef.
- V. Polshettiwar and R. S. Varma, Chem.–Eur. J., 2009, 15, 1582 CrossRef CAS.
- S. E. García-Garrido, J. Francos, V. Cadierno, J. M. Basset and V. Polshettiwar, ChemSusChem, 2011, 4, 104 CrossRef.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2012, 48, 6220 RSC.
- (a) E. J. Gasson and D. J. Hadley, US Pat., US2904552, 1959 Search PubMed; (b) S. K. Roy, S. C. Ray and P. K. Ray, J. Indian Chem. Soc., 1980, 57, 195 CAS; (c) H. Miura, K. Sugiyama, S. Kawakami, T. Aoyama and T. Matsuda, Chem. Lett., 1982, 11, 183 CrossRef; (d) E. N. Zil'berman, Russ. Chem. Rev., 1984, 53, 900 CrossRef; (e) S. C. Roy, P. Dutta, L. N. nandy, S. K. Roy, P. Samuel, S. M. Pillai, V. K. Kaushik and M. Ravindranathan, Appl. Catal., A, 2005, 290, 175 CrossRef CAS; (f) Y. Gangarajula and B. Gopal, Chem. Lett., 2012, 41, 101 CrossRef CAS; (g) M. Tamura, H. Wakasugi, K.-I. Shimizu and A. Satsuma, Chem.–Eur. J., 2011, 17, 11428 CrossRef CAS; (h) K. Yamaguchi, Y. Wang, H. Kobayashi and N. Mizuno, Chem. Lett., 2012, 41, 574 CrossRef CAS.
- (a) K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., Int. Ed., 2004, 43, 1576 CrossRef CAS; (b) K. Yamaguchi and N. Mizuno, Synlett, 2010, 2365 CAS.
- K. Mori, K. Yamaguchi, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Commun., 2001, 461 RSC.
- G. K. S. Prakash, S. B. Munoz, A. Papp, K. Masood, I. Bychinskaya, T. Mathew and G. A. Olah, Asian J. Org. Chem., 2012, 1, 146 CrossRef CAS.
- For reviews on the Beckmann rearrangement, see: (a) R. R. Kumar, K. A. Vanithan and M. Balasubramanian, in Name Reactions for Homologations – Part 2, ed. J. J. Li, John Wiley & Sons, Hoboken, 2009, pp. 274–292 Search PubMed; (b) R. E. Gawley, Org. React., 1988, 35, 1 CAS.
- (a) E. C. Horning and V. L. Stromberg, J. Am. Chem. Soc., 1952, 74, 5151 CrossRef CAS; (b) D. S. Hoffenberg and C. R. Hauser, J. Org. Chem., 1955, 20, 1496 CrossRef CAS; (c) L. Field, P. B. Hughmark, S. H. Shumaker and W. S. Marshall, J. Am. Chem. Soc., 1961, 83, 1983 CrossRef CAS.
- (a) A. J. Leusink, T. G. Meerbeek and J. G. Noltes, Recl. Trav. Chim. Pays-Bas, 1976, 95, 123 CrossRef CAS; (b) A. J. Leusink, T. G. Meerbeek and J. G. Noltes, Recl. Trav. Chim. Pays-Bas, 1977, 96, 142 CrossRef CAS; (c) C. L. Allen, S. Davulcu and J. M. J. Williams, Org. Lett., 2010, 12, 5096 CrossRef CAS.
- (a) T. Setsuda, Jpn. Kokai Tokkyo Koho, JP 52128302, 1977 Search PubMed; (b) S. Komiya, H. Shimazu and T. Tamashima, Jpn. Kokai Tokkyo Koho, JP 2003342245, 2003 Search PubMed.
- (a) S. K. Sharma, S. D. Bishopp, C. L. Allen, R. Lawrence, M. J. Bamford, A. A. Lapkin, P. Plucinski, R. J. Watson and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 4252 CrossRef CAS; (b) G. Saidulu, N. Anand, K. S. R. Rao, A. Burri, S.-E. Park and D. R. Burri, Catal. Lett., 2011, 141, 1865 CrossRef CAS; (c) A. Martínez-Asensio, M. Yus and D. J. Ramón, Tetrahedron, 2012, 68, 3948 CrossRef.
- R. S. Ramón, J. Bosson, S. Díez-González, N. Marion and S. P. Nolan, J. Org. Chem., 2010, 75, 1197 CrossRef.
- (a) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599 CrossRef CAS; (b) D. Gnanamgari and R. H. Crabtree, Organometallics, 2009, 28, 922 CrossRef CAS; (c) J. F. Hull, S. T. Hilton and R. H. Crabtree, Inorg. Chim. Acta, 2010, 363, 1243 CrossRef CAS; (d) P. Kumar, A. K. Singh, R. Pandey and D. S. Pandey, J. Organomet. Chem., 2011, 696, 3454 CrossRef CAS.
