Mechanochemical degradation of lignin and wood by solvent-free grinding in a reactive medium†
Tillmann
Kleine
,
Julien
Buendia
and
Carsten
Bolm
*
Institute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52056 Aachen, Germany. E-mail: Carsten.Bolm@oc.rwth-aachen.de
First published on 1st November 2012
Abstract
A mechanochemical approach for the cleavage of β-O-4-linkages in lignin is reported. The method is transition metal- and solvent-free, requires only inexpensive reagents, and tolerates the presence of water and standard reagent impurities.
1. Introduction
Recently, mechanochemistry has attracted much attention because it allows promotion of reactions under solvent-free conditions.1 Even solids react quickly and quantitatively in the absence of a solvent, avoiding (or at least reducing) the use of those often environmentally problematic, or even hazardous, fossil-derived liquids. Mechanochemical synthesis is mainly done in ball mills, which leads to an efficient mixing of all reagents and provides a significant energy input.2 Compared to other techniques, such as ultrasound or microwave, the energy efficiency of ball milling is higher.3,4 When hard material is ground in a ball mill the particle size decreases. On a large scale, crushing ores is well established using this technique.5 With respect to molecular crystals mechanochemical reactions have been suggested to be driven by an unusual alternation of the electron distribution by molecular deformation.6Depleting fossil carbon sources have generated strong pressure on the oil-based industries, and a shift to renewable resources is considered essential for sustaining industrial growth.7–9 The usage of feedstock-dedicated plants is controversial, because of the low efficiency of land usage, collateral effects on the nourishment supply and increased greenhouse gas emissions.8–10 In the light of these disadvantages, the exploitation of whole-plant material or waste biomass appears desirable. It provides the biopolymers cellulose, hemicellulose, and lignin in large quantities (cellulose: 25–50%, hemicellulose: 25–30%, lignin: 15–30% of dry biomass),11,12 and consequently, those compounds are cheap representing attractive sources for platform chemicals.13,14 Unfortunately, their poor solubility, complexity and structural diversity hamper their use as raw materials for new valuable carbon sources. Of particular interest in this context is lignin. It is a three-dimensional amorphous material consisting of methoxylated phenyl propane units, which are interlinked by various binding motifs. Among them, the β-O-4-linkage is the most abundant [∼60% in hardwood, ∼45% in softwood14]. Although ubiquitously in nature and known for decades, the technical use of lignin is limited by its poor solubility and structural complexity.15 Degradation or solvation processes generally require harsh reaction conditions, which follow a mechanical pretreatment.14,16–18 For unmodified wood, these issues are even more severe.9 Understanding wood and lignin processing on a molecular level appears essential for improving their degradation efficiencies.14,19,20
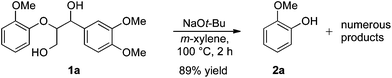
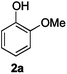
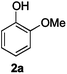
For the determination and analysis of lignin transformation pathways it is common to start with lignin model compounds, which contain the most significant binding motifs of the natural biopolymer.14,21,22 Along those lines, numerous reports have recently focused on selective oxidative or reductive degradation processes catalysed by transition metals.23,24 However, these methods generally require high catalyst loadings, and they suffer from incompatibilities with air, moisture and typical impurities present in the natural products. A simple and efficient method has recently been described by Hartwig, who reported the cleavage of lignin model compound 1a. Treatment of 1a in xylene with an excess of sodium tert-butoxide at 100 °C led to phenol 2a together with various unidentified by-products (Scheme 1).24
 | ||
| Scheme 1 | ||
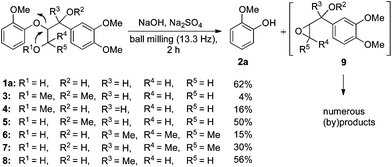
Searching for alternative approaches, we wondered about mechanochemical strategies for the degradation of lignin. In general, those have been known for a long time, but commonly they were applied for particle size reduction as a pretreatment for a chemical depolymerisation process performed under reaction conditions involving solvents in combination with elevated pressure and/or high temperature.12,16,17 Because strongly basic solutions have also been applied in lignin cleavage reactions,20,25,26 we wondered about a combination approach and questioned if solvent-free ball milling in the presence of a (solid) base could be used for lignin degradation (Scheme 2). To the best of our knowledge, this has never been studied although it appeared as an obvious advantageous option. Here, we describe the initial fine-tuning of the ball milling process using lignin model compounds and demonstrate the applicability of the method for the degradation of both technical organosolv lignin and beech wood. The latter two transformations are particularly interesting because they illustrate the potential of the process to overcome the major difficulties in lignin and wood conversions associated with their inherent insolubilities in common solvents.
 | ||
| Scheme 2 | ||
2. Results and discussion
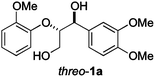
For the initial reactivity search and the subsequent optimisation of the reaction conditions, dilignol erythro-1a was chosen as a model substrate. It is structurally well defined and can readily be accessed in a diastereomerically homogeneous form following established routes developed in our laboratories.22 Unfortunately, our first attempts to degrade erythro-1a by grinding with sodium carbonate (as base and grinding auxiliary) in a planetary ball mill remained unsuccessful, and no conversion was observed after 2 hours (Table 1, entry 1). Assuming that a stronger base was needed, metal hydroxides were applied [in combination with sodium sulfate as a grinding auxiliary (Table 1, entries 2–5)27]. Under those conditions, dilignol erythro-1a reacted, and the formation of 2-methoxy phenol (2a) and a phenyl propane fragment resulting from C–O-bond cleavage was observed. Whereas with lithium hydroxide and potassium hydroxide the yield of 2a was low (only 16% and 29%, respectively), the use of sodium hydroxide gave 2a in remarkable 62% yield.|
|
|||||
|---|---|---|---|---|---|
| Entry | No. of balls | Freq. (Hz) | Base (equiv.) | Time (min) | Yieldb (%) |
| a Use of 0.2 mmol of 1a and 1 g of Na2SO4 (as grinding auxiliary). b Determined by GC using 4-methoxyphenol as an internal standard. c No additional auxiliary. d Used as a 60% suspension in mineral oil. | |||||
| 1 | 30 | 13.3 | Na2CO3 (9.5)c | 120 | 0 |
| 2 | 30 | 13.3 | LiOH (3.5) | 120 | 16 |
| 3 | 30 | 13.3 | KOH (3.5) | 120 | 29 |
| 4 | 30 | 13.3 | NaOH (3.5) | 120 | 62 |
| 5 | 30 | 13.3 | Ca(OH)2 (3.5) | 120 | 0 |
| 6 | 30 | 13.3 | NaH (3.5)d | 120 | 47 |
| 7 | 30 | 13.3 | NaOt-Bu (3.5) | 120 | 70 |
| 8 | 30 | 13.3 | NaOH (1.0) | 120 | 27 |
| 9 | 30 | 13.3 | NaOH (2.0) | 120 | 40 |
| 10 | 30 | 13.3 | NaOH (3.0) | 120 | 52 |
| 11 | 30 | 13.3 | NaOH (5.0) | 120 | 71 |
| 12 | 30 | 13.3 | NaOH (10.0) | 120 | 76 |
| 13 | 30 | 13.3 | NaOH (3.5) | 5 | 2 |
| 14 | 30 | 13.3 | NaOH (3.5) | 15 | 16 |
| 15 | 30 | 13.3 | NaOH (3.5) | 60 | 43 |
| 16 | 30 | 13.3 | NaOH (3.5) | 200 | 70 |
| 17 | 30 | 13.3 | NaOH (3.5) | 720 | 82 |
| 18 | 30 | 13.3 | NaOH (10.0) | 720 | 94 |
| 19 | 15 | 13.3 | NaOH (3.5) | 120 | 37 |
| 20 | 45 | 13.3 | NaOH (3.5) | 120 | 54 |
| 21 | 60 | 13.3 | NaOH (3.5) | 120 | 52 |
| 22 | 30 | 10.0 | NaOH (3.5) | 120 | 23 |
| 23 | 30 | 16.7 | NaOH (3.5) | 120 | 56 |
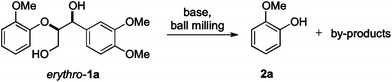
Calcium hydroxide was ineffective. Also sodium hydride and sodium tert-butoxide could be applied leading to 2a in 47% and 70% yield, respectively (Table 1, entries 6 and 7). However, considering the future perspective of lignin and wood conversions, the latter strong bases appeared less attractive for potential scale-ups. Thus for further studies, sodium hydroxide was selected as a base.