- (a) S. Park, Y. Choi, H. Han, S. H. Yang and S. Chang, Chem. Commun., 2003, 1936 RSC; (b) M. Kim, J. Lee, H.-Y. Lee and S. Chang, Adv. Synth. Catal., 2009, 351, 1807 CrossRef CAS.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73 CrossRef CAS.
- (a) A. Mishra, A. Ali, S. Upreti and R. Gupta, Inorg. Chem., 2008, 47, 154 CrossRef CAS; (b) C. L. Allen, C. Burel and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 2724 CrossRef CAS.
- Selective dehydration of aldoximes to nitriles with the aid of transition metals is a well-known process. Recent examples can be found in: (a) H. S. Kim, S. Kim and J. N. Kim, Tetrahedron Lett., 2009, 50, 1717 CrossRef CAS; (b) N. Jiang and A. J. Ragauskas, Tetrahedron Lett., 2010, 51, 4479 CrossRef CAS; (c) Y.-T. Li, B. S. Liao, H.-P. Chen and S.-T. Liu, Synthesis, 2011, 2639 CAS.
- Catalytic hydration of nitriles to amides using aldoximes as the water source has been described in: (a) E. S. Kim, H. S. Kim and J. N. Kim, Tetrahedron Lett., 2009, 50, 2973 CrossRef CAS; (b) J. Lee, M. Kim, S. Chang and H.-Y. Lee, Org. Lett., 2009, 11, 5598 CrossRef CAS; (c) E. S. Kim, H. S. Lee, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2010, 51, 1598 Search PubMed; (d) E. S. Kim, Y. M. Kim and J. N. Kim, Bull. Korean Chem. Soc., 2010, 31, 700 CrossRef CAS; (e) Á. Kiss and Z. Hell, Tetrahedron Lett., 2011, 52, 6021 CrossRef; (f) X.-Y. Ma, Y. He, Y.-L. Hu and M. Lu, Tetrahedron Lett., 2012, 53, 449 CrossRef CAS; (g) X. Ma, Y. He, P. Wang and M. Lu, Appl. Organomet. Chem., 2012, 26, 377 CrossRef CAS.
- C. L. Allen, R. Lawrence, L. Emmett and J. M. J. Williams, Adv. Synth. Catal., 2011, 353, 3262 CrossRef CAS.
- (a) H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 5202 CrossRef CAS; (b) H. Fujiwara, Y. Ogasawara, M. Kotani, K. Yamaguchi and N. Mizuno, Chem.–Asian J., 2008, 3, 1715 CrossRef CAS.
- R. García-Álvarez, A. E. Díaz-Álvarez, J. Borge, P. Crochet and V. Cadierno, Organometallics, 2012, 31, 6482 CrossRef.
- For a recent review on oxidative esterification and amidation of aldehydes, see: K. Ekoue-Kovi and C. Wolf, Chem.–Eur. J., 2008, 14, 6302 CrossRef CAS.
- N. C. Ganguly, S. Roy and P. Mondal, Tetrahedron Lett., 2012, 53, 1413 CrossRef CAS.
- B. K. Allam and K. N. Singh, Tetrahedron Lett., 2011, 52, 5851 CrossRef CAS.
- R. R. Gowda and D. Chakraborty, Eur. J. Org. Chem., 2011, 2226 CrossRef CAS.
- (a) N. Raja, M. U. Raja and R. Ramesh, Inorg. Chem. Commun., 2012, 19, 51 CrossRef CAS; (b) R. N. Prabhu and R. Ramesh, RSC Adv., 2012, 2, 4515 RSC.
- M. A. Ali and T. Punniyamurthy, Adv. Synth. Catal., 2010, 352, 288 CrossRef CAS.
- For a recent review, see: M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108 CrossRef.
- (a) R. Tang, S. E. Diamond, N. Neary and F. Mares, J. Chem. Soc., Chem. Commun., 1978, 562 RSC; (b) A. Nishinaga, T. Shimizu and T. Matsuura, J. Chem. Soc., Chem. Commun., 1979, 970 RSC; (c) K.-I. Tanaka, S. Yoshifuji and Y. Nitta, Chem. Pharm. Bull., 1988, 36, 3125 CrossRef CAS.
- (a) J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 47, 9249 CrossRef CAS; (b) Y. Wang, H. Kobayashi, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 2642 RSC; (c) K. Yamaguchi, Y. Wang and N. Mizuno, Chem. Lett., 2012, 41, 633 CrossRef CAS; (d) W. Xu, Y. Jiang and H. Fu, Synlett, 2012, 801 CAS.
- (a) K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42, 1480 CrossRef CAS; (b) N. Mizuno and K. Yamaguchi, Catal. Today, 2008, 132, 18 CrossRef CAS.