Until then, 3.5 equiv. of the base in combination with a standard quantity of the grinding auxiliary had been applied. With less sodium hydroxide the yield of 2a was lower (Table 1, entries 8–10). Although increasing the amount of the base to 5 and 10 equiv. improved the yield of 2a (up to 76%), it was decided to continue the study with the original base quantity (3.5 equiv.). Longer reaction times led to higher yields (Table 1, entries 13–17), and after 12 h, 2a was obtained in 82% yield. Also here, increasing the base loading to 10 equiv. had a positive effect and 2a could be isolated in 94% yield (Table 1, entry 18).
Finally, the protocol was optimised by improving the mechanical grinding properties of the ball milling process. Three points, which were expected to have an influence on the degradation process, were addressed: first, the grinding bowl topology, second, the number of balls, and third, the grinding speed. The best result was achieved when the reaction was performed in the grinding bowl with a diameter of 4.8 cm using 30 balls and a rotational speed of 13.3 Hz. Use of a grinding bowl with a smaller diameter (2.6 cm), applying more or less balls (Table 1, entries 19–21), or reducing the rotational speed (to 10.0 Hz; Table 1, entry 22) gave 2a in lower yields. At 16.7 Hz more by-products were formed (Table 1, entry 23). Apparently, the reaction required both mechanical stress/pressure (represented by the kinetic energy in the system) and a specific temperature range (resulting from friction). The former was achieved by using an appropriate bowl size, which guaranteed a high motional freedom of the grinding balls. The temperature range was reached by fine-tuning of the rotational speed, which promoted particle interactions and frictions leading to a process temperature of ca. 50 °C.
An agglutination of molten reactants could be prevented by using a grinding auxiliary. Sodium sulfate proved the best. Whereas sodium chloride could also be used as a grinding auxiliary, the presence of calcium chloride inhibited the reaction. Silica, basic aluminum oxide and molecular sieves led to more unselective side reactions.
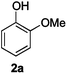
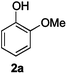
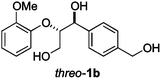
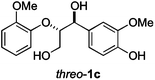
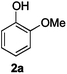
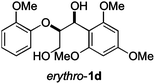
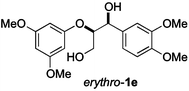
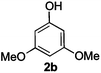
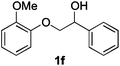
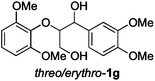
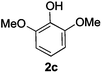
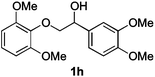
Next, the degradation method was applied to other lignin model compounds such as dilignols 1a–e, g and the less complex monolignols 1f and 1h (Table 2). Because product 2a was volatile and unstable on silica, the yields were initially determined by GC analysis for reactions on a 0.2 mmol scale. The results showed that neither steric hindrance nor the presence of multi-methoxy groups significantly affected the cleavage reactions. The solid dilignols erythro-1a and threo-1b and monolignols 1f and 1h gave higher yields than resin-like threo-1a, threo-1c, and erythro-1d, e, g. Performing reactions on a 0.8 mmol scale (Table 2, entries 1, 8 and 9) allowed us to isolate the products, and in this manner, 2a was obtained in 67% yield from erythro-1a (Table 2, entry 1). Starting from erythro-1g and 1h product 2c was isolated in 60% and 90% yield, respectively (Table 2, entries 8 and 9).
| Entry | Model compound | Product | Yieldb (%) |
|---|---|---|---|
| a Use of 0.2 mmol of 1, 3.5 equiv. of NaOH, 1 g of Na2SO4, 30 balls, 720 min, 13.3 Hz. b Determined by GC using 4-methoxyphenol as an internal standard. The values given in parentheses refer to the amount of isolated phenol obtained from reactions performed on a 0.8 mmol scale. | |||
| 1 |
|
|
82 (67) |
| 2 |
|
|
57 |
| 3 |
|
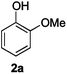
|
91 |
| 4 |
|
|
57 |
| 5 |
|
|
67 |
| 6 |
|
|
57 |
| 7 |
|
|
84 |
| 8 |
|
|
(60) |
| 9 |
|
|
(90) |
The data presented in Table 2 allow a first mechanistic interpretation, which we intended to validate by studying specifically designed model compounds 3–8. Compared to the originally applied mono- and dilignol derivatives 1, various potential deprotonation sites were blocked by the presence of methyl groups in those derivatives. Assuming that the degradation process began with a base-mediated deprotonation of the acidic alcoholic hydroxyl groups, the results obtained with methyl ethers 3–5 were particularly important. As shown by the data in Fig. 2, the yield of 2a was significantly reduced to 4% and 16%, respectively, when the primary hydroxyl group was methylated as in 3 and 4. In contrast, methylation of the secondary hydroxyl group had only a minor impact as revealed by the degradation of 5, which gave 2a in 50% yield. The impact of C-methylations remained inconclusive. The high yield of 2a (56%) obtained in the cleavage of substrate 8 showed that a substitution at the benzylic position had only a minor effect on the degradation efficiency. In contrast, additional methyl groups in the side chain (as in compounds 6 and 7) significantly reduced the yield of 2a (to 15% and 30%, respectively). In those cases, however, the data interpretation is more complex because the reduced cleavage rate could also be due to a conformational change induced by the buttressing dimethyl-substitution at the former methylene group. In summary, all data suggested that the initial deprotonation at the primary hydroxyl group was followed by intramolecular cleavage of the β-ether bond leading to two products: first, an unstable epoxide 9 that underwent subsequent transformations affording numerous unidentified (by)products, and second, phenolate 2a, which could be isolated as a single component in pure form. This mechanistic proposal was in agreement with the one suggested for the classical kraft pulp process.18 Although the protons at the hydroxyl-bearing carbons of the lignol derivatives 1 were not essential for the cleavage, they had a significant influence on the reaction rate indicating the occurrence of subsequent deprotonation steps, which could trigger further inter- or intramolecular reactions.28
Next, the degradation of more complex systems was attempted. For this purpose, two samples of organosolv lignin (A and B) from different sources were applied as starting materials. The structural changes and the degradation progress under standardized reaction conditions (100 mg of lignin, 100 mg of sodium hydroxide, 2.5 g of sodium sulfate, 30 balls, 12 h at 13.3 Hz) were determined by 2D NMR (HSQC) analysis of the samples before and after the grinding. The spectra interpretation was based on the excellent report by Sun et al.17 The signal intensities before and after the treatment of the compounds were normed relative to the content of aromatic protons (δF1 = 100–120 ppm, δF2 = 7.5–6.0 ppm). Those signals were locally isolated and their intensity was expected to remain unchanged during the cleavage reaction. For the quantification the following isolated signals were used: A-α, A-β, B-α, B-β, C-α, C-β (Fig. 1a–f).