- (a) M. E. C. Biffin, J. Miller and D. B. Paul, in The Chemistry of the Azide Group, ed. S. Patai, Interscience Publishers, New York, 1971, pp. 57–190 Search PubMed; (b) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef; (c) Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, Chichester, 2010 Search PubMed.
- See, for example: M. Lamani, P. Devadig and K. R. Prabhu, Org. Biomol. Chem., 2012, 10, 2753 CAS and references cited therein.
- J. He, K. Yamaguchi and N. Mizuno, J. Org. Chem., 2011, 76, 4606 CrossRef CAS.
- (a) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS; (b) D. Milstein, E. Balaraman, C. Gunanathan, B. Gnanaprakasam and J. Zhang, PCT Int. Appl., WO 2012052996, 2012 Search PubMed.
- For review articles, see: (a) D. Milstein, Top. Catal., 2010, 53, 915 CrossRef CAS; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS; (c) C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20 RSC.
- (a) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559 CrossRef CAS; (b) J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550 CrossRef; (c) Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917 CrossRef CAS; (d) K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544 CrossRef CAS; (e) T. Ishida, H. Watanabe, T. Takei, A. Hamasaki, M. Tokunaga and M. Haruta, Appl. Catal., A, 2012, 85, 425–426 Search PubMed; (f) R. Nie, J. Shi, S. Xia, L. Shen, P. Chen, Z. Hou and F.-S. Xiao, J. Mater. Chem., 2012, 22, 18115 RSC.
- Y. Wang, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7250 CrossRef CAS.
- For a very recent review on the Ritter reaction, see: A. Guérinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 19 CrossRef.
- See, for example: (a) M. Mukhopadhyay and J. Iqbal, J. Org. Chem., 1997, 62, 1843 CrossRef CAS; (b) B. Anxionnat, A. Guérinot, S. Reymond and J. Cossy, Tetrahedron Lett., 2009, 50, 3470 CrossRef CAS.
- E. Callens, A. J. Burton and A. G. M. Barrett, Tetrahedron Lett., 2006, 47, 8699 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and A. Khoshnood, Catal. Commun., 2008, 9, 529 CrossRef CAS.
- (a) V. R. Kartashov, K. V. Malkova, A. V. Arkhipova and T. N. Sokolova, Russ. J. Org. Chem., 2006, 42, 966 CrossRef CAS; (b) V. R. Kartashov, A. V. Arkhipova, K. V. Malkova and T. N. Sokolova, Russ. Chem. Bull., 2006, 55, 387 CrossRef CAS.
- G.-R. Qu, Y.-W. Song, H.-Y. Niu, H.-M. Guo and J. S. Fossey, RSC Adv., 2012, 2, 6161 RSC.
- R. I. Khusnutdinov, N. A. Shchadneva, Y. Y. Mayakova, L. F. Khisamova and U. M. Dzhemilev, Russ. J. Org. Chem., 2011, 47, 1682 CrossRef CAS.
- S.-I. Murahashi, T. Naota and E. Saito, J. Am. Chem. Soc., 1986, 108, 7846 CrossRef CAS.
- C. L. Allen, A. A. Lapkin and J. M. J. Williams, Tetrahedron Lett., 2009, 50, 4262 CrossRef CAS.
- C. J. Cobley, M. van den Heuvel, A. Abbadi and J. G. de Vries, Tetrahedron Lett., 2000, 41, 2467 CrossRef CAS.
- M. Tamura, T. Tonomura, K.-I. Shimizu and A. Satsuma, Appl. Catal., A, 2012, 417–418, 6 CrossRef CAS.
- A. J. M. van Dijk, T. Heyligen, R. Duchateau, J. Meuldijk and C. E. Koning, Chem.–Eur. J., 2007, 13, 7664 CrossRef CAS.
- A. J. M. van Dijk, R. Duchateau, E. J. M. Hensen, J. Meuldijk and C. E. Koning, Chem.–Eur. J., 2007, 13, 7673 CrossRef CAS.
- For reviews on this topic, see: (a) R. Skoda-Foldes and L. Kollar, Curr. Org. Chem., 2002, 6, 1097 CrossRef CAS; (b) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef; (c) R. Skoda-Foldes and L. Kollar, Lett. Org. Chem., 2010, 7, 621 CrossRef CAS; (d) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867 CrossRef CAS.
- See, for example: Y. Uozumi and T. Watanabe, J. Org. Chem., 1999, 64, 6921 CrossRef CAS.
- X. Wu and M. Larhed, Org. Lett., 2005, 7, 3327 CrossRef CAS.
- X. Wu, J. K. Ekegren and M. Larhed, Organometallics, 2006, 25, 1434 CrossRef CAS.
- O. Lagerlund and M. Larhed, J. Comb. Chem., 2006, 8, 4 CrossRef CAS.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295 CrossRef CAS.