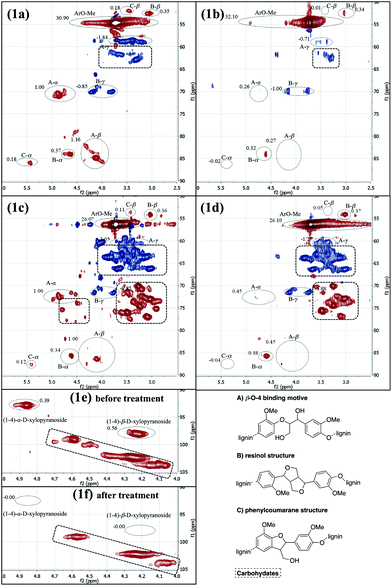 | ||
| Fig. 1 HSQC NMR spectra of lignin samples A and B; (1a) organosolv lignin A; (1b) organosolv lignin A after mechanochemical treatment; (1c) organosolv lignin B; (1d) organosolv lignin B after mechanochemical treatment; carbohydrate section of sample B before (1e) and after (1f) mechanochemical treatment; all spectra were recorded in DMSO-d6. For additional information see ESI.† | ||
Organosolv lignin sample A had been extracted from beech wood in a neutral ethanol/water organosolv process. It was then washed three times with hot water to remove the cellulose and hemicellulose residues. Consequently, the NMR spectrum showed that the carbohydrate content was very low (Fig. 1a). The ratio of binding motifs (β-O-4 : resinol : phenylcoumaran = 3.05![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.00:0.50) revealed that the β-O-4 linkage was the most abundant one in this sample. After treatment under standard reaction conditions (see above for details), the original ratio was changed to 0.80:1.00:0.04 (Fig. 1b). Hence, 76% of the β-O-4 binding motifs present in the untreated organosolv lignin A were cleaved during the process. The resinol binding motifs appeared to remain unaffected by the treatment.
:1.00:0.50) revealed that the β-O-4 linkage was the most abundant one in this sample. After treatment under standard reaction conditions (see above for details), the original ratio was changed to 0.80:1.00:0.04 (Fig. 1b). Hence, 76% of the β-O-4 binding motifs present in the untreated organosolv lignin A were cleaved during the process. The resinol binding motifs appeared to remain unaffected by the treatment.
Also the second organosolv lignin (sample B) originated from beech wood. It was obtained in the same manner as sample A, but it lacked the subsequent purification steps. Consequently, the NMR spectrum showed a significant poly-/oligomeric carbohydrate (cellulose, hemicellulose) content (Fig. 1c and 1e). Before the mechanochemical treatment the ratio of the binding motifs β-O-4 : resinol : phenylcoumaran was 2.83:1.00:0.33 (Fig. 1c), which was similar to the one of sample A. After the grinding, the ratio was 1.21:1.00:0.12 (Fig. 1d) indicating that 55% of the β-O-4 linkages had been cleaved. Interestingly, also the glycosidic bonds (CH–O–CH) had been broken, as revealed in Fig. 1f. Apparently, the grinding process also led to depolymerization of the carbohydrate content of sample B.
Encouraged by the finding that the linkages of both lignin and carbohydrates were cleaved, the applicability of the mechanochemical process for the degradation of naturally occurring dried beech wood was tested. Because the milled wood sample was insoluble in DMSO, it could not be analyzed by NMR before the mechanochemical treatment. After the grinding, however, the residue easily dissolved in DMSO or a mixture of THF and water. Analysis by 2D NMR (HSQC) revealed the typical lignin/carbohydrate fingerprint described by Sun.18 The ratio of the binding motifs for β-O-4 : resinol : phenylcoumaran was 1.73:1.00:0.00 (Fig. 2a), which was similar to the one of the treated organosolv lignin samples A and B. Although a high carbohydrate content was found (Fig. 2a), NMR signals of typical glycoside bindings were lacking (Fig. 2b). Apparently, also in this wood sample, the cellulose and hemicellulose had been degraded to low-molecular weight carbohydrates. Overall these observations supported our initial hypothesis that base-assisted grinding could also be applied for the degradation of natural, untreated wood.
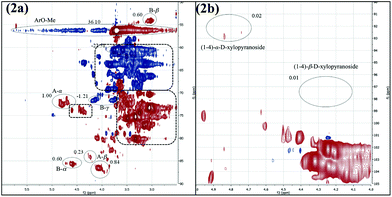 | ||
| Fig. 2 HSQC NMR spectra of milled beech wood. (2a) Section containing β-O-4 binding, resinol motifs, phenylcoumaran motifs and carbohydrates; (2b) carbohydrate section; all spectra were recorded in DMSO-d6. Assignments according to Fig. 1; for additional information see ESI.† | ||
A schematic representation of the suggested course of the mechanochemical degradation of hemicellulose and lignin in the original wood structure based on the reported spectroscopic and mechanistic results is shown in Scheme 3.
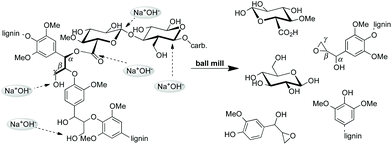 | ||
| Scheme 3 | ||
3. Conclusions
We have developed a base-assisted ball milling process and demonstrated its potential as a mechanochemical technique for the degradation of lignin and wood. Bonds in lignin are cleaved to provide fragments of lower molecular weight, and cellulose and hemicellulose are depolymerised to yield monomeric carbohydrates. Studying model compounds led to an understanding of the underlying cleavage processes, which in lignin predominantly involve the breaking of β-O-4 linkages. NMR spectroscopy indicated that also cellulose and hemicellulose in organosolv lignin and beech wood were degraded. The findings are in good agreement with those of solvent-based procedures,16,18 and they expand the existing technology to a new dimension of biomass degradation. Under transition metal- and solvent-free conditions bonds in both lignin and carbohydrates are cleaved using inexpensive, readily available reagents. Currently, the applied base quantity is still too large to be applied on an industrial scale, but we envision that it can be reduced by subsequent reaction process optimisations.4. Experimental section
4.1. Synthetic procedures
Substrates 1a–e were prepared by following a reported procedure.22 An alternative protocol was used for the synthesis of 1f.23b The syntheses and structural characterisations of dilignol 1g, monolignol 1h and dilignol derivatives 3–8 are described in the ESI.†4.2. Procedure for the cleavage of lignin model compounds in the ball mill
4.3. Degradation of organosolv lignin samples A and B in the ball mill
Organosolv lignin sample B was supplied by the “Aachener Verfahrenstechnik (AVT)”, Aachen (Serafin Stiefel, Simon Roth) in collaboration with the “Max Planck Institute”, Mühlheim. It was derived from an organosolv process using beech wood and ethanol–water = 50/50 without an acid catalyst. The lignin was precipitated with water and used without further purification.
4.4. Procedure for the degradation of milled beech wood in the ball mill
Beech wood toothpicks were dried in a vacuum for one week at room temperature. Subsequently, they were cut and ground in a planetary ball mill. The grinding procedure constitutes as follows: the grinding bowl (20 mL, ZrO2) was charged with the wood material and with 30 balls (5 mm, ZrO2). The material was ground in 40 intervals, each 5 min grinding at 8.3 Hz, 5 min pause, to prevent a temperature increase.A grinding bowl (20 mL, ZrO2) was charged with NaOH (150 mg), milled beech wood (100 mg), Na2SO4 (2.5 g) and 30 balls (5 mm, ZrO2). Then, the mixture was ground in a planetary ball mill (12 h, 13.3 Hz) and subsequently neutralized with aqueous acetic acid (1 M). After the slurry was ground for 1 min (13.3 Hz), it was dissolved in water (10 mL) and THF (20 mL). In a round bottom flask the solvent was removed in a vacuum and DMSO-d6 (5 mL) was added. The mixture was stirred vigorously for 20 min at 60 °C and filtered through a pad of silica gel to remove inorganic salts and water. The silica gel was washed with DMSO-d6 (3 × 1 mL). Subsequently, the mixture was concentrated to 0.7 mL and transferred to an NMR tube to be analyzed by NMR spectroscopy.