- P. J. Tambade, Y. P. Patil, M. J. Bhaushali and B. M. Bhanage, Synthesis, 2008, 2347 CAS.
- Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012 DOI:10.1016/j.cattod.2012.03.039 , in press.
- K. Nakagawa, H. Onoue and K. Minami, Chem. Commun., 1966, 17 RSC.
- (a) T. Naota and S.-I. Murahashi, Synlett, 1991, 693 CAS; (b) J. W. W. Chang and P. W. H. Chan, Angew. Chem., Int. Ed., 2008, 47, 1138 CrossRef CAS; (c) S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Cheng, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002 CrossRef CAS.
- (a) Y. Tamaru, Y. Yamada and Z.-I. Yoshida, Synthesis, 1983, 474 CrossRef CAS; (b) K. Ekoue-Kovi and C. Wolf, Org. Lett., 2007, 9, 3429 CrossRef CAS; (c) Y. Suto, N. Yamagiwa and Y. Torisawa, Tetrahedron Lett., 2008, 49, 5732 CrossRef CAS.
- (a) A. Tillack, I. Rudloff and M. Beller, Eur. J. Org. Chem., 2001, 523 CrossRef CAS; (b) J. Chan, K. D. Baucom and J. A. Murry, J. Am. Chem. Soc., 2007, 129, 14106 CrossRef CAS.
- (a) S. Y. Seo and T. J. Marks, Org. Lett., 2008, 10, 317 CrossRef CAS; (b) C. Qian, X. Zhang, J. Li, F. Xu, Y. Zhang and Q. Shen, Organometallics, 2009, 28, 3856 CrossRef CAS; (c) Q. Qian, X. Zhang, Y. Zhang and Q. Shen, J. Organomet. Chem., 2010, 695, 747 CrossRef.
- G.-L. Li, K. K.-Y. Kung and M.-K. Wong, Chem. Commun., 2012, 48, 4112 RSC.
- W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064 CrossRef CAS.
- K.-I. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785 CrossRef CAS.
- (a) L. U. Nordstrøm, H. Vogt and R. Madesen, J. Am. Chem. Soc., 2008, 130, 17672 CrossRef; (b) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643 CrossRef CAS; (c) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667 CrossRef CAS; (d) S. C. Ghosh and S. H. Hong, Eur. J. Org. Chem., 2010, 4266 CrossRef CAS; (e) Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374 CrossRef CAS; (f) J. H. Dam, G. Ostrovszky, L. U. Nordstrøm and R. Madsen, Chem.–Eur. J., 2010, 16, 6820 CrossRef CAS; (g) J. Zhang, M. Senthilkumar, S. C. Ghosh and S. H. Hong, Angew. Chem., Int. Ed., 2010, 49, 6391 CrossRef CAS; (h) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162 CrossRef CAS; (i) H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159 CrossRef CAS.
- (a) K.-I. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977 CrossRef CAS; (b) J. Zhu, Y. Zhang, F. Shi and Y. Deng, Tetrahedron Lett., 2012, 53, 3178 CrossRef CAS.
- P. Preedasuriyachai, H. Kitahara, W. Chavasiri and H. Sakurai, Chem. Lett., 2010, 39, 1174 CrossRef CAS.
- Isolated examples of amide formation in an aqueous medium from azides and thioacids, promoted by RuCl3, and from silylketene pyridylthioacetal and imines, promoted by Sc(OTf)3, have also been described: (a) F. Fazio and C.-H. Wong, Tetrahedron Lett., 2003, 44, 9083 CrossRef CAS; (b) C. Biaggi, M. Benaglia and A. Puglisi, J. Organomet. Chem., 2007, 692, 5795 CrossRef CAS.
- See, for example: (a) B. Gabriele, G. Salerno, L. Veltri and M. Costa, J. Organomet. Chem., 2001, 622, 84 CrossRef CAS; (b) B. E. Ali and J. Tijani, Appl. Organomet. Chem., 2003, 17, 921 CrossRef; (c) D. J. Knapton and T. Y. Meyer, Org. Lett., 2004, 6, 687 CrossRef CAS; (d) K. M. Driller, S. Prateeptongkum, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 537 CrossRef CAS.
- W.-K. Chan, C.-M. Ho, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 14796 CrossRef CAS.
- (a) S. H. Cho, E. J. Yoo, I. Bae and S. Chang, J. Am. Chem. Soc., 2005, 127, 16046 CrossRef CAS; (b) M. P. Cassidy, J. Raushel and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 3154 CrossRef CAS.
- S. H. Cho and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1897 CrossRef CAS.
- S. H. Cho, S. J. Hwang and S. Chang, Org. Synth., 2008, 85, 131 CAS.
| This journal is © The Royal Society of Chemistry 2013 |