Acknowledgements
The DFG funding within the Cluster of Excellence “Tailor Made Fuels from Biomass” (TMFB) at RWTH Aachen University is greatly appreciated. We thank Dr. Jürgen Puls from the Institut für Holztechnologie und Holzbiologie (HTB), Hamburg, Dr. Roberto Rinaldi from Max Planck Institut für Kohlenforschung, Mülheim a.d.R., Prof. Dr.-Ing. Antje Spieß, Prof. Dr.-Ing. Matthias Wessling, Serafin Stiefel and Simon Roth from the Aachener Verfahrenstechnik (AVT), RWTH Aachen University, for the organosolv lignin samples A and B, respectively. For contributions to the optimization process, we acknowledge Marc Schmitz, and we thank Dr. Christoph Raeuber (RWTH Aachen University) for supporting the NMR investigations.Notes and references
- For very recent reviews, see: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. Ch. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) T. Friscic, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- For overviews on the use of ball milling in organic synthesis, see: (a) B. Rodríguez, A. Bruckmann, T. Rantanen and C. Bolm, Adv. Synth. Catal., 2007, 349, 2213–2233 CrossRef; (b) A. Bruckmann, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131–1141 RSC; (c) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondrushka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC; (d) A. Stolle, B. Ondrushka, A. Krebs and C. Bolm, in Innovative Catalysis in Organic Synthesis, ed. P. G. Andersson, Wiley-VCH, Weinheim, Germany, 2012, pp. 327–349 Search PubMed.
- (a) R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767–772 RSC; (b) F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894–1899 RSC; (c) R. Thorwirth, F. Bernhardt, A. Stolle, B. Ondruschka and J. Asghari, Chem.–Eur. J., 2010, 16, 13236–132421 CrossRef CAS; (d) For a more general review, see: R. B. N. Baig and R. S. Varma, Chem. Soc. Rev., 2011, 40, 1559–1584 Search PubMed.
- For recent work on processing of lignocellulosic material under microwave irradiation, see: J. Xu, J. Jiang, C. Hse and T. F. Shupe, Green Chem., 2012, 14, 2821–2830 RSC.
- Z. V. Todres, Organic Mechanochemistry and its Practical Applications, Taylor & Francis, Boca Raton, 2006 Search PubMed.
- (a) J. J. Gilman, Science, 1996, 274, 65 CrossRef CAS; (b) H. Watanabe, E. Matsui, Y. Ishiyama and M. Senna, Tetrahedron Lett., 2007, 48, 8132–8137 CrossRef CAS.
- (a) R. A. Kerr, Science, 2011, 331, 1510–1511 CrossRef CAS; (b) D. J. Murphy and C. A. S. Hall, Ann. N. Y. Acad. Sci., 2011, 1219, 52–72 CrossRef; (c) A. Demirbas, Energy Convers. Manage., 2009, 50, 2239–2249 CrossRef CAS; (d) G. H. Vogel, Chem. Eng. Technol., 2008, 31, 730–735 CrossRef CAS.
- C. Somerville, H. Youngs, C. Taylor, S. C. Davis and S. P. Long, Science, 2010, 329, 790–792 CrossRef CAS.
- (a) F. Cherubini and A. H. Stroemman, Energy Fuels, 2010, 24, 2657–2666 CrossRef CAS; (b) E. S. Jensen, M. B. Peoples, R. M. Boddey, P. M. Gresshoff, H. Hauggaard-Nielsen, B. J. R. Alves and M. J. Morrison, Agron. Sustain. Dev., 2012, 32, 329–364 CrossRef CAS; (c) C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935–5940 CrossRef CAS.
- H. Michel, Angew. Chem., Int. Ed., 2012, 51, 2516–2518 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- (a) J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS; (b) F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS; (c) J. C. Serrano–Ruiz, R. Luque and A. Sepúlveda–Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC; (d) Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387–1390 CrossRef CAS.
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS.
- (a) For the treatment of wood, see: A. Björkman, Nature, 1954, 174, 1057–1058 CrossRef; (b) For mechanochemical reactions of lignin model compounds, see: Z.-H. Wu, M. Sumimoto and H. Tanak, Holzforschung, 1994, 48, 395–399 CrossRef CAS.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS.
- F. S. Chakar and A. Ragauskas, J. Ind. Crop. Prod., 2004, 20, 131–141 CrossRef CAS.
- (a) C. Somerville, S. Bauer, G. Brininstool, M. Facette, T. Hamann, J. Milne, E. Osborne, A. Paredez, S. Persson, T. Raab, S. Vorwerk and H. Youngs, Science, 2004, 306, 2206–2211 CrossRef CAS; (b) H. Nimz, Angew. Chem., Int. Ed., 1974, 13, 313–321 CrossRef.
- S. Rabe, M. Nachtegaal, T. Ulrich and F. Vogel, Angew. Chem., Int. Ed., 2010, 49, 6434–6437 CrossRef CAS.
- (a) S. Kawai, K. Okita, K. Sugishita, A. Tanaka and H. Ohashi, J. Wood Sci., 1999, 45, 440–443 CrossRef CAS; (b) T. Kishimoto, Y. Uraki and M. Ubukata, Org. Biomol. Chem., 2008, 6, 2982–2987 RSC.
- J. Buendia, J. Mottweiler and C. Bolm, Chem.–Eur. J., 2011, 17, 13877–13882 CrossRef CAS.
- (a) S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS; (b) J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS; (c) S. K. Hanson, R. Wu and L. A. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS; (d) B. Sedai, C. Díaz–Urrutia, R. T. Baker, R. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS; (e) S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, A. D. Sutton and D. L. Thorn, J. Am. Chem. Soc., 2009, 131, 428–429 CrossRef CAS; (f) P. Kelley, S. Lin, G. Edouard, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 5480–5483 CrossRef CAS; (g) S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, L. A. Silks and D. L. Thorn, J. Am. Chem. Soc., 2010, 132, 17804–17816 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS.
- (a) C. Hagström-Näsi and E. Sjöström, J. Wood Chem. Technol., 1988, 8, 299–311 CrossRef; (b) J. E. Miller, L. Evans, A. Littlewolf and D. E. Trudell, Fuel, 1999, 78, 1363–1366 CrossRef CAS; (c) S. Karagöz, T. Bhaskar, A. Muto and Y. Sakata, Fuel, 2004, 83, 2293–2299 CrossRef; (d) A. Vigneault, D. K. Johnson and E. Chornet, Can. J. Chem. Engin., 2007, 85, 906–916 CAS; (e) Y. Chen, R. R. Sharma-Shivappa, D. Keshwani and C. Chen, Appl. Biochem. Biotechnol., 2007, 142, 276–290 CrossRef CAS; (f) S. Nenkova, T. Vasileva and K. Stanulov, Chem. Nat. Compd., 2008, 44, 182–185 CrossRef CAS; (g) U. Wongiriwan, Y. Noda, C. Song, P. Prasassarakich and Y. Yeboah, Energy Fuels, 2010, 24, 3232–3228 CrossRef; (h) V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem.–Eur. J., 2011, 17, 5939–5948 CrossRef CAS.
- For base cleavage studies involving lignin model compounds, see: (a) G. Miksche, Acta. Chem. Scand., 1973, 27, 1355–1368 CrossRef CAS; (b) K. Poppius, Acta. Chem. Scand., 1984, 38, 611–616 CrossRef.
- The amount of grinding auxiliary cannot be reduced on a small scale, because the grinding beaker needs a minimum filling for optimal performance. We assume that less can be used upon scale-up or by altering the reactor design.
- (a) E. L. Clennan and G. L. Pan, Org. Lett., 2003, 5, 4979–4982 CrossRef CAS; (b) A. Nagaki, E. Takizawa and I. Yoshida, J. Am. Chem. Soc., 2009, 131, 1654–1655 CrossRef CAS; (c) V. Capriati, S. Florio and R. Luisi, Chem. Rev., 2008, 108, 1918–1942 CrossRef CAS; (d) Y. Ma, Heteroat. Chem., 2002, 13, 310–315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36456e |
| This journal is © The Royal Society of Chemistry 2013 |